
Abstract
Inflammatory bowel diseases (IBD) comprise a distinct set of clinical symptoms resulting from chronic or relapsing immune activation and corresponding inflammation within the gastrointestinal (GI) tract. Diverse genetic mutations, encoding important aspects of innate immunity and mucosal homeostasis, combine with environmental triggers to create inappropriate, sustained inflammatory responses. Recently, significant advances have been made in understanding the interplay of the intestinal epithelium, mucosal immune system, and commensal bacteria as a foundation of the pathogenesis of inflammatory bowel disease. Complex interactions between specialized intestinal epithelial cells and mucosal immune cells determine different outcomes based on the environmental input: the development of tolerance in the presence of commensal bacterial or the promotion of inflammation upon recognition of pathogenic organisms. This article reviews key genetic abnormalities involved in inflammatory and homeostatic pathways that enhance susceptibility to immune dysregulation and combine with environmental triggers to trigger the development of chronic intestinal inflammation and IBD.
Introduction
The intestinal microbiota serves as an auxiliary organ of the body, linking the gastrointestinal tract to the external environment. The mucus barrier, a boundary that normally separates the microbiota from the mucosal surface of the intestine, insulates the intestinal immune system from overt contact with the microbiota. 1 Interaction of the microbiota with the host across the mucus layer or by specific sampling mechanisms like the sub-epithelial dendritic cells and microfold (M) cells, establishes a co-dependent system that determines health or disease. Alterations to any of the three components of the intestinal system, such as dysbiosis (alteration of the make up of the intestinal microbiota), disruption of the mucus barrier, or dysregulated immune system activity, may foreshadow inflammatory bowel disease (IBD). The interplay between genetic mutations and environmental factors ultimately influences the development of IBD.
The gastrointestinal tract harbors trillions of bacteria, from all three domains of life (i.e. Archaea, Prokarya and Eukarya). 2 The gut contains 1000–5000 different species, with 90% coming from the phyla Bacteroidetes or Firmicutes.2,3 Individual human microbiota (enterotypes in the small intestine or faecotypes in the colon) are affected by dietary preferences, including carbohydrate content or animal fat.3,4 A diet heavy in total fats and meat increases odds of developing IBD, whereas high fiber and fruit intake decreases the risk of Crohn’s disease (CD). 5 Atypical compositions of enterotypes and faecotypes have been implicated as both cause and effect of IBD. The relationship between dysbiosis and IBD remains obscure, but it is clear that healthy individual microbiotas differ from those with IBD. 6
IBD associated microbiotas show decreased levels of common Clostridium species, or increased levels of common infectious species, including Campylobacter, Shigella and Escherichia. 7 There is no consensus regarding a definitive dysbiotic microbiota; this uncertainty is compounded by significant heterogeneity between studies. 7
In health, the intestinal microbiota positively influences immune function, maintains intestinal epithelial cell (IEC) integrity, and protects against IBD by fermenting dietary fiber to short-chain fatty acids (i.e. acetate, propionate and butyrate). Bacteroidetes produce high levels of acetate and proprionate, whereas Firmicutes produce high amounts of butyrate. 8 Butyrate has been shown to reduce TNF-α levels in circulation and at the gut-mucosal interface in CD patients. 9 Butyrate down-regulates NF-κB expression and inhibits LPS stimulation of pro-inflammatory cytokines. 9 Additionally, the reduction of butyrate producing Clostridium species results in a decrease of regulatory T cells, possibly contributing to inflammation. 10 The keystone pathogen hypothesis holds that perturbations of the normal gut environment lead to dysbiosis. When a less prevalent commensal bacterium (i.e. a pathobiont) expands its presence in the community, its abundance initiates the transformation of a healthy microbiota into a dysbiotic state. The delicate balance that exists between the environment, host and intestinal flora must be maintained to preserve intestinal epithelial integrity, modulate conflicting immune responses and prevent clinical manifestations of IBD.
The intestinal epithelium has a surface area of approximately 400 m2 made from a single layer of IECs largely adapted to perform metabolic and digestive functions. However, their influence on the development and function of the mucosal immune system has garnered considerable attention in relation to IBD. The intestinal epithelium exerts a critical immunoregulatory influence via IEC specialization that forms both physical and biochemical barriers to intestinal microorganisms. The normal epithelium coordinates appropriate immune responses, including tolerance and pathogen specific immunity (Figure 1) by initiating innate and adaptive immune systems. IECs regulate responses to luminal bacteria while maintaining intestinal homeostasis by delicately balancing both antimicrobial and immunoregulatory functions.
