
Abstract
Innate immunity is an evolutionarily ancient form of host defense that serves to limit infection. The invading microorganisms are detected by the innate immune system through germline-encoded PRRs. Different classes of PRRs, including TLRs and cytoplasmic receptors, recognize distinct microbial components known collectively as PAMPs. Ligation of PAMPs with receptors triggers intracellular signaling cascades, activating defense mechanisms. Despite the fact that Gram-negative bacteria and parasitic protozoa are phylogenetically distant organisms, they express glycoconjugates, namely bacterial LPS and protozoan GPI-anchored glycolipids, which share many structural and functional similarities. By activating/deactivating MAPK signaling and NF-κB, these ligands trigger general pro-/anti-inflammatory responses depending on the related patterns. They also use conservative strategies to subvert cell-autonomous defense systems of specialized immune cells. Signals triggered by Gram-negative bacteria and parasitic protozoa can interfere with host homeostasis and, depending on the type of microorganism, lead to hypersensitivity or silencing of the immune response. Activation of professional immune cells, through a ligand which triggers the opposite effect (antagonist versus agonist) appears to be a promising solution to restoring the immune balance.
Introduction
Microorganisms are so widespread in the world that one can even be tempted to say that we live in a sea of microorganisms. From the first moments of life, when we take the first breath of air and swallow the first sip of milk, microorganisms enter our body to accompany us to the end of our lives. Both outside and inside, the human organism is colonized by a huge array of microorganisms: mostly bacteria, but also fungi, viruses, and other microbes. Together these are referred to as the human microbiota, and all the genes of the human microbiota form the microbiome; however, colloquially the two terms are often used interchangeably. Based on the results of many studies, it has been established that the microbiota influences both the duration and quality of human life by providing nutrients and vitamins for cells, preventing colonization by harmful bacteria and viruses, or “programming the immune system.” But this ally community, which is subjected to constant changes over time and under the influence of environmental conditions, stress, or drugs delivered to the body, can become enemies and be linked to a plethora of morbid conditions, from obesity to anxiety, asthma or autoimmune diseases, to some infections.1–5
At the basis of these processes, the mechanisms of mutual recognition in host–microbe interactions have been developed over centuries, allowing the parties to detect one another. From the host’s perspective, the function of these mechanisms is to activate the immune system. More specifically, the host has been armed with germline-encoded sensors, so-called PRRs, which recognize PAMPs. Because PAMPs have evolved into conserved molecules on pathogens, they have unique molecular structures which are recognized by dedicated PRRs. As a consequence of PAMP–PRR interactions downstream signal transduction pathways are triggered which ultimately result in the activation of gene expression and synthesis of a broad range of molecules, including cytokines, chemokines, cell adhesion molecules, and immunoreceptors, aimed at limiting the survival of the intruders. 6
According to the Global Burden of Disease (GBD) Study, the vast majority of one million of parasites-related deaths result from protozoan infections. One of the world’s most prevalent infectious diseases is malaria, caused by the parasitic protozoa Plasmodium spp. Malaria was the major killer in the group of protozoan diseases in the year 2016, with around 720,000 deaths, followed by leishmaniasis, which caused the deaths of 20,000 people. 7 These infections are endemic in areas inhabited by over one billion people. Also, infections caused by resistant Gram-negative bacteria are becoming increasingly prevalent, posing a serious threat to public health worldwide, because they are difficult to treat and are associated with high morbidity and mortality rates. 8 While much effort is being made toward a better understanding of the sensing of Gram-negative bacteria by specialized phagocytic cells, relatively few corresponding studies have been conducted on protozoa.
This review highlights current knowledge of the major glycoconjugates associated with Gram-negative bacteria and protists, i.e. LPS and GPI-anchored glycolipids, respectively, and the response of phagocytic cells against these Ags. The possibility of using these glycoconjugates and their chemical derivatives in restoring host homeostasis, disturbed during pathogen-related diseases, is also considered.
Structure, features, and function of LPS and GPI glycolipids
Although bacterial LPS and protozoan GPI-anchored glycolipids are different general “patterns,” they share some similarities in structure, function, and physicochemical properties.
LPS is a major component of the external leaflet of the outer membrane of Gram-negative bacteria. Chemically, it is a large amphipathic molecule consisting of a polar polysaccharide part, which includes both the O-Ag and the core region, and the nonpolar lipid A component. LPS that contain all three regions are called smooth (S)-form LPS, while LPS lacking the O-Ag are referred to as rough (R)-form LPS or LOS.
9
The O-specific chain is the outermost part of the LPS, built of repeating oligosaccharide subunits containing from one to eight carbohydrate residues. The O-Ag, in addition to neutral sugars, i.e. hexoses and hexosamines, also contains uronic acids, 6-deoxyhexoses, and 3,6-dideoxyhexoses.10–13 The structures of the repeating subunits (determined by their stereo configuration), their sequences, the positions of bonds, and the location of non-sugar substituents condition intrastrain diversity within a species and create the characteristic somatic Ags unique to each strain.
14
,
15
The core oligosaccharide, located in the middle of LPS and linked to both the O-Ag and lipid A (via a 3-deoxy-
A tremendous diversity of glycoconjugates are abundant and ubiquitous on the surface of many protozoan parasites, GPI-anchored glycolipids have been proposed as their prominent representatives. 31 The basic structure of the GPI anchor is highly conserved. The anchor is built of a lipophilic portion coupled via an intermittent inositol phosphate to a core tetrasaccharide glycan with the sequence Man(α1-2)Man(α1-6)Man(α1-4)GlcN(α1-6)-myo-inositol-P. This backbone is common to all GPIs found in eukaryotes, but in protozoan parasites, it can be variously modified by the presence of: (i) distinct lipid components (diacyl glycerol, alkyl/acyl glycerol, lyso-alkylglycerol or ceramide type), (ii) additional sugar moieties on the third and/or first Man, (iii) extra ethanolamine phosphate (EthP) groups on the carbohydrate moiety, (iv) an acyl moiety on C2 of inositol, and/or (v) aminoethylphosphonate (AEP) on GlcN.31–33 The non-N-acetylated GlcN is a unique component of the glycan part of GPIs. 32 Generally, the aliphatic residues are responsible for embedding GPIs into the outer leaflet of the plasma membrane, but their unique chemical composition, which is specific to different cell types, organisms and stages of their life cycle, endows them with the ability to exert different biological activities. 34 In a majority of eukaryotes, GPI modification of proteins is essential for their proper function, as it allows proteins to associate with membrane microdomains or lipid rafts. 32 In contrast to eukaryotic GPIs, many protozoan GPIs are attached to glycopolymers, e.g. the lipophosphoglycans (LPGs) of Leishmania and Trichomonads, the lipoarabinogalactan (LAG) of Crithidia fasciculata, or the lipophosphonoglycan (LPG-Ac) of Acanthamoeba. Alternatively, they can exist in the membrane as free glycophospholipids, e.g. the glycoinositolphospholipids (GIPLs) of Leishmania, Trypanosoma cruzi, Leptomonas, Herpetomonas, Phytomonas, and Toxoplasma. 31 , 33 , 35
The best characterized LPGs are those of Leishmania promastigotes. They have a conserved glycan core region of Gal(α1-6)Gal(α1-3)Galf(β1-3)[Glc(α1)-PO4]Man(α1-3)Man(α1-4)-GlcN(α1) linked to a 1-O-alkyl-2-lyso-phosphatidylinositol anchor. A conserved domain consisting of a Gal(β1-4)Man(α1)-PO4 backbone of repeat units (n = ∼15–30) is attached to this moiety. 36 A distinguishing feature of LPGs which is responsible for the polymorphisms among Leishmania spp. is the variable sugar composition and sequence of branching sugars attached to the repeat units and cap structure. 37 Similarly to Leishmania LPG, the LPG of trichomonads consists of at least three regions: a ceramide-containing anchor, a glycan core, and oligosaccharide repeat units. 31 , 38 The recently determined structure of the glycan core indicates that it is unique among protozoa as it is built of a rhamnan backbone substituted with long side chains and contains an inositol residue at the reducing end, linked to a 4-linked α-glucuronic acid (GlcA) residue. 39
GIPLs, which are surface Ags of Leishmania amastigotes, are low molecular mass molecules similar to LPG in sharing a common lipid backbone and a glycan motif containing up to seven sugars. 40 Leishmania GIPLs can be classified as type I, II, and hybrids. Type I (Man-rich) GIPLs are found in Leishmania infantum, L. donovani, L. tropica, and L. aethiopica. The Gal-rich type II GIPLs are common in L. braziliensis, L. major, L. mexicana, and L. panamensis. Finally, hybrid GIPLs have mixed structural features of type I and II. They are found in L. mexicana and L. donovani. 41
