
Abstract
Complement is an important arm of the innate immune system. Recent studies have shown that products of complement pathway activation can interact directly with other innate immune signaling molecules, including TLRs and inflammasome family members, during some infectious and chronic inflammatory disorders. Activation of the complement system generates anaphylatoxins, such as C3a and C5a, which modulate inflammation. However, the biological effects of interactions between the anaphylatoxins with their receptors may vary across species. In this study, we demonstrate that human complement and rat complement differ in the way they modulate the inflammatory response to the human pathogen, Neisseria gonorrhoeae, as well as purified pathogen-associated ligands, such as LPS. While rat serum down-regulates MyD88-dependent pro-inflammatory cytokine responses in macrophages, human serum has no effect, or in some cases an enhancing effect. Further, the inhibitory effect of rat serum on otherwise pro-inflammatory stimuli is mediated by complement, specifically C3a–C3a receptor interactions, via an undefined signaling mechanism that down-regulates the transcription factor, NF-κB and NLRP3 inflammasome-mediated caspase-1 activation. This study highlights important functional differences between rodent and human complement that could explain some of the differences in immune responses between these two species. Understanding the crosstalk between complement and other arms of the innate immune system will facilitate the development of better anti-inflammatory therapeutics.
Introduction
Complement is an essential part of the innate immune system and is often the first line of defense against many pathogens.1,2 In the fluid phase, the complement system comprises >30 proteins that include activators and inhibitors. Activation of complement occurs through three major pathways: the classical pathway, the alternative pathway and the lectin pathway. All three pathways converge at the formation and activation of C3 convertases; the classical and lectin pathways form C4bC2a, whereas the alternative pathway forms the C3bBb complex. Addition of another C3b molecule to each of the C3 convertases to form C4b2aC3b and C3bC3bBb imparts C5 convertase activity. Cleavage of C5 is the first step in assembly of the membrane attack complex (MAC; C5b-9). The byproducts generated during complement activation can be divided into three major groups, based on their function. They are the anaphylatoxins, C3a, C4a and C5a, potent pro-inflammatory and chemoattractant molecules, which mediate leukocyte activation and recruitment; opsonins, such as C3b, iC3b and C3d, which coat the surface of pathogens, enhancing uptake by antigen presenting cells such as neutrophils, macrophages and dendritic cells; and, finally, components of the membrane attack complex, MAC, a pore that kills Gram-negative pathogens and altered ‘self’ cells by direct lysis. Several recent studies have highlighted the role of these complement proteins or byproducts in cross-regulating other signaling pathways in the immune system.3,4
The mechanism by which complement regulates the immune response is unclear, and may vary according to the particular complement protein examined and the experimental system. This has led to a number of seemingly contradictory reports in the literature. While most of the studies performed with human complement show a positive or pro-inflammatory role for complement in modulating the immune response, studies in mice suggest that complement negatively regulates inflammation. For example, the anaphylatoxins C5a and C3a increase inflammasome activation by LPS in human monocytes. 5 C5a also up-regulates cholesterol crystal-induced NLRP3 inflammasome activation, IL-1β, TNF-α and reactive oxygen species production in macrophages, 6 and up-regulates IL-6 and the pro-inflammatory Th-17 pathway. 7 In contrast, studies in mice suggest that C5a down-regulates the TLR4 and CD40-mediated induction of IL-12 family cytokines, 8 and down-regulates the IL-17/IL-23 axis by induction of IL-10 through a phosphoinositide 3-kinase-ERK1/2-mediated mechanism. 9
One paradox in microbial pathogenesis is that microorganisms can both activate and evade the innate immune system. This is evident with Neisseria gonorrhoeae, a sexually transmitted Gram-negative pathogen that activates multiple pro-inflammatory signaling pathways, including TLRs and the inflammasome,10,11 while at the same time both activating and evading complement activation.12,13 We surmised that N. gonorrhoeae would be an ideal model pathogen to study the interaction of the complement and TLR signaling pathways, and specifically to examine the species-specific effects the host-derived complement proteins given the conflicting data in human and rodent systems. We investigated the effects of complement from two different species, humans and rats, on the inflammatory response in macrophages treated with N. gonorrhoeae or purified TLR ligands. Our data demonstrate that species-specific differences in functionality could determine the outcome of complement-mediated regulation of innate immune responses in humans compared with rodent models of infection and immunity.
Materials and methods
Bacterial strains
Neisseria gonorrhoeae strains MS11, FA1090, FA19 and F62 were used in this study.14,15 The bacteria were streaked onto chocolate agar plates and grown overnight (∼15 h) at 37℃ in an atmosphere enriched with 5% CO2. Bacteria were suspended in GCB media containing Isovitalex to an OD600 of 0.2 and grown at 37℃, with shaking (225 rpm) until an OD600nm of 0.6 was reached. The bacteria were then diluted to an OD600 of 0.3 (∼108 bacteria/ml) for use in experiments. All N. gonorrhoeae–macrophage experiments were done at a MOI of 5:1.