11
IECs form not only a physical and biochemical barrier that separates luminal bacteria from the mucosal immune system, but can also serve as early contributors to an immune response. Commensal bacteria are required for normal intestinal function, including fermentation of indigestible sugars into short-chain fatty acids, enhancing mucin production and developing tolerance. Secretory GCs and Paneth cells secrete mucus and antimicrobial peptides that prevent mucosal penetration by commensals or pathogens. When the composition of the microbiota changes, resulting in dysbiosis and the potential for invasion by bacteria increases, a pro-inflammatory state may develop.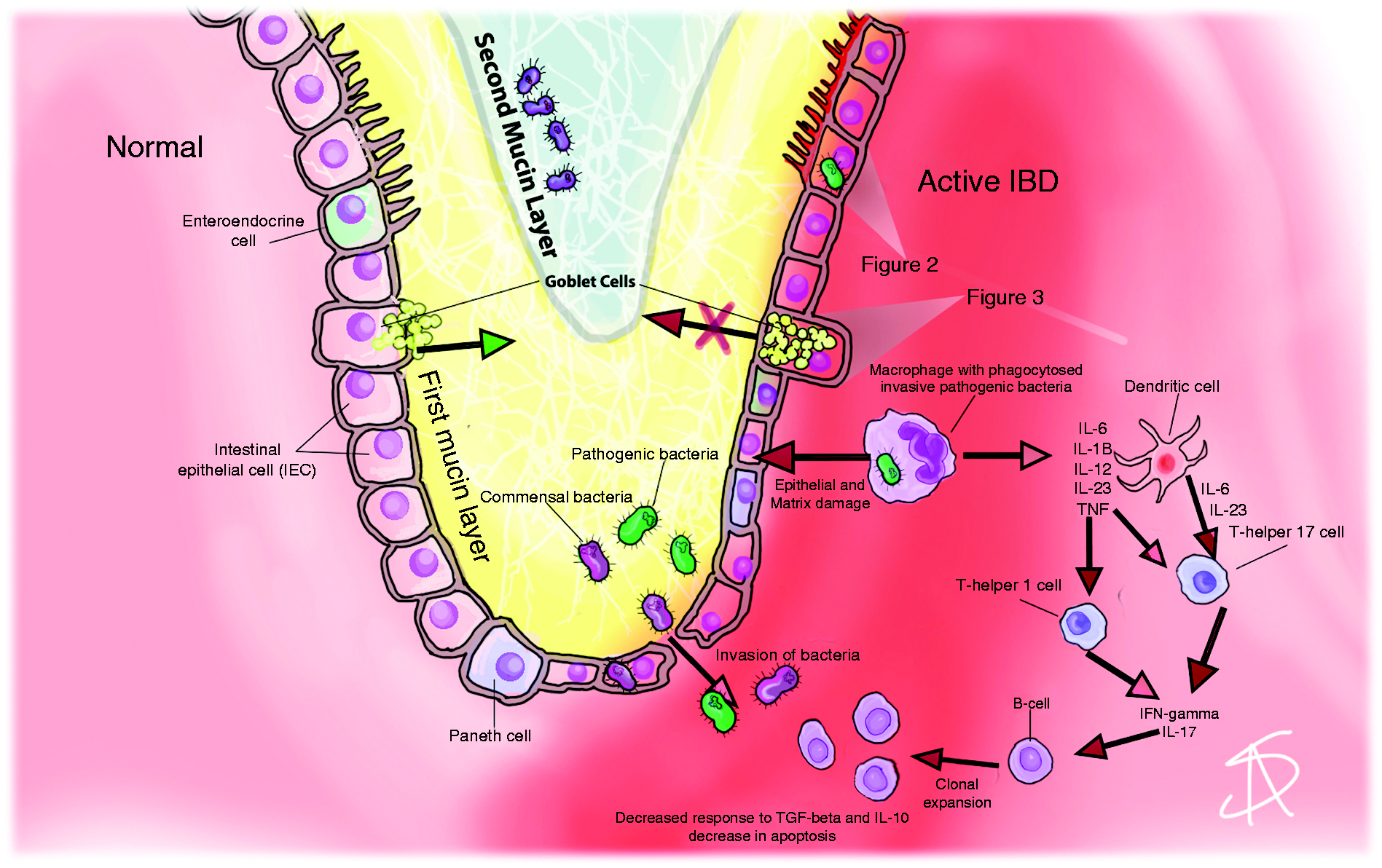
IECs rely on a family of sensors called PRRs to interpret microbial signals. PRRs detect evolutionarily conserved structures from microbes called PAMPs or microbial-associated molecular patterns (MAMPs). Activation of these sensory mechanisms initiate the first line of defense against invading microorganisms. PRRs expressed by IECs discern the luminal microbial composition, help differentiate between commensal and pathogenic microbes, and influence mucosal immune responses to establish tissue homeostasis when a healthy microbiota is present. The influence of PRRs as promoters of tolerance can swiftly change to drive the immune system into a state of activation, and then ultimately shape the adaptive immune response. The engagement of PRRs by either PAMPs or MAMPs triggers a dichotomy of intracellular signaling cascades, with PAMPs culminating in the expression of pro-inflammatory cytokines and MAMPs triggering immunoregulatory responses. PRRs consist of three receptor families: TLR, NLR and RIG-I-like receptors. These receptors differentially recognize MAMPs or PAMPs, discerning friend from foe. Rakoff-Nahoum et al. found that TLRs detect commensal bacteria under steady-state conditions, a critical step in maintaining intestinal epithelial homeostasis and protecting against gut injury. 12 These effects occurred by the binding of commensal bacterial components to TLRs, followed by downstream signaling and production of cytokines (i.e. IL-6, TNF-α and KC-1), epidermal growth factor receptor ligands and heat-shock proteins at a balanced level when the epithelium was intact.12,13 Next, they demonstrated that mice deficient of an adaptor molecule essential for TLR-mediated cytokine induction or commensal bacteria were highly susceptible to chemically induced inflammation. These findings highlighted a mechanism whereby chronic intestinal inflammation resulted from defective TLR interactions with commensal bacteria. Furthermore, absence of the transcription factor complex NF-κB, a major downstream regulator of TLR receptor signaling, resulted in impaired antimicrobial peptide expression, increased bacterial translocation into the mucosa and onset of chronic intestinal inflammation. 