As it appears from the above descriptions of the chemical structures of LPS and GPI-anchored glycolipids, they share some similarities as they both are amphiphilic molecules consisting of a nonpolar lipid part linked to polar sugar moieties. The phosphorylated repeating units of the glycan constituent in LPGs resemble LPS O-Ags found in Gram-negative bacteria. LPS and GPI glycolipids, which come in contact with the external environment, provide a barrier against surrounding stress factors, which makes them indispensable for bacterial and protist survival in various distinct ecosystems. 9 , 31 Moreover, both these glycoconjugates take part in microbial self-defense against the protective action of the host immune system, in particular, counteracting complement activity, phagocytosis, and oxidative burst. 29 , 38 ,42–46 This activity is confirmed by the fact that mutants defective in expressing those molecules or with a changed glycan length are highly susceptible to extra- and intracellular killing. 29 , 43 , 45
The bactericidal action of compliment is based primarily on the activation of the membrane attack complex (MAC), which causes cell lysis. The MAC complex binds at the tips of polysaccharide chains, and due to its hydrophobic character easily penetrates bacteria with hydrophobic surfaces, i.e. rough forms (R). 10 The hydrophilic surface of smooth bacteria (S) with O-specific LPS significantly impedes or even prevents MAC deposition on the outer membrane. Moreover, the long O-Ags projecting outwards from the overall bacterial cell surface may limit the incorporation of MAC components into their target site. In protozoa, a similar function is played by the phosphoglycan (PG) moiety, which forms part of LPG or proteophosphoglycans (PPGs) of Leishmania spp. LPG moieties of metacyclic promastigotes of Leishmania with very long PG repeats constitute a protective barrier against complement, while PPGs, which have a lower number of PG moieties, activate this extracellular mechanism of host defense. 43 , 47
Another common feature of LPS and GPI anchors of proteins is that they bind with high affinity to cholesterol-containing raft model membranes. 48 The possibility of association with lipid-like detergent resistant membranes has also been demonstrated for LPG and GIPLs of Leishmania. 49 What differentiates LPS molecules and GPI anchored glycolipids most is that they have distinct conservative “patterns” which include lipid A or the glycan core, respectively. The presence of these conservative elements suggests these conjugates have different ways of alerting phagocytic cells. 50
Immune sensing of LPS in professional phagocytes
Induction of antibacterial defense by triggering inflammatory host responses is a crucial function of the innate immune system in response to invading bacteria. This reaction prevents the spreading of pathogens and suppresses their growth. 9 After recognizing PAMPs, PRRs induce several extracellular activation cascades and downstream signaling, which ultimately leads to the expression of a plethora of genes and promotes secretion of pro-inflammatory cytokines, as well as activating oxidative burst and the production of antimicrobial peptides (AMPs). 9 , 51
As an archetypal PAMP and a major recognition marker of Gram-negative bacteria, LPS triggers the activation of specialized phagocytes during bacterial invasion. It is also the target of bactericidal peptides and complement components. 48 Studies in recent years have uncovered a diverse set of eukaryotic receptors that recognize LPS: TLRs, G-protein-coupled receptors, integrins, receptor-like kinases, and caspases. 52 However, responses to extracellularly localized LPS molecules depend mainly on TLR4, which is the only unequivocally confirmed TLR receptor sensing this ligand, while the sensing of cytosol-localized LPS relies on caspases. 53 TLR4 is commonly expressed at low levels in non-immune cells, but a relatively high-level expression can be observed in innate immune cells, such as macrophages (Mϕs), dendritic cells (DCs), monocytes, and neutrophils. 54 , 55 Although these specialized phagocytes arise from common myeloid precursors, they show distinct responses to TLR4 activation induced by LPS, which underpin the balance between innate and adaptive immunity. 56 , 57 Mϕs are considered to be the primary targets of LPS. When activated, they first initiate inflammatory processes and then persist in the tissue, switching to an anti-inflammatory phenotype to restore tissue homeostasis. Conventional DCs, upon contact with LPS, acquire the ability to migrate, produce pro-inflammatory cytokines, undergo terminal differentiation with Ag presentation, and die by apoptosis. 55 Monocytes and neutrophils have also been reported as highly LPS-responsive cells. They are involved, through TLR4 signaling pathways, in both oxidative burst and induction of NO. 57
The sensing of the LPS molecule by PRRs can be localized on the cell membrane of professional phagocytes (e.g. TLR4, DC-SIGN, C-lectins), in their cytoplasm (murine caspase-11 or human -4, -5), and in body fluids (LPS-binding protein (LBP) and mannose-binding lectin (MBL)). In addition to LPS sensing, PRRs perform diverse other functions such as the initiation of major signaling pathways, presentation of PAMPs to other PRRs, and promotion of microbial uptake by phagocytosis. 58 To sum up, it is obvious that binding of LPS to different receptors activates distinct signal transduction pathways. However, binding of LPS to the receptors of the same type but localized on different immune cells triggers signal transduction pathways, both common for all cells and cell-type-specific ones.
Pathways of activation of TLRs by LPS
In higher organisms, extracellular LPS monomers which have detached from the aggregates present in body fluids or from the surface of bacteria are captured by LBP. LBP binds a variety of LPS chemotypes from rough and smooth strains of Gram-negative bacteria, circulating as micelles or free molecules, or incorporated into outer membrane vesicles (OMVs). 9 , 59 , 60 For signal transduction to occur, LPS/LBP complexes must be bound to cluster of differentiation 14 (CD14). 59 , 61 The glycoprotein CD14 acts as an LPS receptor either when anchored in the cell membrane (mCD14) or as a soluble molecule (sCD14) in serum. 62 CD14 relays the LPS/LBP complex to myeloid differentiation factor 2 (MD2), which is associated with TLR 4. Binding of LPS to TLR4/MD2 results in homodimerization of the receptor complex and initiation of an intracellular signaling cascade that drives broad MyD88- and TIR domain-containing adaptor inducing IFN-β (TRIF)-dependent gene transcription. 63 The delivery of LPS to TLR4 stimulates the synthesis of phosphatidylinositol 4,5-biphosphate (PIP2) to promote myddosome formation, i.e. the recruitment of adaptor proteins TIRAP and MyD88 via homophilic TIR–TIR interactions. 58 The MyD88-dependent pathway leads to the canonical production of transcription factors NF-κB and AP-1, followed by the expression of inflammatory cytokines with the participation of IFN regulatory factor 5 (IRF5), which is critical for the process. 52 LPS-induced TLR4 signaling can also occur via an alternative, MyD88-independent pathway (Figure 1). In this case, CD14 stimulates internalization of the activated TLR4 into endosome compartments. There, TLR4 interacts with the adaptor proteins TRAM and TRIF to induce the expression of NF-κB/IRF3-dependent type I IFN. 52 , 64 Some Gram-negative pathogens, such as Legionella pneumophila, Francisella tularensis, Helicobacter pylori, Coxiella burnetti, and Porphyromonas gingivalis, synthesize atypical LPSs which are poorly recognized by human TLR4, and can thus activate harmful proinflammatory responses.65–67 Signaling through the TLR2/MyD88 pathway has been reported for some of the above-mentioned bacteria. However, these results must be treated with great caution, since they rather resulted from the contamination of LPS preparations than are a rule. 65 ,68–70
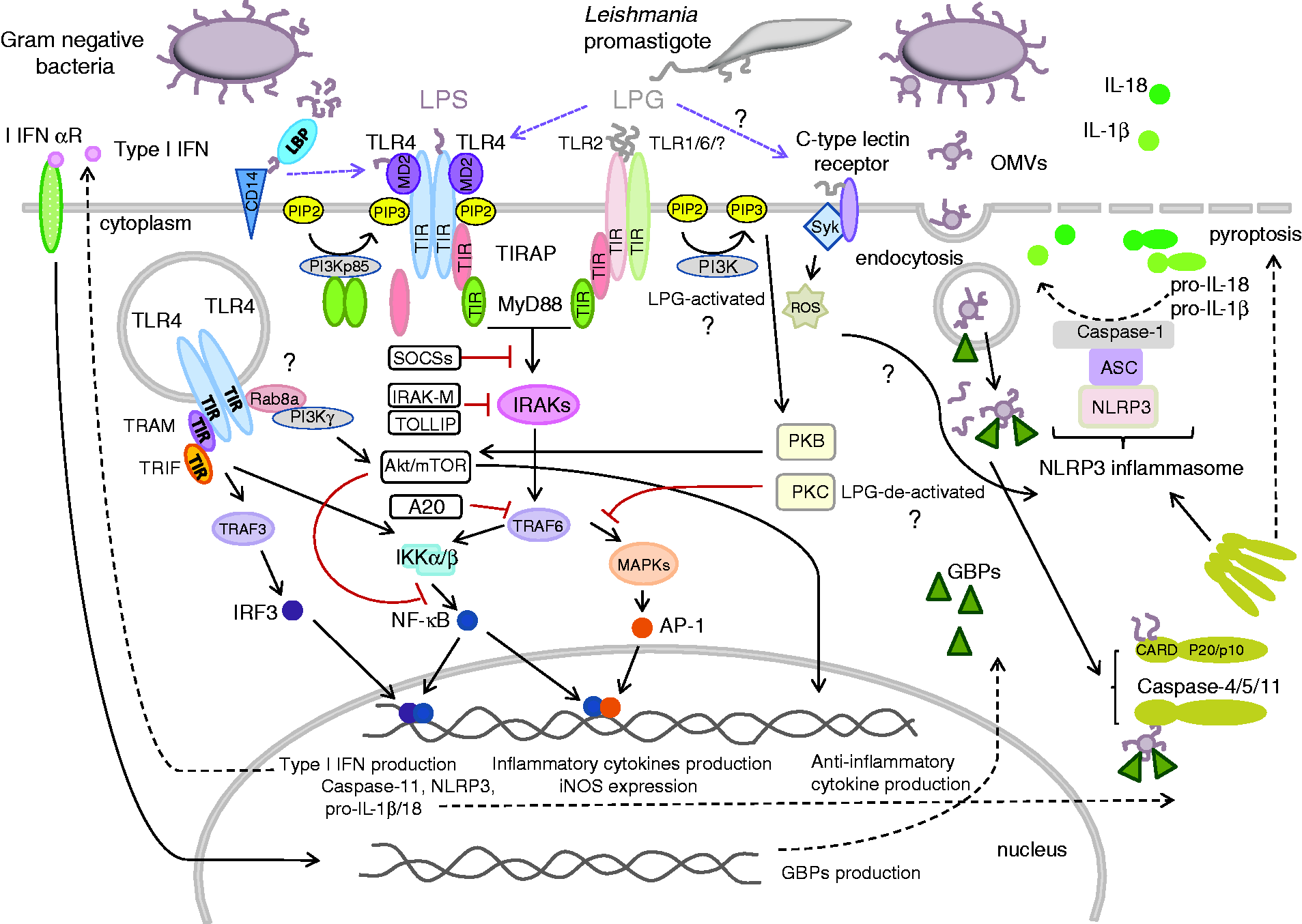
Main TLR-mediated signaling pathways triggered upon stimulation by LPS and LPG. Downstream of TLR4 and TLR2 receptors, both LPS and LPG can generate signals through a common MyD88-dependent pathway, while a MyD88-independent pathway is peculiar to TLR4 signaling. The activation of the canonical pathway results in early phase activation of the NF-κB factor and production of inflammatory cytokines. By contrast, signaling through the non-canonical pathway leads to late phase transformation of inactive NF-κB to an active form and production of type I IFNs. In primed Mϕs, both LPS and LPG can be sensed through NLRP3 inflammasome. Excessive immune response, which can be detrimental to the host, is regulated by negative feedback. The main role in the regulation of TLR2/4 signaling, and, hence, the production of IL-12 and excessive Th1 polarization, is played by the endogenous suppressors PI3Ks. The activation of the PI3K/Akt/mTOR pathway by both LPS and LPG results in the production of anti-inflammatory cytokines. Negative regulation of TLR2 and TLR 4 signaling also involves other suppressor molecules. For more details, see text.