Cell culture
Peripheral blood mononuclear cells from de-identified human blood (NY Biologics, New York, NY, USA) were isolated using Ficoll Hypaque. The cells were cultured in RPMI 1640, 10% FBS, 10% human Ab serum (source for human M-CSF), 100 mM HEPES (pH 7) and pencillin/streptomycin for 5 d. On d 6 the cells were harvested in cold 1 × PBS and re-plated for experiments. Mouse bone marrow-derived macrophages (BMDMs) were isolated from wild type, C3a receptor-deficient (C3aR−/−), 16 C5a receptor-deficient (C5aR−/−) 17 mice in C57BL/6 background. The BMDMs were cultured in RPMI 1640, 10% FBS, 10% L929 cell culture media and penicillin/streptomycin for 6 d. Cell culture media was usually replenished on d 4. On d 7, the cells were harvested in cold 1 × PBS and re-plated for experiments in RPM I 1640 with only 10% FBS and no antibiotic.
Serum
Sprague Dawley rat serum was purchased from Innovative Research (Novi, MI, USA). Human serum pooled from six healthy donors was obtained under a protocol approved by the Institutional Review Board at the University of Massachusetts Medical School (Worcester, MA, USA).
Serum bactericidal assays
N. gonorrhoeae harvested from an overnight culture on chocolate agar plates were diluted to an OD600 of 0.3 (108 bacteria/ml) in Hanks Blank Salt Solution (HBSS) media containing 1 mM CaCl2 and 0.2 mM MgCl2 (HBSS++). Approximately 100 µl of the suspension (107 bacteria) was incubated with serum at the concentrations specified for each experiment. Aliquots of 25 μl of the reaction mixtures were serially diluted 1:10 and plated onto chocolate agar in duplicate at the beginning of the assay (t0) and again after incubation at 37℃ for 10 min (t10). Percentage killing was calculated as (CFU at t0 – CFU at t10) ×100/CFU at t0.
Serum based assays
N. gonorrhoeae strain MS11 was propagated as described above. For serum-based assays, the final dilution of sub-culturing of bacteria was done in HBSS++. The bacteria were incubated with a designated amount of rat or human serum for 10 min, which permits sufficient complement deposition for opsonization, but without significant loss of bacterial viability (Figure S1). C3 Fragment (C3b/iC3b) deposition on the surface of bacteria was confirmed by flow cytometry as follows. Following incubation with human or rat serum for 10 min at 37℃, gonococci were treated with FITC-labeled polyclonal goat anti-human C3 (AbD Serotec/BioRad, Hercules, CA, USA). The bacteria were washed in HBSS, fixed with 1% paraformaldehyde in PBS, and analyzed by flow cytometry on a BD FACScan (BD Biosciences, San Jose, CA, USA) using FlowJo software (Tree Star, Ashland, OR, USA). Following treatment with serum, bacteria were added to a monolayer of 0.5 × 106 cells human monocyte-derived macrophages (MDM) or mouse BMDM at a MOI of 5:1 and incubated overnight at 37℃ in a 5% CO2 incubator.
Chemical inhibitors and peptides
C3a receptor inhibitor, SB290157 was purchased from Sigma Aldrich (St. Louis, MO, USA). The macrophage culture was pre-incubated with the inhibitor SB290157 at low nanomolar concentrations for 1 h before stimulation with N. gonorrhoeae or LPS from Escherichia coli; at high concentrations, the inhibitor act as an agonist rather than an antagonist. 18 Purified recombinant human C3a was obtained from R&D Systems (Minneapolis, MN, USA).
Cytokine expression
Macrophage cultures plates in 24-well plates at a density of 0.5 × 106 cells per well in RPMI 1640 (10% FBS) were incubated with N. gonorrhoeae strain MS11 (MOI: 5:1) or stimulated with LPS (100 ng/ml) from E. coli overnight. Stimulation with Pam3CSK4 (100 ng/ml), poly I:C (5 µg/0.5 × 106 cells) and poly I:C (2 µg/0.5 × 106 cells) with lipofectamine were carried out similarly. The tissue culture supernatants following overnight stimulation were collected and analyzed for pro-inflammatory cytokine/chemokine levels using the following commercially available ELISA kits: human ΙL-1β, IL-6, CCL5 and CXCL10, and mouse IL-1β and CCL5 (R&D Systems); human and mouse TNF-α (eBiosciences, San Diego, CA, USA). The cells treated with LPS overnight were also treated with ATP for 30 min prior to harvesting supernatants for inflammasome activation.