14 Activation of an additional TLR pathway, tied to TLR2/1, has been shown to be an important mechanism for immune responses against intracellular Mycobacterium tuberculosis. 15 This antimicrobial response depends on vitamin D to promote production of cathelicidin, 16 and provides a possible linkage between vitamin D receptor (VDR) polymorphisms and IBD.17,18 These data highlight how interactions between TLRs, commensal bacteria and downstream signals regulate intestinal homeostasis, and how disruptions compromise tissue repair and promote chronic inflammation, and, ultimately, IBD.
A chief virulence factor, i.e. the ability to breach the basolateral membrane during the process of invasion, is generally not exhibited by commensal bacteria. Commensals generally remain within the lumen and interact passively with the apical membrane. Commensal stimulation of apical sensors by Ag prevents ubiquination of inhibitory NF-κB (IκB) in the cytoplasm, which restrains NF-κB activation.
19
Protection from pro-inflammatory cytokines supports tolerance and maintenance of intestinal homeostasis (Figure 2).
19
In contrast, bacterial Ags sensed by cellular receptors on the basolateral membrane evoke the opposite response, rapidly releasing NF-κB from IκB,20,21 leading to release of pro-inflammatory cytokines and initiation of an innate immune response. Similar responses accompany activation of intracellular NOD2 receptors, which detect the presence of bacterial peptides within the cytoplasm. Intracellular peptidoglycan binding to NOD2 results in an alternative pathway for activating NF-κB and downstream inflammatory cascade. NOD2 mutations within the leucine-rich repeat microbial sensing domain are strongly associated with CD, contributing to CD pathogenesis by paradoxically inhibiting NF-κB activation and subsequently down-regulating antimicrobial peptide production.
22
Though NOD2 is most commonly associated with CD, the broader family of intracellular NOD-like receptors form an important link between intracellular sensing of pathogenic proteins and inflammasome activation. The inflammasome is a multi-protein complex that combines with various NLRs to govern critical mucosal immune responses and intracellular surveillance. NLRs detect invading pathogens and initiate IEC secretion of pro-inflammatory cytokines IL-1β and IL-18 by activating caspase-1, an important effector of the inflammasome (Figure 2).