It is known that both forms of LPS, rough and smooth, can signal through TLR4, but only the latter requires the involvement of CD14. In in vitro experiments with mouse mast cells and in a mouse model in vivo, R-LPS readily activated signaling through the TLR4/MD-2 CD14-independent pathway, while the S-form needed the assistance of both LPS-binding proteins: CD14 and LBP. 71 Since human neutrophils either express low amounts of mCD14 or lack it altogether, only the R-form LPS can activate them in the canonical pathway. The activation of neutrophils with S-form LPS is also possible, but only in a serum environment containing high concentrations of soluble CD14. 57
Recently, it has been shown that intracellular cytosolic LPS can also be positively sensed by PRRs. 19 The recognition occurs in the non-canonical inflammasome pathway. 72 The signal transduction cascade is directly mediated by the cytosolic LPS receptor murine caspase-11 or its human orthologs caspase-4 and -5. In this case, LPS functions as a molecular platform for the recruitment and activation of these inflammatory caspases. The lipid A component of LPS binds to the CARD motif of pro-caspase-11 and promotes its oligomerization and activation leading to: (i) pyroptosis, an inflammatory programmed cell death mode, and (ii) the secretion of pro-inflammatory cytokines such as IL-1β and IL-18. The cytokines begin to be produced upon activation of the canonical NLRP3 inflammasome. 60 , 63 , 73 Until recently, the ways by which LPS gains access to the cytosol have been poorly understood. Several hypothesis have been formulated to explain this event, including: (i) membrane damage, inflicted in the host cells by virulence-associated secretion systems or pore-forming toxins, allowing the entry of pure LPS moieties, (ii) the delivery of LPS with remnants of bacterial cells disintegrated by phagocytosis, and (iii) the transfer of LPS inside OMVs internalized via endocytosis. 52 , 74 , 75 The participation of OMVs has lately been confirmed, and the question how they are delivered into the cytosol has been solved. 76 Unexpectedly, the non-canonical inflammasome intracellular LPS-sensing pathway, commonly considered to be TLR4-independent, was found to rely indirectly on the activation of the TLR4/TRIF signaling pathway. 60 Gu et al. reported that the TRIF-mediated cytosolic delivery of LPS from OMVs depends on the production of I IFNs and the expression of guanylate-binding proteins (GBPs). 77 Together, these findings provide novel insights into how the host coordinates extracellular and intracellular LPS sensing to orchestrate immune responses during Gram-negative bacterial infection. They also show a novel function of GBPs as a hub between cell-autonomous and innate immunity. 60
In most cases, the process of pathogen recognition involves several PRR systems acting simultaneously or sequentially to modulate the TLR4-signaling pathway. Although LPS responses rely mainly on a membrane spanning complex formed by TLR4/MD-2, several molecules have been shown to act as co-receptors and/or accessory molecules. One such co-regulator, contributing to the LPS signaling cluster, is complement receptor 3 (CR3), expressed on the surface of DCs and Mϕs. 78 In DCs, which express lower levels of CD14 and TLR4 than do Mϕs, the CD11b integrin (component of CR3), capable of binding LPS, serves as a positive regulator of TLR4-induced signaling pathways. CD11b has been shown to be indispensable in two steps of the MyD88-independent pathway: the initial TLR4 endocytosis and the subsequent TRIF-mediated signaling in the endosomes. The integrin has also been found to play a role in MyD88-dependent signaling in DCs. To sum up, CD11b controls the trafficking and signaling functions of TLR4 in a cell-type-specific manner, being a key mediator of the adjuvant effect of LPS in DCs and a strong inhibitor of TLR responses in Mϕs. 56 Monocytes and neutrophils substantially upregulate the expression of CD11b under LPS stimulation regardless of its form (smooth or rough). 57
Negative regulation of the TLR4 signaling pathway
Dysregulated or excessive cytokine secretion may have serious or even fatal consequences in acute conditions such as sepsis, and in chronic inflammatory and autoimmune diseases. To ensure that this potentially destructive pathway is strictly regulated, the host responds to LPS-mediated activation of TLR4 in Mϕs and monocytes with coordinated release of pro-inflammatory cytokines and regulatory mediators. 79 TLR signaling can be controlled by intracellular regulators, down-regulation of the transcription and translation of TLR genes, or degradation of the proteins that build them (Figure 1). 80
Soluble TLRs (sTLR) have been proposed to constitute a first-line negative regulatory mechanism. The sTLR4 isoform has been identified by screening a mouse Mϕs cDNA library, and its role in the negative regulation of NF-κB activation and TNF-α production has been confirmed in vitro. The second line of defense consists of several intracellular negative regulators such as MyD88s (the short form of MyD88), IRAKM, SOCS1, NOD2, PI3K, TOLLIP, and A20, which are either expressed constitutively or induced by LPS. 80
Phosphatidylinositol-3-phosphate kinase (PI3K), expressed constitutively by many cells, has been reported to play a negative regulatory role in LPS signaling in the canonical and alternative pathways. 79 , 81 , 82 Laird et al. proposed a model for the formation of a complex involved in the regulation of signals downstream of the LPS-activated TLR4/TIRAP pathway. 81 According to this model, the subunits of the complex start to assemble upon TLR4-independent dimerization of MyD88, ultimately leading to the recruitment of class of IA PI3K kinase. In response to LPS-mediated receptor activation, this complex is subsequently recruited to TLR4 via TIRAP, and phosphorylated by a Src-kinase. The activated PI3K kinase consumes PIP2 in the membrane, thereby down-regulating TLR4 signaling by preventing the recruitment of TIRAP to the complex and impeding the formation of the myddosome. 81 In the alternative pathway involving TLR4/TRAM regulated endocytosis, IB PI3Kγ is recruited by GTPas Rab8a to dorsal ruffles on the surface of Mϕs. This complex is responsible for the activation of the Akt/mTOR pathway and decreasing the production of pro-inflammatory and increasing the production of anti-inflammatory cytokines. 82
Upon contact with LPS, monocytes can produce Myd88s, which is an antagonist of the IRAK phosphorylation pathway, and, as such, may play an important role in the regulation of the cellular response to IL-1- and LPS-induced NF-κB activation. 83 Other inhibitors of IRAK phosphorylation, include splice variants of IRAK kinases (IRAKM, IRAK2c, IRAK2d). Their inhibitory activity has been demonstrated in Mϕs from IRAKM-/- mice stimulated with LPS, which displayed intensive NF-κB and MAP kinase activation. 80 , 84 Likewise, Mϕs from SOCS1-deficient mice developed enhanced phosphorylation of STAT1, IκBα, p38, and JNK (c-Jun N-terminal kinase) in response to LPS stimulation. 80
LBP, which is the co-receptor of TLR4, has a dual, concentration-dependent role in the pathogenesis of Gram-negative bacterial sepsis. It has been proposed that when the LBP level in body fluids is low, the protein intercalates into cytoplasmic membranes of mononuclear cells (MNC) before it binds LPS. The association of the LBP/LPS complex with the membrane is necessary to enhance the activation of MNCs and the secretion of TNF-α, IL-1, IL-6, and other potent mediators. However, in the acute phase, a significant rise in serum LBP concentration leads to quick complexation with free ligands, a process that protects the complex from being incorporated into the membrane, thereby inhibiting the activation of MNCs. In this way, LBP neutralizes LPS and protects the host from septic shock. 59
LPS-mediated activation of PRRs other than TLRs
In addition to LPS-activated TLR4-mediated pathways, there exist other ways in which LPS can be directly detected by distinct PRRs. This applies mainly to DCs and an alternative way of LPS signaling through mCD14. In some DC cells, but not in Mϕs, the co-receptor CD14 (apart from transferring LPS to the TLR4/MD-2 complex and promoting its internalization into endosomal compartments) can initiate a transduction pathway which activates Src-family kinase and phospholipase C (PLC)g2, increases intracellular Ca2+ concentration, and initiates the translocation of calcineurin-dependent NF of activated T cells (NFAT) to the nucleus. The activation of this pathway is totally independent of TLR4 and other TLRs. Its main role is to induce apoptotic death of terminally differentiated DCs in order to prevent autoimmunity. 85 This provides a new role for CD14: the regulation of DC life cycle through LPS-stimulated activation of NFAT.