Western blotting
Human macrophages were plated in six-well plates overnight at density of 1 × 106 cells/ml RPMI (10% FBS, penicillin/streptomycin). The cells were then stimulated with either N. gonorrhoeae MS11 at a MOI of 5:1 or E. coli LPS (100 ng/ml) for 5 min in RPMI (10% FBS, no antibiotic). The cells were harvested in cold 1 × PBS and lysed in 1 × RIPA buffer (complete protease inhibitor cocktail, 1 mM EDTA, sodium orthovanadate, sodium fluoride, PMSF, 1 mM DTT) for 1 h on ice. The cell lysates are then collected and measured for protein concentration using Pierce BCA assay (ThermoFisher Scientific, Waltham, MA, USA). For measuring NF-κB activation and caspase-1 activation, the cell lysates are run on a 10% Bis-Tris Gel and transferred to a PVDF membrane followed by Western blot with IκB, caspase-1, caspase-1 p20 and GAPDH (all from Cell Signaling Technology, Danvers, MA, USA) and secondary goat anti-rabbit IgG-HRP Ab (EMD Millipore, Temecula, CA, USA).
Caspase-1 activation and apoptosis assays
Human MDM were incubated with N. gonorrhoeae strain MS11 at 37℃, 5% CO2 for 4 h. The cells were then harvested in cold PBS and active caspase-1 stained using the FAM-FLICA Caspase-1 Assay Kit from ImmunoChemistry Technologies (Minneapolis, MN, USA), according to the manufacturer's protocol. Fluorescence was measured by flow cytometry using BD FACScan and the FlowJo data analysis platform at the Boston University Flow Cytometry Core.
Immunofluorescence
Human MDM were seeded overnight on sterile cover slips in 24-well plates at a density of 0.3 × 106 cells/well in RPMI 1640, 10% FBS. N. gonorrhoeae strain MS11 (OD600 0.3) was stained with CFSE stain for 30 min in HBSS++ at 37℃ and 5% CO2. The CFSE-stained bacteria were then treated with human or rat serum (20% v/v) for 10 min at 37℃, and used to infect human MDM at a MOI (5:1) for 3 h at 37℃ and 5% CO2. The infected macrophages on the coverslips are washed with warm 1 × PBS and fixed with 100 μl 4% paraformaldehyde for 10 min at room temperature (20–22℃). The cells were then stained with mouse monoclonal anti-H.8 Ab supernatant, 2C3 (1:2 dilution) and goat anti-mouse IgG-PE (1:2000) for 1 h at room temperature, for visualizing cell surface adhered extracellular bacteria (red/orange bacteria/Texas Red). Intracellular bacteria are not stained with 2C3 Ab, and hence green in color (CFSE staining/FITC). The cells were washed with warm 1 × PBS and stained with Evans Blue (macrophage surface staining/Texas Red) and DAPI (nuclear staining). The cover slips are mounted on glass slides and the macrophages and level of N. gonorrhoeae infection is determined by fluorescence microscopy using Olympus Microscope BX50.
Statistics
Fold changes in cytokine induction relative to N. gonorrhoeae or LPS stimulation alone is plotted as mean of multiple independent experiments with SEs. An unpaired two-tailed Student t-test was done to determine the significance of differences, with P-values as indicated.
Results
Rat serum, but not human serum, down-regulates cytokine secretion in response to N. gonorrhoeae and TLR ligands
We first determined the effects of human and rodent serum on the induction of pro-inflammatory cytokines/chemokines in human MDM in response to N. gonorrhoeae strain MS11 (Figure 1). Bacteria were pre-incubated with either 20% or 50% human or rat serum for 10 min at 37℃, and added the mixture to the human macrophage culture at a MOI of 5:1. This incubation period ensured opsonization of bacteria, measured as C3 fragment deposition, but did not permit bactericidal activity (data not shown). In this case, the experimental treatment resulted in both bacterial bound, as well as soluble serum, factors generated during complement activation. We observed that while human serum had no effect on, or in some cases augmented, cytokine induction, rat serum significantly down-regulated N. gonorrhoeae induced IL-1β (Figure 1, upper panel) and TNF-α (Figure 1, middle panel) secretion. Interestingly, rat serum had no such down-regulatory effect on the production of the chemokine CCL5, (Figure 1, lower panel). However, when the bacteria were washed after pre-incubation with serum to remove all soluble serum factors, the inhibitory effect of rat serum on both IL-1β and TNF-α production in macrophages in response to N. gonorrhoeae was abrogated. Additionally, this inhibitory effect of rat serum on pro-inflammatory cytokine induction was lost upon heat inactivation of serum.