23
Although traditionally tied to inflammation, aberrant inflammasome signaling can disrupt homeostasis by promoting pro-inflammatory factor secretion while inhibiting release of homeostatic protective factors. This circumstance creates an imbalance in the factors that promote tolerance and maintain barrier function, weakening the epithelial barrier and increasing susceptibility of the mucosal immune system to inappropriate activation. These dynamics may promote progressive epithelial barrier damage, chronic inflammation, or induction of full-blown inflammation in those genetically susceptible to IBD.
24
The intestinal epithelium functions as a sentry, maintaining vigilance against invading bacteria. TLR signaling via NF-κB is an important component not only of pathogen sensing, but also in maintaining epithelial homeostasis. Commensal (purple bacteria) antigenic stimulation of apical TLRs results in inhibition of NF-κB activation thereby decreasing the release of pro-inflammatory cytokines. Invasive (green bacteria) bacterial Ags sensed by TLRs on the basolateral membrane evoke the opposite response, by activating a rapid release of NF-κB and promoting the release of pro-inflammatory cytokines. NLRs detect invading pathogens and initiate intestinal epithelial secretion of the pro-inflammatory cytokines IL-1B and IL-18 by activating caspase-1, an important activator of the inflammasome. NLRs can also stimulate NF-κB through an alternative pathway.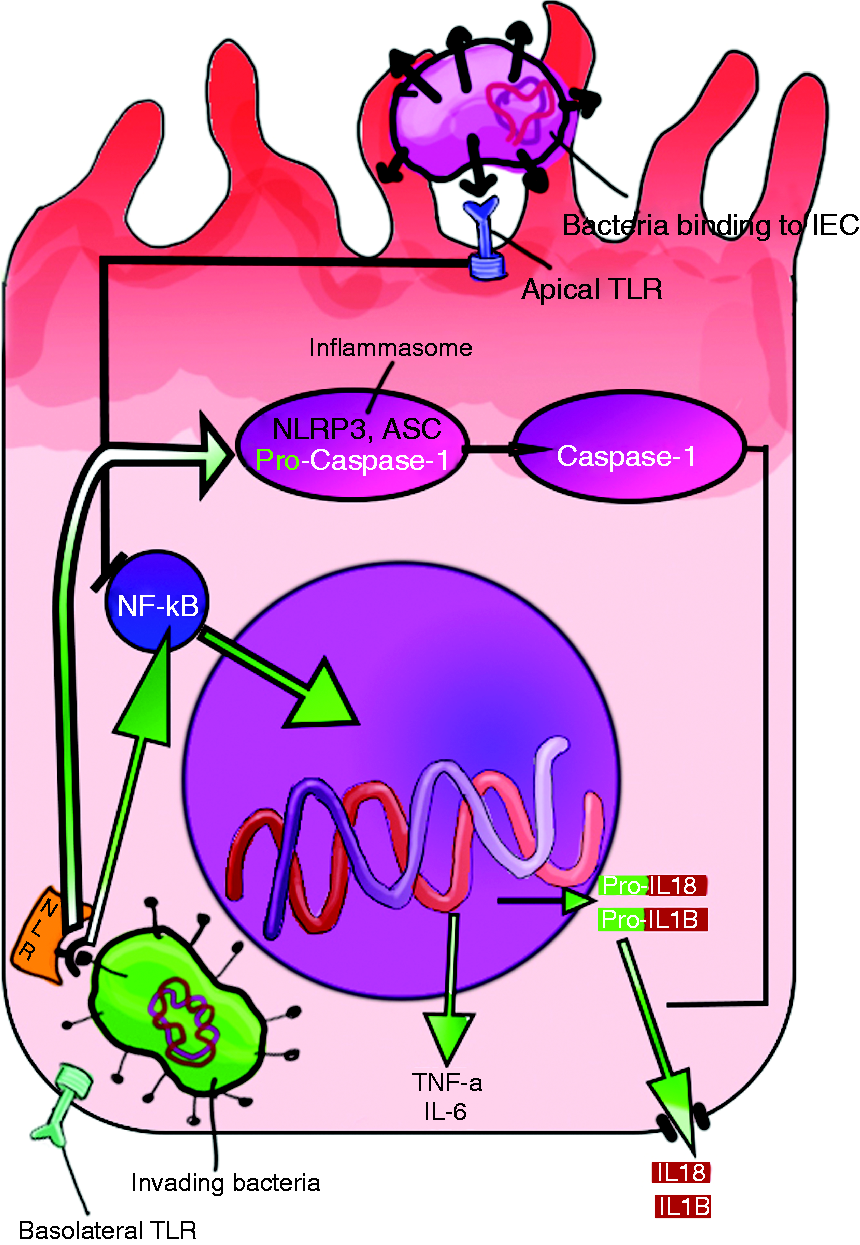
A basic premise for understanding CD and ulcerative colitis starts with the premise that the intestinal lining is the gateway to IBD. In accordance with that, all roads to inflammation start with IECs. Specifically, IECs constitute a crucial part of the innate immune system. IECs generally act as sentries, maintaining vigilance against invading hordes of bacteria while promoting intestinal homeostasis and tolerance with normal flora. These complex tasks are simultaneously accomplished by specialization of IECs into the form of goblet cells, M-cells and Paneth cells. Goblet cells primarily secrete mucins and create the first line of defense against microbial attachment and invasion. Intestinal commensals depend on mucus for anchorage and nutrients, in addition to undigested carbohydrates. The by-products of energy production enhance intestinal epithelial functions such as mucin synthesis and secretion via NLR-3-mediated pathways. 25 Goblet cells secrete mucin plus the proteins trefoil factor 3 (TFF-3) and resistin-like molecule-B. 11 TFF-3 plays an important role in the stabilization, maintenance and repair of the intestinal epithelial barrier. 26 Resistin-like molecule-B promotes mucin secretion and regulates macrophage and adaptive T-cell responses during inflammation. 11 Intestinal M cells deliver Ags to immature professional Ag presenting dendritic cells (DC), dampening responses to commensals and maintaining immune tolerance. 27
Contributors to inflammation and homeostasis
DCs link innate and adaptive immunity. They dwell in the lamina propria as immature cells to interact with the microbiota. Immature DC are highly phagocytic and avidly seek Ags. At this stage, they ineffectually stimulate adaptive T-cell responses, but once activated by sensing microbial Ags through their PRRs, they evolve into mature DCs. PRR activation also initiates cytokine secretion and induces migration of maturing dendritic cells to draining lymph nodes, where they finish maturating and serve as professional APCs to stimulate naïve T cells. The type of MAMPs and PAMPs that are encountered during maturation determine DC immune tone after maturation. 28 M cells facilitate wholesale transfer of luminal Ags and intact microorganisms to underlying mucosal immune cells, which are hungrily waiting to process these representatives of the microbiota. 