Innate immunity, which serves to limit microbial infection, triggers not only pro-inflammatory signaling cascades but also internalization of the microbes. An example of a receptor involved simultaneously in LPS-triggered signaling and phagocytosis is a C-type lectin, DC-SIGN present on the surface of both Mϕs and DCs, but not monocytes. 86 Bacteria such as H. pylori and certain strains of Klebsiella pneumoniae interact with DC-SIGN through outer core OS structures of LPS which contain Lex [Gal(β1-4)(Fucα1-3)GlcNAcβ] or mannose sugar residues. 87 This binding interaction mediates the clearance of pathogenic organisms and potentially harmful glycoconjugates by activating phagocytosis. Recent studies have also shown that apart from pathogen binding, DC-SIGN may play a role in signal transduction by modulating TLRs. In DCs, the ligation of C-lectins by TLR resulted in the binding and internalization of pathogens for Ag processing and presentation to T-cells. 88 , 89 Additionally, in renal tubular epithelial cells, an LPS-induced interaction of DC-SIGN with TLR4 affected the MyD88-independent pathway of TLR-4-induced NF-κB (p65) activation. 90
The impact of LPS structure on signaling events
Opportunistic pathogens growing inside the host develop adaptive mechanisms in order to evade immune recognition. Gram-negative bacteria can modulate the host’s response by regulating LPS synthesis. Binding of properly modified LPSs leads to no or reduced recognition by specialized immune cells, which may finally result in latent or chronic infections. For example, persistent Pseudomonas aeruginosa isolates obtained from the respiratory tract of patients with cystic fibrosis had shorter O-Ag chains and lipid A modified. In turn, the human gastric pathogen H. pylori may remain asymptomatic during the lifetime of colonized individuals or eventually lead to gastric ulcer and atrophic gastritis due to expression of Lewis determinants terminating O-specific oligosaccharide, that mimic host cell Ags. An example of an extreme remodeling of LPS leading to reduce its detection by the immune system is F. tularensis, a highly infectious category A human pathogen. Under infection conditions this bacterium can synthesize LPS without O-Ag and with drastically reduced core-oligosaccharide. These adaptive changes, although not shared in all bacteria are a conserved theme in infections, irrespective the type of pathogen or the site of infection.91–93
The structure of LPS, especially the chemistry of lipid A and the presence of both Kdo and Kdo-Hep in the inner core, is crucial for binding by LBP and MD-2 and creation of the active LPS/CD14/TLR4 complex. 94 The exact structure of LPS determines its immunogenicity and, hence, its function. 66 , 68 Lipid A from enteric commensal and some pathogenic E. coli is highly immunogenic, even at low doses, and is thus a model agonist in TLR4 signaling. It possesses a hexa-acylated lipid A moiety substituted with two phosphates. 9 Recognition of this type of endotoxin is mediated by the sixth acyl chain that protrudes from the MD-2 hydrophobic pocket and bridges TLR4/MD-2 to the neighboring TLR4 ectodomain, driving receptor dimerization via hydrophobic interactions. 95 A change in the degree of FA substitution in the lipid A disaccharide moiety usually drastically reduces the immunostimulatory potential of the agonist or can even completely block pro-inflammatory reactions, making LPS an antagonist of human TLR4 signaling. 96 This phenomenon is caused by the difference in the binding mode of the changed LPS with MD-2, which is not productive and does not promote the formation of the signaling complex. 94 Examples of other bacteria which avoid recognition by immune cells by producing weakly activating LPS include P. aeruginosa, a producer of a penta-acylated endotoxin, and H. pylori, which synthesis a tetra-acylated endotoxin. 67 , 97 The recognition of hypo-acylated LPSs, however, is species-specific, which means that they can still activate the TLR4/MD-2 complex in some mammal species (e.g. mouse, rat, horse). 95 Quite similar rules apply to triggering responses through the caspase-4/5/11 system. Generally, binding of penta- and hexa-acylated lipid As by the CARD domain of caspases leads to their oligomerization and activation, while tetra-acylated ones do not induce pyroptosis despite recognition by the receptor. 19 , 72 , 73 Exceptionally, a similar effect is evoked binding of penta-acylated LPS from Rhodobacter sphaeroides, also known as an antagonist of TLR4 signaling. 73
The arrangement of FAs in LPS and their length are also of great importance in inducing the host immune response. The 3 + 3 “symmetric” location of FA chains in the LPS from Neisseria meningitidis was associated with a stronger endotoxic activity than the “asymmetric” 4 + 2 arrangement found in E. coli. 94 Of these two features, however, the length of FA chains is the more important. For instance, the lipid A of L. pneumophila LPS has the same FA arrangement as E. coli, however, it is a weaker inducer of the immune response. Non-enterobacterial species often express lipid As containing long FA chains that either fail to trigger activation or antagonize the TLR4 receptor. 65 , 67 The LPS of L. pneumophila, for instance contains in its structure an extremely long N-acyloxyacyl 27-hydroxyoctacosanoic acid [28:0(27-OH)], which probably plays a decisive role in lowering its immunogenicity. This seems to be confirmed by the fact that the hypo-acylated LPS of Rhizobium, which contained the same FA, showed a similar activity. 68 The influence of FA chain length on monocyte activation was also studied for a mutant derived from commensal E. coli strain JM83 expressing P. gingivalis acyltransferase (htrB-htrBPg+) in which a 12:0 FA in the lipid A has been replaced with 16:0. Such a small change in the length of the alkyl chain reduced the ability of monocytes to secrete IL-8. 98 Synthetic monosaccharide-based TLR4 modulators allow to extend the agonism/antagonism rules to the monosaccharide scaffold and set the structure–activity relationship (SAR). 99 , 100 Among the synthetic triacylated monosaccharide lipid A analogs, some compounds with three tetradecanoyl (14:0) groups or those containing a dodecanoyl (12:0) group acted as agonists of TLR4, while other analogs with a decanoyl (10:0) or hexadecanoyl (16:0) group were antagonistic and inhibited IL-8 and TNF-α production. 99 A computational analysis of docking calculations of different new FP7 derivatives, which were synthetic diacylated monosaccharide ligands, showed they were still able to bind to the TLR4/MD-2 complex and to CD14. All binding experiments consistently provided the same order of affinity between hMD-2 and the synthetic molecules: FP12(12:0) > FP7(14:0) > FP10(10:0) > FP116(16:0). The antagonist potency of molecules with 10, 12, and 14 carbon chains was demonstrated as they were able to block LPS-activated TLR4 downstream signals in human and murine cells. The longer the chain, the smaller was the solubility of FP and their activity. 100 This finding indicates that only LPS with a specific number of FA of a specific length can exert an agonistic activity.
Subtle chemical variations in the sugar part of the inner core, manifested as a decreased number or lack of Kdo residues, also cause dramatic changes in endotoxin activity and are responsible for the switch from TLR4 agonism to antagonism. Despite the inability of the core sugars to bind to MD-2, which is crucial for the endotoxic effect of LPS, their interactions with the TLR4 receptor were found to be important for increasing LPS binding affinity and specificity to the MD-2/TLR4 heterodimer. 94
The phosphorylation state of lipid A also influences the immunogenicity of LPS. A deletion of a phosphate group can result in a loss of endotoxic activity. 9 For example, LPSs from F. tularensis and H. pylori, which lack the 4′-phosphate group, exhibited low immunogenicity, although, exceptionally, the same effect was observed for the LPS of L. pneumophila, substituted with both phosphate groups at positions 1’ and 4’ of the disaccharide backbone. 65 , 67 , 68 The role of phosphates was also demonstrated in monocytes challenged with dephosphorylated LPS derived from a Salmonella enterica mutant. They were found to secrete significantly lower levels of IL-6, IL-1β, and TNF-α in comparison to monocytes activated with wild type LPS. 101 Just as in the case of hypo-acylated LPSs, the effect of the phosphorylation state of lipid A on endotoxic activity is species-specific. Oblak and Jerala’s research indicated that the phosphate group facilitated the appropriate positioning of the endotoxin in the hydrophobic pocket of murine but not human MD-2 through interaction with amino acid residue 122. 95
Altogether, this shows that mainly lipid A modifications are strategies used by extra- and intracellular pathogens to evade recognition or activation of receptors and thus provide protection against host innate immunity.
Receptor recognition of GPI-anchored glycolipids of parasitic protozoa
The sensing of protozoan infections involves a strong innate immune response followed by a predominantly Th1 response. Cells such as neutrophils, Mϕs, and DCs constitute the first line of defense, sensing the invaders mainly through TLR2 and, to a lesser extent, TLR4 and TLR9. 102 , 103 Purified protist PAMPs containing the GPI motif, such as the LPGs of T. cruzi, Leishmania and Trichomonads, and the GIPLs of Leishmania and Toxoplasma gondii, are generally sensed by TLR2, but to some extent also by TLR4; some of them also ligate to heterodimeric TLR2/TLR1 and TLR2/TLR6 complexes or to the TLR4 receptor alone. 40 , 102 , 104 , 105 Usually, signal transduction is Myd88-dependent, but in some cell lines and for some species, it can occur via the Myd88-independent pathway. 34 , 106 , 107 TLR signaling, followed by recruitment of different adaptor molecules, activates various transcription factors, such as NF-κB or IRF3/7, to induce the production of pro-inflammatory cytokines and type I INFs (Figure 1). Moreover, activation of Mϕs and DCs leads to the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS), which limit pathogen survival. Furthermore, Ag presentation by activated DCs enhances the adaptive immune response. 104
Leishmania spp. are obligate intracellular protozoans which infect mononuclear phagocytes. They are etiological agents of leishmaniasis in people, a disease that manifests itself in different forms depending on the Leishmania species involved. 108 Mϕs serve as critical host cells for Leishmania, because it is in those cells that the parasite undergoes amastigotes transformation and replication. 109 Leishmania triggers phagocytic cell signaling, causing a pro- or an anti-inflammatory response, depending on the Ags, ligands, stage of infection, and signaling cascades involved. These responses respectively decrease or increase intracellular parasite replication.