Effect of rat serum and human serum on cytokine secretion in human macrophages. Human MDMs were infected with N. gonorrhoeae (GC) strain MS11 (MOI 5:1) for 16 h. GC were pre-incubated with 20% or 50% (volume/volume) or heat-inactivated (HI), 20% (v/v) pooled normal human serum (solid gray bars) or corresponding concentrations of fresh or heat-inactivated rat serum (white bars) as indicated on x-axis for 10 min and then added to the human macrophages. Serum containing bacteria were added either directly (solid gray and white bars) or after washing with HBSS++ to remove soluble complement fragments (gray or black striped bars, for human and rat sera, respectively). Stimulation with GC alone (without any serum pre-treatment) is shown by the solid black bar. Stimulation of macrophages with LPS (100 ng/ml) and ATP without (solid black bar) or with human (solid gray bar) or rat serum (white bar) were used as controls. The y-axis shows the fold change in production of IL-1β (top graph), TNF-α (middle graph) and CCL5 (bottom graph) by human macrophages upon each treatment relative to GC or LPS alone, respectively, was calculated. Mean (SEM) of three independent experiments is shown; *P < 0.05; **P < 0.005. n.s.: non-significant.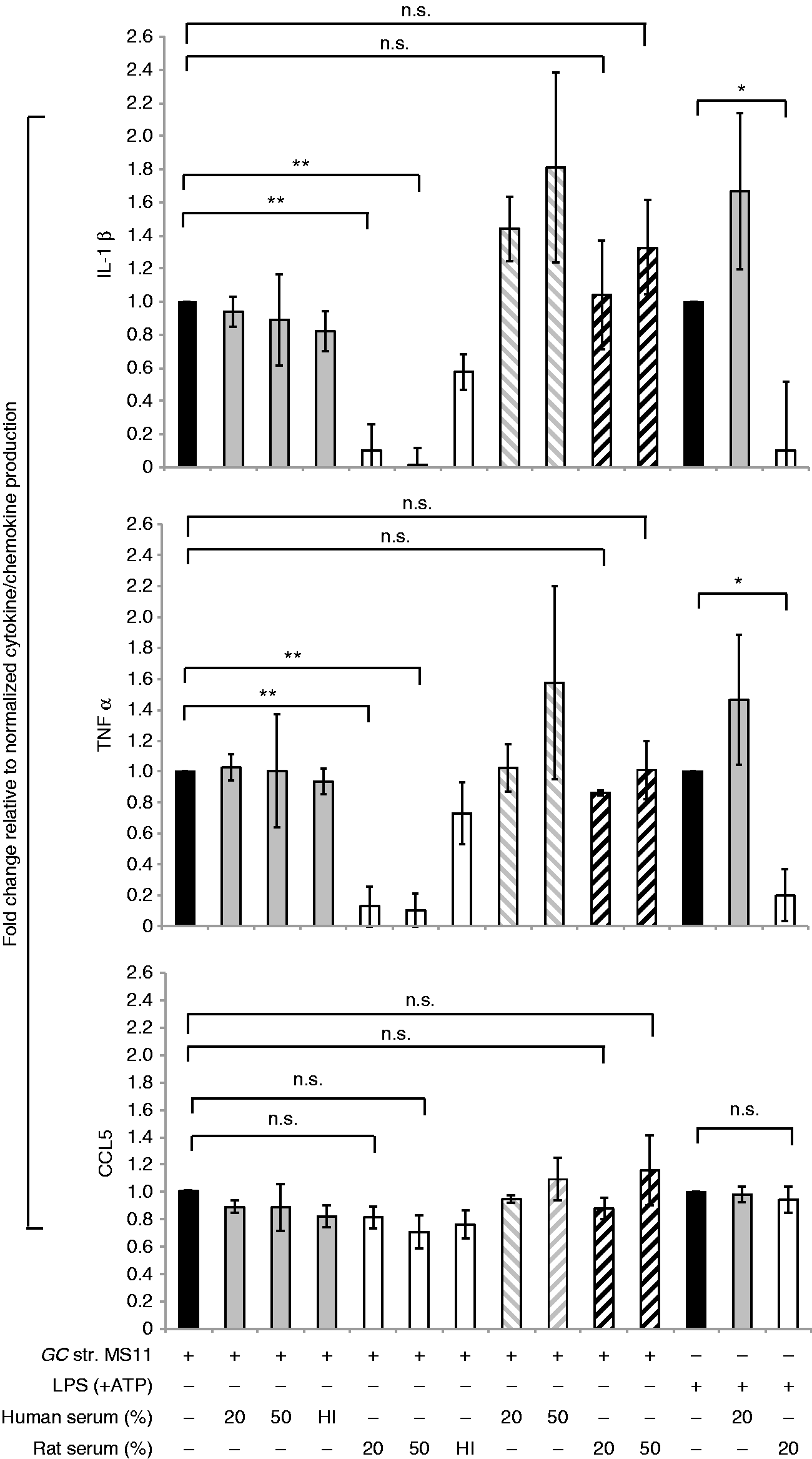
The down-regulatory effect of rat serum on pro-inflammatory cytokine production in macrophages was not due to increased bactericidal activity against N. gonorrhoeae because there was no statistically significant difference in bacterial killing by human or rat serum under these experimental conditions (Figure S1). The inhibitory effect of rat serum on macrophage inflammatory responses was not due to an effect on macrophage cell viability since rat and human serum had similar effects on macrophage viability, as measured by propidium iodide staining (data not shown). We further tested the ability of human and rat serum to regulate pro-inflammatory cytokine induction in human macrophages in response to other strains of N. gonorrhoeae, specifically FA1090, F62, FA19 and MS11—strains that differ in their ability to resist complement.19,20 As shown in Figure S2, rat serum down-regulates both IL-1β and TNF-α induction in human macrophages in response to all strains of N. gonorrhoeae.