29 M cells are capable of both non-specific and specific receptor-mediated microbial uptake, subsequently delivering Ag to underlying immune cells. Another, perhaps more prominent route of transepithelial Ag transfer, occurs via small-bowel goblet cells (GC). These cells have been observed to shuttle protein Ags across the epithelium through transient intracellular channels to waiting CD103+CD11c+ lamina propria DCs, a subset of APCs known for promoting IgA production, imprinting lymphocytes for gut homing and inducing regulatory T cells. 30 DC juxtaposed to GCs actively forming intracellular GC-associated Ag passages (GAPs) have been visualized taking up various labeled substances. 30 Subsequently, ingested Ag is processed and presented in the context of MHC to activate T cells in the draining lymph nodes (LN). 31 Acetylcholine, acting on the muscarinic acetylcholine receptor, drives GAP formation during basal conditions, mainly in goblet cells lining the small intestine. 31 Interestingly, the presence of bacterial commensals appears to limit the propensity of colonic GC to form GAPs, as the absence of bacterial components or TLR signaling pathways actually induces colonic GAP formation. 31 In fact, the administration of antibiotics promoted the translocation of colonic bacteria from the lumen to draining mesenteric LN via GAPs and CX3CR1+CD11b+CD103– DCs, 32 which are major inducers of effector T cells. 33 Additionally, sub-epithelial mononuclear phagocytes also sample luminal Ags via transepithelial dendrites. 34 The impact of these specific cells on the systemic immune system remains unclear, though they are one pathway known to activate naïve immune cells. Crypt-based Paneth cells, however, clearly reduce immune cell exposure to microbial Ags by secreting broad-spectrum antimicrobial peptides, such as alpha-defensins, cathelicidins and lysozymes. 35 While commensal Ags drive the deposition of this chemical barrier, 36 the final driver of unstirred water layer (bounded by the bottom of the mucin layer and the apical surface of the epithelium) sterility comes from active transport of secretory IgA across the epithelial barrier. 37 IECs carry the dimeric IgA made by lamina propria plasma cells into the lumen via the polymeric immunoglobulin receptor (pIgR). Down-regulation of IEC expression of pIgR, which occurs during IBD-related intestinal inflammation, potentially contributes to IBD pathogenesis. When aberrant cellular localization of pIgR disrupts dimeric IgA transcytosis, IgA subsequently accumulates in the lamina propria, leading to epithelial damage and barrier disruption. Diminished levels of luminal IgA levels can no longer neutralize microbes or prevent their breakdown products from penetrating the mucin layer. This defect in normal mucosal function adds yet another failure mode to the list of intestinal barrier defects that contribute to mucosal inflammation and disrupted microbial homeostasis; subsequent immune tolerance disintegrates and the dysregulated inflammation characteristic of IBD ensues. 37
Role of tight junctions in barrier function
In addition to the chemical and humoral barriers constructed by the IECs, a physical barrier is erected by connections between the cells themselves. The apical junction complex (AJC) is a dynamic gateway between intestinal epithelial cells. The AJC plays a critical role in resisting molecular movements across the epithelial barrier. This gateway is comprised of both apical tight junctions (TJ) and adherens junctions (AJ). The AJC initiates and maintains cell–cell contact, functions in cell–cell communication and regulates paracellular permeability. 