LPG is a key virulence factor in the promastigote form of Leishmania and the main ligand recognized by Mϕs in the early hours of mammalian infection. It occurs both on the parasite surface as well as in the secretory form; these two forms are structurally similar but differ in the average number of phosphorylated oligosaccharide repeat units and in the type of sugar moieties present in the glycan chain. 102 Depending on the strain and/or biochemical features, LPG can have different immunomodulatory properties. 110 As shown in Table 1, LPGs derived from different strains of Leishmania generally triggered signaling through TLR2, but also TLR4, or both, with varying intensities. 37 , 41 , 105 , 110 , 111 Also, the ligation of LPG from L. major with TLR2 indirectly decreased TLR9 expression in in vitro experiments and promoted a host-protective response in Mϕs. 46 In contrast to the above observations, LPG of L. mexicana was not the major mediator of TLR2 activation in in vivo experiments. 112
Published studies evaluating the effect of TLRs stimulation by the investigated ligands.
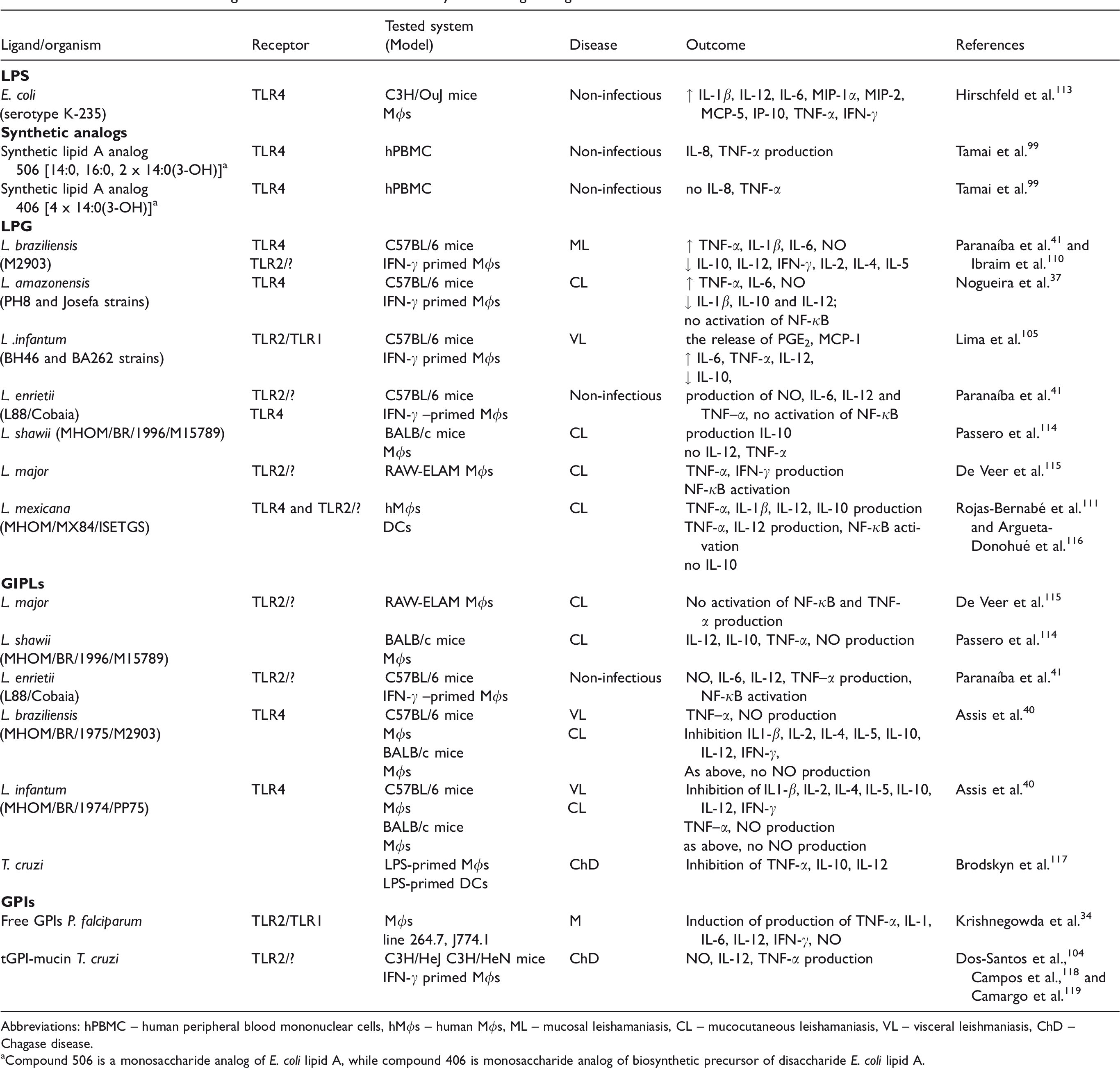
Abbreviations: hPBMC – human peripheral blood mononuclear cells, hMϕs – human Mϕs, ML – mucosal leishamaniasis, CL – mucocutaneous leishamaniasis, VL – visceral leishmaniasis, ChD – Chagase disease.
aCompound 506 is a monosaccharide analog of E. coli lipid A, while compound 406 is monosaccharide analog of biosynthetic precursor of disaccharide E. coli lipid A.
Usually, infection of Mϕs with parasites leads to the activation of phosphorylation events, mediated by protein kinase C (PKC), which regulate respiratory burst and the production of cytokines and NO. 120 However, L. donovani promastigotes inside naive Mϕs failed to activate the phosphorylation of MAPK kinases and the degradation of IκB-α. 121 In turn, an LPG-defective mutant derived from the wild type-induced rapid and transient activation of ERK1/2. 122 In earlier studies, LPG had been shown to negatively regulate PKC, suggesting possible involvement of a molecular mechanism in the inhibition of the process. 123 Since successful healing of leishmaniasis and killing of the parasites is initiated by CD4+ T lymphocytes secreting IFN-γ and TNFs, which act on Mϕs via specific receptors, studies with activated Mϕs have been conducted by numerous research teams. 47 Lodge and Descoteaux demonstrated that the internalization of the parasite resulted in the activation of the ERK1/2 and p38 MAPK pathways. 122 Likewise, the use of purified molecules of the LPG polymer to activate TNF-α- or INF-γ-primed Mϕs confirmed that LPG-impelled TLRs activated PKC/ERK/JNK pathways. However, the ability of particular LPG moieties to modulate signaling was variable and, perhaps, species-specific. 105 , 110 , 112 For example, MAPK signaling triggered by the LPG of L. braziliensis activated NF-κB while that triggered by LPGs from L. infantum and L. amazonensis did not. 37 , 41 , 110 The LPG from L. infantum, after ligation with TLR1/2, induced a range of pro-inflammatory responses, including IL-12 production, PGE2 and NO release, increased lipid droplet formation, and the expression of COX-2 and peroxisome proliferator-activated receptor gamma (PPAR-γ). The activation of PPAR-γ down-regulates an LPG-induced inflammatory response. 105 Moreover, the expression of PPAR-γ and PGE2 is necessary for polarization of M2 Mϕs, which play a role in tissue repair and are permissive to Leishmania replication. These facts suggest that L. infantum took advantage of M2 Mϕs to replicate in the host.124–126 A distinct effect was observed after stimulation of Mϕs with the LPGs of L. braziliensis and L. amazonensis, which, on the one hand, elicited the production of NO and, on the other, decreased pro-inflammatory IL-12. In yet another study, the interaction of L. major-LPG with TLR2 induced the expression of TLR9 and the production of the anti-inflammatory cytokine IL-10, which can inhibit MAPK-mediated IL-12 production in Mϕs and be used by the parasites to avoid or suppress the host-protective immune response. A similar cytokine production profile was observed after ligation of LPG from L. shawii with TLR2 (Table 1). 114
Although LPG has a huge potential to activate the pro-inflammatory response in Mϕs, most visceral and some cutaneous strains of Leishmania have been shown to selectively suppress IL-12 expression in Mϕs but not in DCs. 116 , 127 Thus, LPG-activated DCs were the principal source of IL-12 and played a key role in the induction of cell-mediated immunity to intracellular Leishmania by triggering the production of INF-γ in NK and T cells. 116 , 128 Despite its pro-inflammatory activity, LPG has been shown to inhibit the progression of inflammatory signaling originating from TLR2/TLR4 by inducing the expression of suppressors of the cytokine signaling (SOCS) family proteins, viz. SOCS-1 and SOCS-3. 115 PI3K signaling activated by Leishmania infection negatively regulated IL-12 production as well (Figure 1). 129
In all Leishmania LPGs, the lipid moiety is conserved, and it is the sugar part of the polymer, which shows greater polymorphism, that seems to be largely responsible for the immunomodulatory properties of this molecule. 37 However, proper signaling through TLRs requires the involvement of the entire intact LPG moiety, since neither the glycan nor the lipid moiety alone are capable of inducing pro-inflammatory responses, as shown in a study of L. infantum LPG. 105 A similar study with LPG from L. major demonstrated that LPG deprived of the lipid constituent could inhibit TNF-α production. 115 The 1-O-alkylglycerol fragment also inhibited the activity of purified PKC, although the PG portion exerted some effect as well. 123 Furthermore, the immune response was affected by the structure of the oligosaccharide moiety, as shown by the fact that an unbranched LPG from the BA262 strain of L. infantum was a stronger activator of pro-inflammatory response in Mϕs than a branched LPG from strain BH46. 105 Likewise, a longer LPG from the L88 strain of Leishmania induced a more pronounced response than a shorter LPG from the Cobain strain. 41 The above data suggest that biochemically distinct LPGs can differentially modulate Mϕ functions. The genetic background of the (murine) host also has a significant influence on the outcome of the infection, as manifested by the opposing effects on PKC-α activity, of LPG-stimulated Mϕs derived from resistant versus susceptible mouse strains. 130 Likewise, the mechanism of pathogenesis of leishmaniasis seems to play a role, with dermotropic species exhibiting a more exacerbated pro-inflammatory profile, and viscerotropic species presenting an immunosuppressive potential following stimulation of Mϕs with LPGs. 105 , 131
GPI family molecules from protozoan parasites play important roles in the establishment of several parasitic infections. They are the major constituents of the amastigote surface and are thus key agents involved in later stages of Leishmania infections in Mϕs. GIPLs derived from L. enrietii, a species that is non-infectious to humans, drive the production of a whole range of pro-inflammatory cytokines, mainly through the TLR2 pathway. Although GIPLs derived from strain L88 activated the production of higher levels of NO, IL-12, and TNF–α than those from the Cobain strain, their LPGs presented similar inducible behaviors. 41 By contrast, GIPLs from visceral leishmaniasis strains such as L. braziliensis and L. infantum were potent inhibitory molecules that did not activate TLR4/ERK/p38/JNK signaling and, consequently, inhibited NO production (Table 1). 40 In the case of T. cruzi (the causative agent of Chagas disease), several glycoconjugates, classified as GIPLs and GPIs, acted, respectively, as either antagonists or agonists of TLRs signaling. In T. cruzi GIPLs, the lipid portion of the molecule is a ceramide that is attached to the oligosaccharide part differently than in Leishmania, in which alkylacyl-PI or lyso alkyl-PI moieties are present. 31 , 118 , 132 In in vitro experiments, T. cruzi GIPLs had a suppressive effect on Mϕs and DCs by impairing TNF-α production, and thus delaying an effective host immune response. The ceramide portion of the GIPL was shown to be responsible for most of the activity exhibited by the whole molecule. 117 On the other hand, GPI-anchored mucin-like glycoproteins isolated from T. cruzi induced the production of NO as well as IL-12 and TNF-α by murine Mϕs. The GPI moiety of T. cruzi glycoproteins, which consisted of AAG and lacked inositol-acylation, was by itself able to trigger cytokine production in monocytic lineage cells. In this it resembled the ceramide from GIPLs, though it acted in the opposite manner. The GPI molecules of Plasmodium, which contained a diacylglycerol moiety in the lipid part were similar to most other protozoan GPI anchors, with the exception that had a unique myristoyl moiety linked to the C2 of the inositol ring. Like other conjugates of this type, they had an immunostimulating activity and acted as agonists of the TLR2/TLR1 MYD88-dependent pathway activating MAPK signaling and pro-inflammatory response. 31 , 34 GPI-associated signaling was also carried out to some extent through the non-canonical TLR4 pathway or other receptors. 34 To sum up, parasite-derived GIPLs and GPI molecules have an immunoregulatory activity, and they can induce both activating and deactivating signal-transducing effects on the murine immune system, contributing to parasite pathogenicity. 118