These data suggested that the species from which the serum was derived could determine the response of human macrophages to these stimuli. We next asked if the species from which the macrophages were derived also influenced the response. As shown in Figure 2, human and rat serum had the same effects on mouse BMDM that were observed in human macrophages, where rat serum dampened the induction of IL-1β and TNF-α secretion, whereas human serum did not; and, as observed in the human macrophages, there was no effect on CCL5 (Figure 2).
Effect of rat serum and human serum on cytokine secretion in mouse macrophages. Mouse BMDMs were infected with N. gonorrhoeae (GC) strain MS11 (MOI 5:1) for 16 h. GC were pre-incubated with either 20% or 50% (v/v) pooled normal human serum or heat-inactivated (HI) human serum (solid gray bars) or rat serum (white bars), and the entire reaction mixture was then added to human macrophages. Stimulation of macrophages with LPS (100 ng/ml) without (solid black bar) or with human (solid gray bar) or rat (white bar) serum were used as controls as indicated on the x-axis. The y-axis shows the fold change in production of IL-1β (top graph), TNF-α (middle graph) and CCL5 (bottom graph) by mouse BMDMs upon each treatment relative to GC or LPS treatment alone, respectively, was calculated. Mean (SEM) of three independent experiments is shown; *P < 0.05; **P < 0.005. n.s.: non-significant.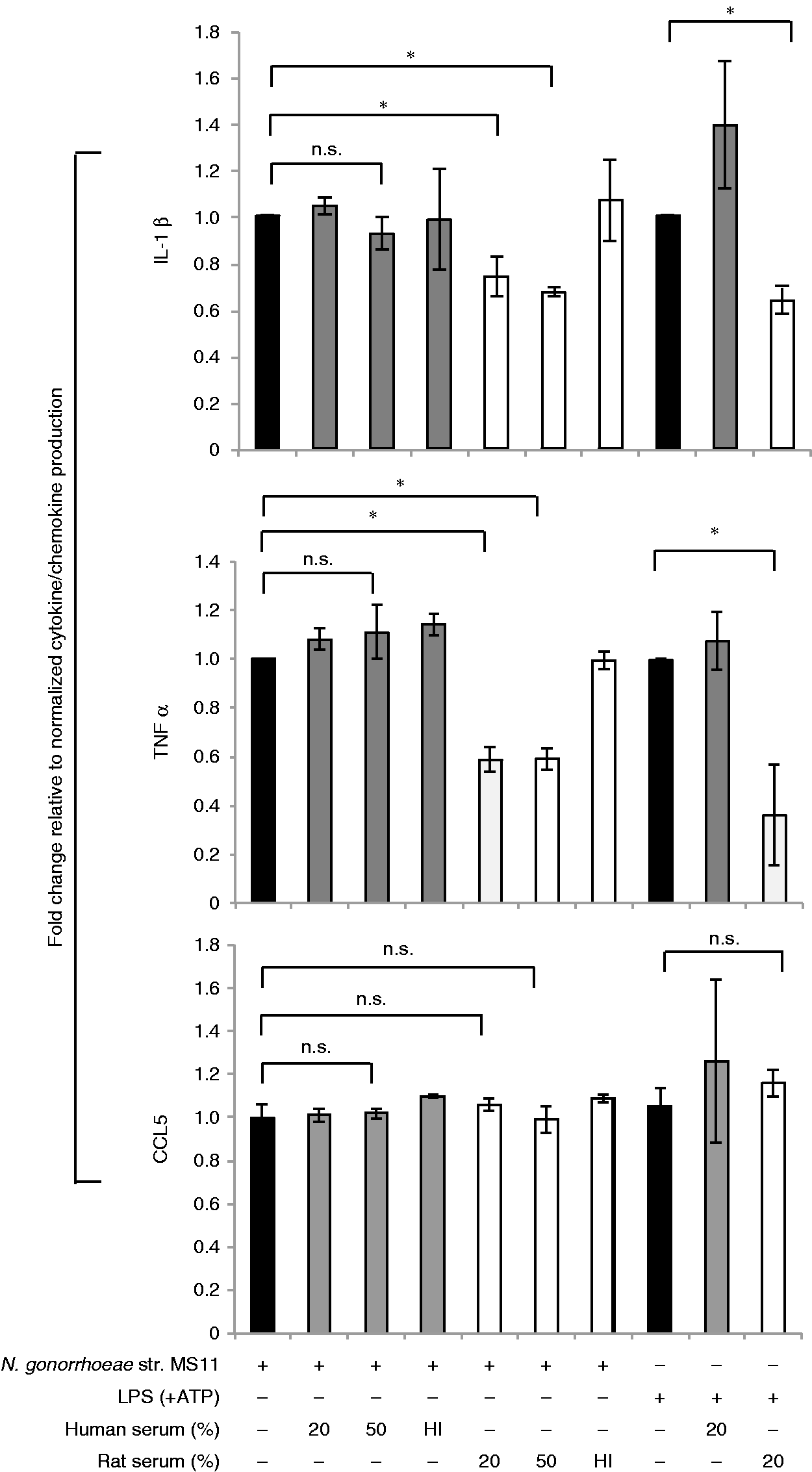