38 The strands of the AJC form a continuous gasket around and between cells. A reduction in TJ strands along with TJ strand breaks give rise to one form of barrier dysfunction seen in IBD patients.39–41 A more global disruption of TJs results from increased pore forming claudin expression, potentiated by the inflammatory cytokines TNF-α, IFN-γ and IL-1β. Increased TNF-α up-regulates production of the pore-forming claudin 2, which enhances TJ permeability to cation flux and contributes to the clinical feature of diarrhea in IBD. 42 These cytokines induce paracellular permeability and intestinal barrier dysfunction, forging a strong link to active IBD. 43 Furthermore, inflamed mucosa from IBD patients exhibit reduced protein expression of the AJ constituents E-cadherin and α-catenin.44–46 These consequences of uncontrolled mucosal cytokine production directly compromise intestinal barrier function and facilitate the development of chronic intestinal inflammation. Closer examination of the interplay between IECs and the innate immune system reveals even deeper complexities. For instance, IEC-expressed E-cadherin plays a central role in maintaining both physical and physiologic barrier functions by providing mechanical connections between IECs. 47 E-Cadherin also promotes proper growth and positioning of goblet and Paneth cells. In fact, loss of E-cadherin compromises the frequency, placement and development of both goblet and Paneth cells. 48 Consequently, the expression of antibacterial peptides diminishes, clearance of enteropathogenic bacteria suffers and intestinal homeostasis is lost. 48 These conclusions, drawn from animal models, illustrate growing understanding of how genetic mutations influence IBD susceptibility.
Genetic contributors to IBD
Almost 200 mutations have been identified as IBD susceptibility loci,49,50 with many regulating the ability of IECs to insulate themselves from direct contact with the intestinal microbiota, or handle the stress of metabolic or environmental factors while maintaining normal cellular functions. Beginning with the apex of the intestinal epithelium, CDH1 gene polymorphisms identified in CD patients result in abnormal intracellular accumulation of E-cadherin and impair its localization to functional sites in intestinal epithelial cell membranes.
51
Another important contribution to the physicochemical barrier function of IECs stems from their secretion of mucins. The importance of mucin secretion to intestinal homeostasis became obvious after observing that mice missing the MUC2 gene developed spontaneous colitis.
52
The colitis appeared to be related to excessive interactions between IECs and bacterial Ags due to the loss of an insulating mucin barrier. An interesting corollary to this experiment was conducted in mice with genetically defective NOD-like receptor family pyrin domain containing 6 (NLRP6), an intracellular sensor of bacterial Ags that initiates innate immune responses through activation of the inflammasome.
53
In actuality, this experiment linked the sensing of commensal bacterial Ags to the secretion of mucin by IECs. Animals with defective NLRP6 were unable to activate the inflammasome complex in the presence of intestinal commensals, which, in turn, prevented activation of caspase-1 and the subsequent conversion of inactive cytoplasmic LC3-I to its active form, LC3-II.
53
LC3 activation promotes fusion of intracellular vesicles such as phagosomes to lysosomes or mucin granules to the apical epithelium of goblet cells (Figure 3). IECs lining the intestine of NLRP6-deficient mice demonstrated highly engorged mucin granules confined to the cytoplasm and an absent mucin layer.
54
The mutant mice developed colitis when exposed to commensal organisms, as the absent mucin layer allowed bacteria to closely associate with IECs and subsequently activate innate immune mechanisms.