It has been shown that myd88−/− mice are still able to produce cytokines upon Leishmania or T. cruzi infection. This finding suggests that, in addition to TLRs, other innate immune receptors are required for host resistance. 133 L. major promastigotes inhibited IL-12 production in Mϕs also through CR3 (CD11b/CD18) receptor engagement and down-regulation of ETS-mediated transcription. 134 Since LPG is thought to bind CR3 directly and is the major C3bi acceptor, it may have modulated the process. 107
Some studies have demonstrated the engagement of caspase-1/Myd88 in the response to parasite infection as well, although the PAMPs involved in the process have not been identified. 135 , 136 However, the results of experiments with LPG-activated Mϕs suggest that this Ag might participate in the classical inflammasome activation. LPGs obtained from L. braziliensis and L. mexicana, but not L. amazonensis, induced the production of IL-1β, which is known to be a part of the inflammasome complex, along with caspase-1 and members of the NLR family of pathogen sensors. 110 , 111 , 133 Conversely, GIPLs from L. braziliensis and L. infantum strongly inhibited the expression of IL-1β. 40 Thus, the effects of inflammasome activity are predicted to differ widely among the various Leishmania species or cell cycle stages. 136
Cell-autonomous defense of phagocytic cells and glycoconjugate-based microbial interference
Among specialized immune cells in metazoans, Mϕs and neutrophils are the major effector cells. They patrol their environment in search of pathogens and parasites for subsequent clearance. Upon recognition of PAMPs, these specialized phagocytes internalize the invading microbes, activate cell-autonomous defenses, and employ conserved strategies to kill the bacteria. 137 Among the cellular processes stimulated downstream of the PR machinery is the initiation of phagocytosis and autophagy, followed by compartment maturation manifested in the activation of a respiratory burst, production of AMPs, and/or secretion of extracellular traps. Concurrently, some microbes have evolved mechanisms not just to avoid being killed but to survive inside hosts and exploit them for resources.
Phagocytosis
Phagocytosis is a key process of the defense system, in which specialized phagocytic cells (Mϕs, DCs, monocytes, and neutrophils) recognize particles (>0.5 µm), bind them to their surface, and internalize them into a plasma membrane-derived intracellular vacuole. 138 What is important, the aim of such internalization can differ depending on cell type. While Mϕs and neutrophils are specialized in intracellular killing, DCs are involved in presenting an Ag to Th lymphocytes. Phagocytosis can be subdivided into three main stages: particle binding, phagosome formation and maturation, and degradation of ingested material. The binding of particles is mostly done by phagocytic receptors, although the participation of TLRs in initiating signaling pathways that elicit rearrangement of the actin cytoskeleton and, in consequence, the formation of the phagosome, have also been reported. 66 , 139 The nascent phagosome is then remodeled in a process involving the trafficking of vesicles, in particular tethering and docking of membranes from early endosomes, late endosomes and lysosomes, which ultimately leads to the formation of the hybrid microbicidal phagolysosome. The components of the endocytic pathway are identifiable biochemically by a distinct set of molecular markers (Rab5, Rab7, and lysosome-associated membrane proteins (LAMPs), respectively). 139 The maturation of phagosomes, apart from the differential recruitment and activation of Rab-family GTPases to the membrane, manifests in decreasing luminal pH and altering the key phosphatidylinositol (PI) lipids through regulation by lipid kinases and phosphatases. The acidification of the lumen is due to the action of a V-type vacuolar H+ ATPase in the membrane bilayer, which pumps hydrogen ions into the lumen. The fusion and fission events also enrich the lumen in hydrolytic enzymes and bactericidal peptides which help degrade the ingested bacteria. 70 , 137 Moreover, the internalized microbes can be destroyed and killed in processes that involve the transport of certain metals or the production of ROS and RNS inside the compartments. 140 , 141
Although most microorganisms are successfully internalized and eliminated by phagocytes, some pathogens have developed survival strategies that interfere with the internalization and/or maturation processes. 138 , 142 , 143 The microbial mechanisms of subverting cell-autonomous defenses likely originated as resistance to predation by single-celled eukaryotes. 137 The O-Ag is the immunogenic portion of LPS, which presents epitopes for immune responses and has the potential to influence the host–parasite interaction at several levels. 10 Its proper structure and/or composition provides protection against phagocytosis by specialized immune cells. 44 This is achieved by prevention of ligation with receptors, modulation of the rate of internalization, or modification of phagosome membrane into a non-fusogenic parasitophorous vacuole providing a protective niche. 29 , 66 , 87 ,144–147 For example, the LPS of L. pneumophila shed in a liquid culture as a non-vesicular fraction was shown to arrest phagosome maturation in monocytic host cells. 148 In another study, LPS liberated to cytosol by proliferating S. Typhimurium cells closed inside vacuolar compartments participated in host cell signaling, possibly facilitating bacterial intracellular lifestyle. 149 These data indicate that LPS protects microbial cells against phagocytosis and that appropriate epitopes are decisive for the intracellular fate of bacteria. 44 Some studies have shown that the same determinants can counteract phagocytosis in different immune cells, however, the response to this protection is not uniform for all professional phagocytes. 146
As a response to intruders, activated phagocytic cells express inducible NO synthase (iNOS), an enzyme responsible for the production of NO, which is an essential mediator of defense. Pathogens can escape being killed by phagocytes by interfering with the expression of iNOS. 140 For example, an LPS rough mutant of Burkholderia pseudomallei, but not the wild type strain with a complete S-LPS, stimulated the phosphorylation of Y701-STAT-1 and the expression of IFN-regulatory factor 1 (IRF-1), both of which are essential transcription factors of iNOS, indicating that O-PS modulated the antimicrobial activity of the Mϕ. 144 Invading microorganisms can also be eliminated in processes that involve NADPH oxidase (NOX), which mediates the release of ROS. ROS promote pathogen clearance mechanisms on many levels but can also potentially contribute to microbe persistence. 141 , 150 For instance, a mutation in the LPS genes of S. enterica reduced the production of the endotoxin, leading to bacterial survival or even replication in murine Mϕs. Compared to the WT strain, the mutation also significantly reduced oxidative response. 45
Since a low pH is not sufficient per se to kill bacteria in an oxygen-independent process, phagosomes acquire a series of proteases, hydrolases, lysozymes, and AMPs which help them to break down some bacterial components or disrupt membrane integrity. 70 , 137 The main biological function of the O-Ag is to protect cells against various harmful factors. In Francisella, its loss resulted in increased susceptibility to killing by membrane active compounds such as serum and AMPs, reducing intracellular survival of the bacteria. Indeed, O-Ags have a major impact on the resistance of bacteria to phagocytosis; however, modifications of the structure of the lipid A and core moieties are also important. Acylation of the lipid A and the presence of a Hep branch in the core part had a protective effect on Klebsiella, defending it against clearance by Mϕs mainly through inhibition of the action of AMPs. 44 , 151 LPS has also been shown to exert both an inhibitory and a stimulatory effect on lysozyme expression, depending on the LPS source, its concentrations, as well as duration of contact with different types of targeted cells. 152
Glycoconjugates of protozoa can also influence the progression of phagocytosis. There are several properties of LPG that give rise to its protective role against the defense system of the host cell. 43 The LPG of Leishmania was able to deactivate human monocytes and block their ability to undergo respiratory burst. 153 , 154 In vitro experiments showed that the membrane of Leishmania-containing phagosomes was enriched with LPG, which interfered with their maturation by blocking the assembly of NOX, fusion with endosomes, and recruitment of ATPases. 38 , 122 , 155 , 156 One of the mechanisms that prevent the recruitment of NADPH oxidase is LPG-induced disruption of lipid microdomains, the possible sites of the assembly of this enzyme complex. 156 LPG is also believed to affect actin recruitment necessary for delaying phagosome maturation since truncation of LPG in mutants impaired this process. 157 , 158 Some reports have also shown that LPG of Leishmania plays a protective role against NO production by iNOS in Mϕs, however, other studies have demonstrated that NO production is reduced mainly due to the presence of GIPLs on amastigotes and inhibition of TLR4/p38 signaling. 40 , 159