Finally, the anti-inflammatory effect of rat serum was not specific to N. gonorrhoeae-induced complement activation or pro-inflammatory signaling because we observed the same effect when cells were stimulated with the TLR4 ligand, LPS (Figures 1 and 2; far right). Similarly, rat serum inhibited TNF-α and IL-6 secretion in response to the TLR2 ligand, Pam3CysSK4 (Figure 3a). However, there was no effect of rat serum on poly I:C treatment, with or without lipofectamine, which directs the ligand to the cytosolic receptor RIG-I or endosomal TLR3, respectively (Figure 3b).
Effect of human and rat serum on MyD88 dependent vs. independent cytokine induction. Human MDM were stimulated for 16 h with either (a) Pam3CysSK4 (100 ng/ml) for TLR2 activation or with (b) poly I:C (2 μg/ml) alone for TLR3 activation and poly I:C (5 μg/ml) with lipofectamine for RIG-I activation. The macrophages were stimulated either with Pam3CSK4 or poly I:C alone (solid black bar) or with either 20% (v/v) pooled normal human serum (solid gray bars) or rat serum (white bars) as indicated on x-axis. The y-axis shows the fold change in production of (a) IL-6 and TNF-α and (b) CXCL10 and CCL5, produced by human macrophages upon each relative to treatment with Pam3CSK4 or poly I:C alone as calculated. Mean (SEM) of three independent experiments is shown; *P < 0.05; **P < 0.005. n.s.: non-significant.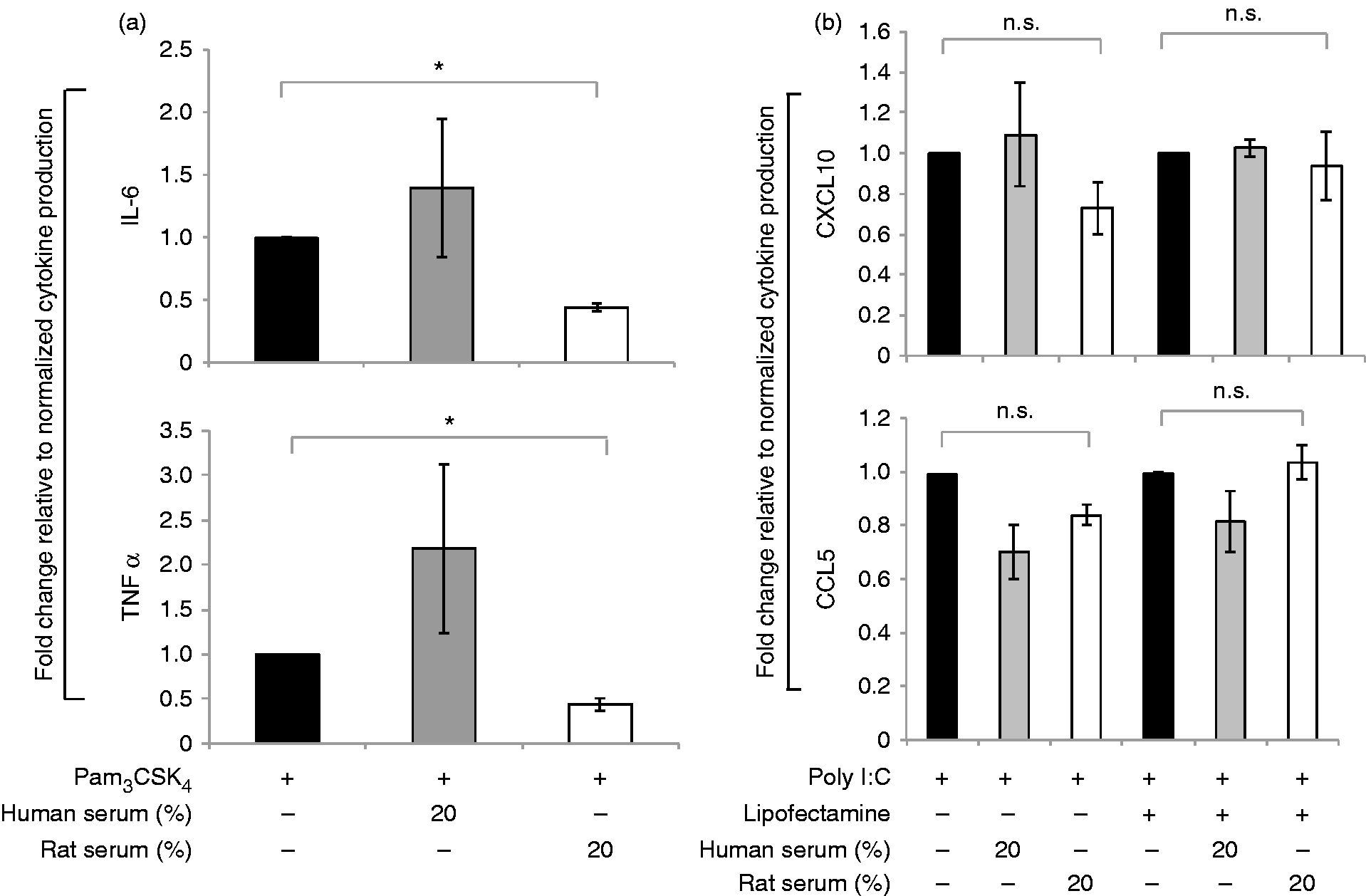
From the data obtained thus far we conclude that a heat-labile, soluble serum factor in rat serum could inhibit MyD88-dependent pro-inflammatory cytokine secretion in macrophages in response to N. gonorrhoeae and MyD88-dependent TLR ligands.
The opposing effects of human and rat serum on cytokine production
Our observation that human and rat sera have opposing effects on the regulation of a cytokine in response to inflammatory triggers led us to ask if the effect of human serum could override the down-regulatory effect of rat serum on cytokine induction in macrophages and vice versa. We stimulated human macrophages with N. gonorrhoeae strain MS11 pre-incubated with a fixed amount (20% v/v) of rat serum either alone or with increasing proportions of human serum (20% or 50% v/v). In the converse experiment, we added increasing amounts of rat serum while maintaining a fixed concentration of human serum. We observed that the effects of the serum from one species could stoichiometrically outcompete the regulatory effects of serum from the other species (Figure 4a, b). Thus, if human serum was in excess the dampening effect of rat serum was lost, and if rat serum was in excess, the neutral or pro-inflammatory effect of human serum was opposed.