Defect in autophagy blocks NLRP6 conversion of LC3 I to LC3 II. LC3 II promotes fusion of intracellular vesicles such as phagosomes to lysosomes or GC mucin granules to the apical epithelium, and its absence prevents mucin granules from fusing with the apical membrane. Thus, an adequate mucin layer is not produced. Now bacteria are able to closely associated with the mucosa in the absence of a mucin layer and promote a state of chronic inflammation.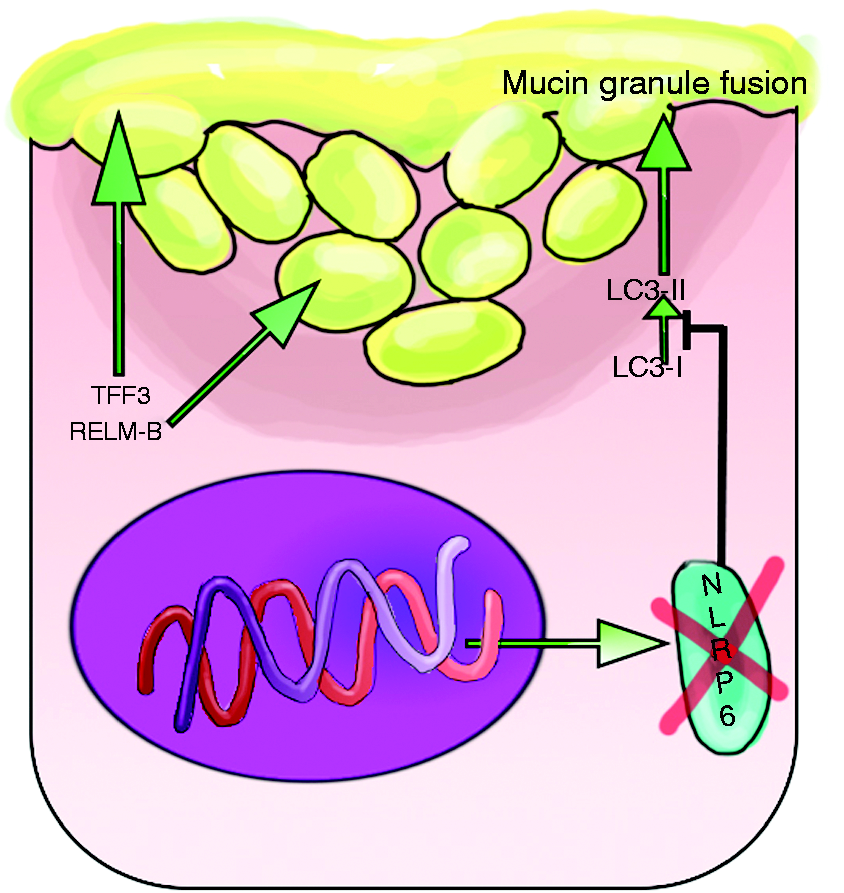
Under other conditions of cellular stress, such as those encountered by IECs in the setting of dysbiosis or overt inflammation, a variety of intracellular processes begin to go awry. The orderly folding of newly formed proteins in the endoplasmic reticulum may become disrupted during the early phases of cellular stress. Accumulating misfolded proteins can quickly glom up normal cellular metabolism and initiate orderly death of the cell through the process of apoptosis. Prior to that tipping point, cells employ a rescue technique called the unfolded protein response (UPR) that involves up-regulation of X-box protein-1 (XBP-1) and subsequent production of protective chaperone proteins that help normalize protein folding. 55 The presence of a mutated form of XBP-1 helps explain why some IBD patients have difficulty maintaining barrier function in the setting of stressful events, such as intestinal infection or dysbiosis. 56 In the event that the UPR misfires or cannot rescue the cell from its dysfunctional state, the autophagy pathway kicks in. Autophagy is a process that allows stressed cells to eliminate damaged proteins or recycle damaged organelles into additional metabolic substrates, or eliminate intracellular pathogens. Defects in the autophagy pathway decrease IEC resilience in the setting of metabolic or environmental stressors. This pathway highlights another important CD susceptibility factor, one encoded by the single nucleotide polymorphism ATG16L1T300A, which causes threonine to be substituted for alanine in this important autophagy protein. This substitution makes the autophagy protein ATG16L1 susceptible to cleavage by caspase-3, which becomes activated during stress scenarios involving starvation-induced metabolic stress, death receptor activation by TNF/TNF-α or intestinal infection by the pathogenic gut bacterium Yersinia enterocolitica. 57 Downstream consequences of inappropriate ATG16L1 cleavage include Paneth cell dysfunction in CD patients and impaired mucin production, mounting endoplasmic reticulum stress and bacterial persistence. 58 Mice engineered to express the mutant ATG16L1 allele exhibit enhanced NF-κB activation and transmural inflammation that develops into a spontaneous enteritis. 59 One final supportive role played by IECs in intestinal homeostasis and immune tolerance comes from the secretion of mucosal support factors. The importance of these factors first became evident in mice genetically incapable of secreting the intestinal homeostasis factor TFF-3. TFF-3 knockout mice are more susceptible to dextran sodium sulfate (DSS)-induced colitis than wild-type animals. The administration of recombinant TFF-3 improves colitis by restoring their capacity for epithelial restitution. 60 These examples highlight the methods IECs use to insulate themselves and the underlying mucosal immune system from direct contact with commensal organisms, and how IECs from genetically susceptible IBD patients malfunction under the stress of infection or dysbiosis. The final variable in the inflammation equation ties abnormal cell function to impaired antimicrobial or growth factors and explains how disturbances in the host–microbe balance at the mucosal interface directly play an important role in IBD pathogenesis.