In recent times, some studies have indicated that PRR signaling could also influence phagosome maturation, but this issue is controversial as different authors report contradictory results. An in vitro study has indicated that phagosomes from MR−/− bone marrow-derived Mϕs (BMDM) stimulated with L. donovani showed a delay in maturation, while phagosomes from TLR2−/− and MyD88−/− acquired the LAMP-1 marker faster than those obtained from WT BMDM. 160 Phagosome maturation has also been shown to be modulated through Myd88 and TLR2-mediated signaling after phagocytosis of E. coli and S. typhimurium, respectively. 161 Cellular activation by cytokines also influenced phagosome maturation. Stimulation of Mϕs with IFN-γ induced delayed phagosomal proteolysis, fusion of phagosomes with lysosomes, and acquisition of maturation markers by phagolysosomes. What is important, the effect of the cytokines was dependent on the type of stimulated cell (DCs, neutrophils, Mϕs) and the type of stimulus used. 162 The above observations suggest that PPR agonists such as LPS and LPG indirectly influence phagosome maturation.
Autophagy
Bacteria which have successfully managed to avoid death in phagosomes can be released into the host cell cytoplasm. This event triggers a more stringent catabolic pathway called autophagy, which serves as an additional defense mechanism preventing colonization and, in some cases, dissemination of invasive pathogens. An autophagic process in which intracellular pathogens and/or their damaged phagosomes are specifically recognized and digested is termed xenophagy. 137 Generally, the released bacteria are subjected to ubiquitination before attaching to the double-membrane phagophore. 163 During xenophagy, the phagophore expands around the cytosolic bacterium to finally engulf it in the autophagosome enriched with microtubule-associated protein 1 light chain 3 (LC3). Next, upon fusion with lysosomes, the phagosome develops into an acidic, degradative compartment, the autolysosome, in which the bacterium and the membranes are digested. 164 Detailed mechanisms of autophagy have been discussed in the reviews by Kimmey and Stallings, Kunz et al., and Calvo-Garrido et al.165–167 Despite the fact that the original function of autophagy is defense against intracellular pathogens, some of them such as C. burnetii, L. pneumophila, Brucella abortus, P. gingivalis, and T. cruzi, are able to subvert or exploit the process for their own purpose. 52 , 108 , 164 , 166 ,168–171 The most important moiety among the PAMPs which trigger autophagy and the production of ROS leading to clearance of bacterial pathogens, is LPS. 52 On the other hand, LPS moieties with specific structures may be involved in the survival of the above-mentioned pathogens, inside autophagic compartments. For example, the LPS of P. gingivalis exists in at least two known forms, O-LPS and A-LPS (characterized by a heterogeneous lipid A structure). A-LPS isoforms (tetra- and pentacylated), depending on the prevalence of lipid A moiety, can modulate autophagy by altering lipidation of LC3 and thereby influencing intracellular bacterial survival. 172
In vivo studies of mice sepsis indicated that intracellular killing of bacteria strongly correlated with autophagy of peritoneal Mϕs. 173 According to another report, autophagy of LPS-activated Mϕs suppressed the level of inflammatory cytokines (TNF-α, IL-1β, IL-6, and IL-12), while LPS-primed human neutrophils, in a similar situation, secreted IL-1β. 174 , 175 This indicates that LPS-stimulated autophagy regulates cytokine production in various ways depending on the type of the immune cell involved. The molecular mechanisms regulating this process in Mϕs have been partly elucidated. The adaptor protein p62, which is one of the core autophagy components that deliver ubiquitinated bacteria to the phagophore and induce autophagosome biogenesis, is also critical for the suppression of cytokine production. 163 , 176 Following assembly of the autophagic machinery, this protein is activated by TANK-binding kinase 1 (TBK1), which belongs to the IKK family, while upstream signals are triggered through the TLR4/TRAF6/p38 pathway upon LPS-stimulation. 169 , 176 , 177
Extracellular traps
Extracellular traps (ETs) liberated from various phagocytes are fragments of decondensed nuclear or mitochondrial DNA decorated by granular enzymes, peptides, and histones. The role of ETs is to ensnare and kill extracellular pathogens and elicit pro-inflammatory responses. 70 A variety of stimuli promote the formation of a neutrophil extracellular trap (NET), among others, bacterial LPS as well as the LPG of Leishmania and the soluble Ag of T. cruzi.178–180 The process of NETosis requires the involvement of intracellular signaling pathways, most commonly Raf/MEK/ERK kinases and p38/MAPK. 181 This microbicidal mechanism, referred to as classical NETosis, has been shown to depend on ROS generation by NOX2. NET release in neutrophils was also shown to occur through an early/rapid ROS-independent mechanism, called early/rapid vital NETosis. 182 It was accompanied by a substantially lower level of ERK activation and a rather moderate level of Akt activation, whereas the activation of p38/MAPK was similar to that found in classical NET formation. 181 L. amazonensis promastigotes were able to trigger both types of NETosis under different system modulation. In the classical process, promastigotes triggered a higher release of DNA than did amastigotes. This was attributed to the fact that LPG was abundant in promastigotes and had a domain similar to other molecules such as acid phosphatase, PPG, and GIP lipids, which possible contribute to NET formation. 182 In L. donovani promastigotes, LPG appeared to be involved in resistance to alternative NET-mediated killing, since the wild type maintained its viability in the presence of NETs. What is more, mutant parasites lacking LPG were efficiently killed by these extracellular structures. On the other hand, LPG and Gp63 were not involved in NET simulation, which was in contrast to the results obtained by Guimarães-Costa et al. in their study of classical ROS-dependent NETosis. 179 , 183 These discrepancies could be explained by carbohydrate modifications inside LPG moieties as well as by the distinct types of stimuli involved. A similar phenomenon was observed for neutrophils activated by LPS derived from various serotypes of E. coli. Depending on the LPS type, “suicidal” (ROS-dependent) or “vital” (ROS-independent) NETosis was induced. The latter required activated platelets and was therefore thought to occur predominantly during sepsis. Findings of Pieterse et al. suggested that LPS sensing by neutrophils might be a critical determinant for restricting NET release to certain Gram-negative bacteria only, which in turn might be crucial for minimizing unnecessary NET-associated immunopathology. 178
In vitro and in vivo studies showed that release of ETs by activated mononuclear phagocytes was also effective against such parasites as Plasmodium falciparum, T. cruzi, and T. gondii. 181
Restoring the immune balance as a treatment strategy in pathogen-related diseases
From the first moments of life, our body is colonized by indigenous microbiota which proliferate to reach counts more or less equal to those of human cells. 184 The “quantity and quality” of the microbes that inhabit environments such as the lung, skin, or gut are decisive for its health, as these microorganisms activate and modulate the immune system to maintain host homeostasis. 3 The importance of microbiota in the development and function of the immune system has been investigated with the use of germ-free animals, 185 , 186 which allowed understanding the mechanisms of microbiota associated-human diseases. However, the question is how this knowledge could be generalized, since it is often uncertain whether disruption in the microbiota associated with a disease is a cause, a contributing factor, or merely a consequence of the disease state. Thus, comparison of the microbiomes of different groups of people, such as those with a particular disease with healthy individuals can shed better light on these issues. For example, dysbiosis in humans caused by both lung and gut microbiota impairment has been shown to be associated with the development of inflammatory as well as infectious diseases. 187 , 188 Recently, increasing evidence suggests that the alterations in the lung microbiome constitution are associated with severity of pulmonary diseases such as asthma or chronic obstructive pulmonary disease (COPD). 189 , 190 These disturbances in microbiota, similarly to infectious diseases, resulted in an imbalance of the Th1/Th2/Th17/Treg cell population. 185 , 191 , 192 The particular subsets of Th cells are classified according to the pattern of cytokines they produce and functional differences. Th1 cells, which are critical to defense against intracellular pathogens, selectively produce IL-2, IFN-γ, and TNF-α. Th2 are a source of IL-4, IL-10, and IL-13, and predominate in response to helminth infestations. Immunosuppressive Tregs potently inhibit effector Th1 and Th2 cells responses by secreting IL-10, IL-35 and TGF-β, and pro-inflammatory Th17 and Th22 responses characterized by production of IL-17 and IL-22, respectively. 193 However, uncontrolled responses of Th1 and Th17 cells have been associated with autoimmune diseases, and those of Th2 and Th17 – with allergic diseases (e.g. asthma). 185 , 188 , 194 The functional differentiation of CD4+ T cells occurs in an appropriate Ag-stimulated environment. In most models, LPS elicits Th1 responses by stimulating IL-12. Ags which induce the release of TNF and IL-1 may promote Th2, while Ags which stimulate the production of IL-6, TNF-α, and IL-1β in a TGF-β-rich microenvironment can promote Th17 polarization. The Treg differentiation pathway is directed by IL-10 or PGE2. 195 Leishmania and T. cruzi Ags activate naïve T cells through T-cell receptor-peptide Ag-major histocompatibility complex II (TCR-Ag-MHC II), and co-stimulatory molecules and cytokines induced by TLRs. 196 , 197 In turn, LPS indirectly induces IFN-γ production by iNKT cells through TCR-Ag-CD1d in combination with cytokines and co-stimulatory molecules. The direct stimulation of IFN-γ by LPS does not require CD1d-mediated presentation of an endogenous Ag and is dependent only on APC-derived cytokines. 198 By enhancing phagosomal maturation, TLRs may affect the subsequent cellular responsiveness that includes Ag processing, antigenic determinant selection, co-stimulatory molecule expression, and cytokine production and thus control T cells polarization. 196
Chronic persistence or repeated exposure to certain microbial pathogens contributes to an overwhelming immune response and systematic release of pro-inflammatory cytokines (IL-12, TNF-α, IFN-γ, IL-6, and IL-17). 199 This hyper-responsiveness to infection can lead to an inflammatory SIRS (systemic inflammatory response syndrome) response, known as sepsis, as well as chronic inflammatory or autoimmune diseases. LPS is recognized as the most potent microbial mediator implicated in the pathogenesis of sepsis and septic shock. Sudden release of large quantities of this moiety into the bloodstream initiates pathogenic reactions. 200 Many different strategies have been proposed to ‘tame the monster’, including inhibition of endotoxin signaling by blocking the TLR4 receptor, inactivation of the endotoxin itself, and blocking of the activities of particular cytokines. While TNF inhibitors have been approved by the US Food and Drug Administration for the treatment of some chronic inflammatory systemic diseases, they have not been successfully used in sepsis therapy. Likewise, numerous attempts to block the endotoxin and its receptor in treating sepsis have given inconsistent and largely negative results. 201 These failures may partly have been due to the fact that in creating anti-sepsis therapies scientists were guided by the old confounding definition of sepsis, in particular the belief that excessive stimulation of the immune system was the exclusive cause of this disorder. Currently, it is known that only patients in the early stages of sepsis show a pro-inflammatory response, in contrast to patients in the later stages, who may present a suppressed immune response. 202 It is also known that immunodeficiency is due to impaired PI3K signaling, whereas autoimmunity and leukemia are accompanied by unrestrained signaling through the PI3K/Akt axis. 203 Given this knowledge, modulation of biological pathways, such as the PI3K pathway, or manipulation of subpopulations of T cells have been suggested as more promising approaches to the treatment of sepsis. 204 , 205 Lapara and Kelly reported that Mϕs infected with L. amazonensis or L. major promastigotes suppressed LPS-induced inflammatory responses, 199 while infection with L. mexicana amastigotes resulted in an inhibition of LPS-induced IL-12 production. 200 , 206 While the pro-inflammatory activity of LPG, the major glycoconjugate of promastigotes, is well-documented, its anti-inflammatory properties are also well-known. 37 , 105 , 110 , 111 , 115 They are associated with the inhibition of the MyD88-dependent pathway and modulation of the PI3K pathway (Figure 1). 115 , 129 In turn, GIPLs, which are highly abundant on amastigotes, suppress IFN-γ-induced expression of many pro-inflammatory genes, thus regulating T-cell signaling. 40 , 115 A great potential for silencing the LPS-induced pro-inflammatory immune response was demonstrated for the GIPL from T. cruzi, and more precisely for its ceramide part. 117 This clearly indicates that GIPLs have a potential for decreasing T-cell signaling by competing for the TLR4 receptor with LPS.
The strategies relying on modulation of the PI3K pathway and/or polarization of iNKT cells, might also be adopted for elimination of intracellular pathogens which suppress or impair immune response by modulating cytokine release, inhibiting MHC II-dependent Ag presentation, or inducing and expanding counterprotective Th cells. 207 Immunostimulatory molecules are promising candidates on which to base new treatment strategies that target infectious diseases. A specific and complex group of such molecules comprises glycolipids and includes serum lipids or microbial lipid Ags that mediate CD1d-restricted activation of iNKT cells. 208 , 209
iNKT are lymphocytes which contribute to antimicrobial host responses in bacterial, parasitic, viral, and fungal infections and also mediate the natural antitumor response. As described earlier, iNKT can be activated indirectly after glycolipid Ag presentation through the CD1d-Ag-TCR cell axis or by sensing APC-released cytokines, or after direct ligation of the Ag with TCR. As a result of activation, iNKT cells can be subdivided into NKT1, NKT2, NKT17, and NKT10 subsets. The NKT1 sub-population is in many ways equivalent to Th1 cells in that it produces IFN-γ upon activation. 210 Synthetic sphingolipid αGalCer was shown to be a strong agonist of TCR for iNKT cells in vivo but not for other lymphocytes. 211 One of earlier studies on this ligand indicated that upon CD1d-mediated stimulation, iNKT cells differentiate into NKT1, which release mainly INF-γ and can also produce IL-4, albeit at lower levels. Recent studies, however, have shown that αGalCer can also stimulate differentiation into the NKT10 subset producing anti-inflammatory IL-10. Also, αGalCer-activated iNKT cells undergo “anergy,” a differentiation step resulting in unresponsiveness, lack of proliferation, and an inability to produce IFN-γ upon re-stimulation. Use of α-GalCer is currently being investigated in a number of clinical trials, however, given the above, the efficacy of such a strategy is questionable. 210 On the other hand, promising immunostimulatory properties against L. major infection, observed as a reduced number of intracellular parasites and induced production of pro-inflammatory cytokines, have been demonstrated for the lipopeptidophosphoglycan (LPPG) of Entamoeba histolytica and its synthetic GPI-anchor analogs. 208 LPPG harbors two PI anchors, EhPIa and EhPIb, which show structural and immunological similarities to αGalCer and are, thus, presented to iNKT by APCs expressing CD1d molecules. Therefore, LPPG and PI molecules have been proposed as candidates for targeting pathogens that invade immune cells to multiply and survive in them. 212 Equally promising immunomodulatory effects can be expected from αGalCer analogs with a changed length of the acyl or the phytosphingosine chain, which have different affinities for the NKT cell TCR, and thus different thresholds of NKT cell activation. 213 So far, no trials have been performed with their use.
Since iNKT activation can also be Ag-independent and driven solely by cytokines (Il-12 and/or IL-18) from innate immune cells, stimulation of TLRs with an alternative ligand, different from the infectious intracellular pathogen, might switch the host’s response to pro-inflammatory. However, since LPS is a strong inducer of inflammation, it seems that experiments should rather be performed with high affinity and low toxicity monosaccharide analogs of lipid A, which allow better control of the level of iNKT activation.
Concluding remarks
As shown in the present review, the signaling pathways triggered by bacterial LPS and GPI-anchored glycolipids of protozoa overlap in many places. It seems that the main and decisive role in directing the immune response is played by TLR-mediated signaling, which influences cytokine production, expression of co-stimulatory molecules, and determinant selection for cross-presentation. Taken together, all these processes are crucial to upholding the balance of subpopulations of Th cells and maintaining host homeostasis. Moreover, TLR signaling, which depends on the nature of the stimulating ligand (agonist versus antagonist), can play a dual role by eliciting pro- or anti-inflammatory responses.
In order to polarize host responses to infections, treatment strategies have to be developed which will take into account the nature of the pathogen and the stage of its life cycle. To be effectively eliminated, intracellular pathogens which avoid or subvert host innate immunity require a strengthening of the pro-inflammatory responses. Conversely, the action of strong microbial ligands which trigger hyper-responsiveness needs silencing. Thus, studies which give a better insight into the mechanisms involved in the abrogation of the inflammatory response provoked by some protozoa parasites and bacterial LPS can be an inspiration for designing a new generation of therapies based on targeted interference with signaling pathways with the use of synthetic analogs of natural glycoconjugates. Co-stimulation with an alternative ligand-conductor might re-tune the immune melody from pathological to protective.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partially supported by National Science Centre, Poland (grant 2017/01/X/NZ1/01153).