Mutations within NOD2, the first susceptibility gene identified in CD patients,61,62 result in reduced Paneth cell secretion of defensins, leading to a weakened antimicrobial barrier through enhanced cytokine secretion, immune activation by either normal or dysbiotic intestinal flora and promotion of ileal inflammation in CD.63–65 Recent studies have revealed that bacterial muramyl dipeptide-mediated NOD2 activation also results in induction of autophagy. 66 However, frame shift mutations within the NOD2 gene result in defective intracellular pathogen killing due to defects in the autophagy response. 67 Impressively, mice homozygous for NOD2 frame shift mutation had failure of ATG16L1 recruitment to the plasma membrane and consequent loss of autophagosome formation for bacterial killing. 68 Hence, NOD2 plays an important role in detecting bacterial and viral pathogens, linking detection to antimicrobial peptide secretion and coordinating autophagy through ATG16L1. Therefore, any disruption of NOD2-mediated influence on normal biological activity puts one at risk for IBD on multiple fronts. Wnt signaling is another pathway involved in Paneth cell positioning, differentiation and maturation. 69 Activation of the Wnt pathway results in the formation of a B-catenin/transcription factor (TCF)-4 complex, where TCF-4 acts as a transcription factor affecting downstream gene expression. Diminished levels of Wnt induce TCF4 assembly directly impacting gene expression of alpha defensins and led to a significant decrease in antimicrobial peptide production in mice.35,70 These findings were independent of the NOD2 genotype and led to the discovery of single nucleotide polymorphism variants of TCF-4 that result in reduced mRNA expression of TCF-4 in many patients with ileal CD. 71 Interestingly, 1,25(OH)2D3 augmentation has been tied to enhanced TCF-4 expression, 70 providing yet another mechanism by which VDR receptor polymorphisms contribute to IBD susceptibility.
Conclusion
To conclude, early observations in both humans and animals called attention to the role of luminal bacteria in IBD. Advances in both genetics and microbiology/immunology have provided tools and targets for sorting out the complex relationship between genes and IBD first documented in twin studies. 72 Since then, accumulating evidence strongly links genetic mutations encoding defects in immune system activities, host defense and intestinal barrier function to altered responses to the intestinal microbiota. These relationships provide great insight into the foundational causes of IBD. This paper has reviewed how key elements within the adaptive and innate arms of the immune system catalyze the IBD phenotype in response to altered environmental cues. The complex interplay between the microbiota, immune tolerance and intestinal barrier function has also been highlighted. Established on a backdrop of genetic predispositions and environmental insults, IBD arises within the context of complex interactions between the intestinal microbiota, a dysregulated immune system, and a dysfunctional intestinal epithelial barrier. Given the number of variables in play, it becomes clear why no singular factor can be identified as a cause of IBD. Instead, the inciting events leading to IBD encompass the interplay of dysbiosis combined with compromised barrier function, to initiate aberrant immune activation. As our understanding of the disease process drives new therapies, methods of elucidating the predominant factor(s) leading to an individual’s IBD susceptibility will become crucial for tailoring the right therapy to the appropriate patient. The customization of personalized treatment plans based on individual susceptibilities holds the greatest promise for future successes.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.