Human and rat serum compete for the same target on macrophages. Human MDM were infected for 16 h with (a) N. gonorrhoeae strain (GC) MS11 (MOI 5:1) alone, or (b) E. coli LPS (100 ng/ml) with ATP; either alone (solid black bars) or pre-treated with a steady concentration of rat serum (10%) mixed with increasing concentrations of human serum (10% or 20%) (white bars) or a steady concentration of human serum (10%) mixed with increasing concentrations of rat serum (10% or 20%) (solid gray bars) as indicated on the x-axis. The y-axis shows the fold change in the concentrations of IL-1β (a, b, top graphs) and TNF-α (a, b, bottom graphs) produced by human macrophages upon each treatment condition relative to treatment with GC or LPS alone as calculated. Mean (SEM) of three independent experiments is shown; *P < 0.05; **P < 0.005.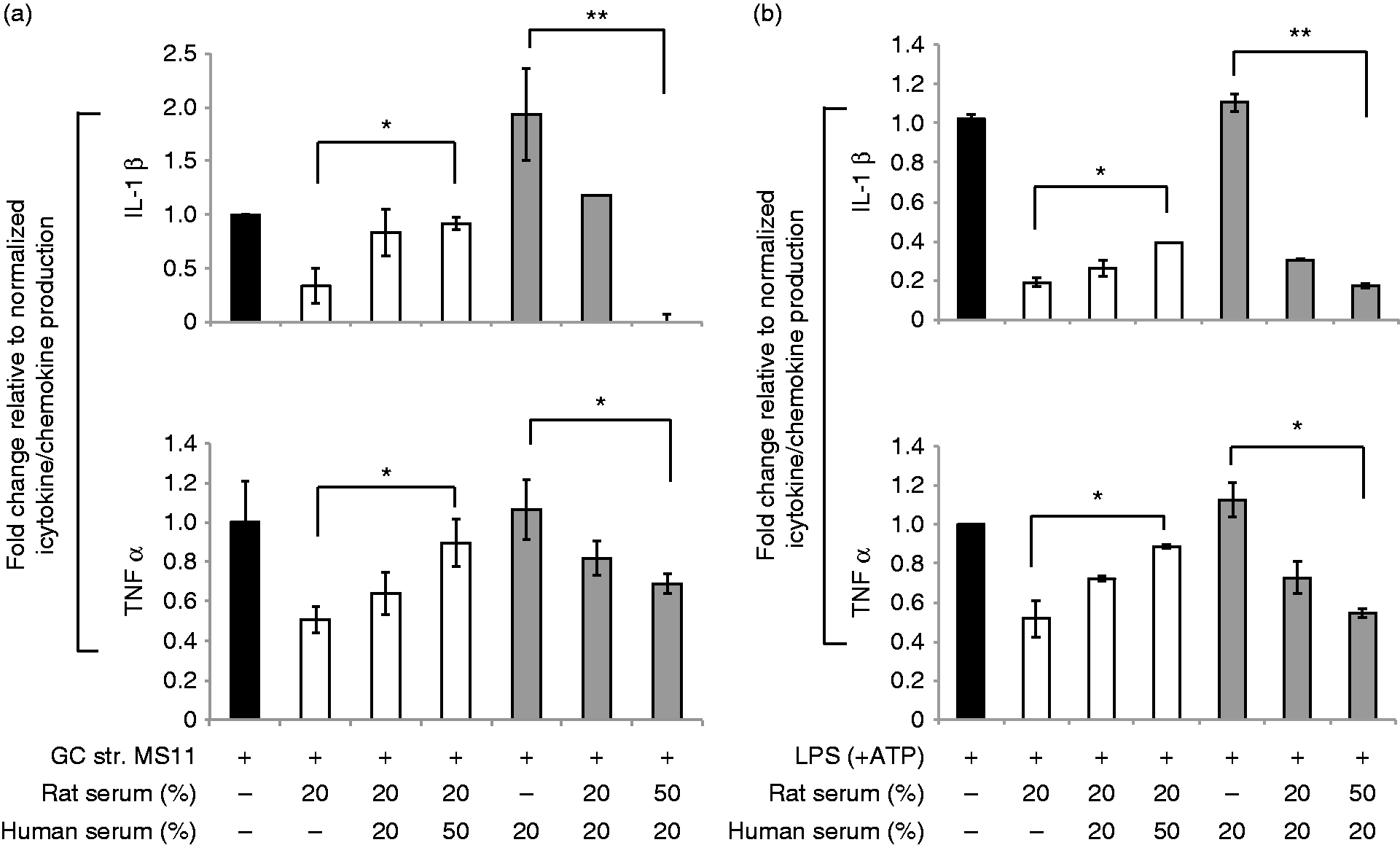
Rat C3a down-regulates pro-inflammatory cytokine responses in macrophages
We next sought to identify if specific complement proteins were responsible for the inhibitory effect of rat serum on production of pro-inflammatory cytokines. Recent studies have indicated that the soluble complement proteins C3a and C5a can regulate innate immune signaling; however, the role of these anaphylatoxins in the innate immune response is complex and varies with the complement source, model system and cellular context.21,22 We hypothesized that the rat serum-dependent phenotype was mediated through the C3a or C5a receptor using three different approaches. First, we examined the requirement for expression of the C3a receptor (C3aR) 16 or C5a receptor (C5aR) 17 using mice deficient in these respective receptors, and compared them with C57BL/6 wild-type mice. While rat serum inhibited the secretion of TNF-α in response to N. gonorrhoeae in the C57BL/6 and C5aR-deficient macrophages in a similar manner, the rat serum had no effect on TNF-α in the C3aR deficient cells (Table S1). There was similar trend with IL-1β secretion although it did not reach statistical significance.
We next used a commercially available inhibitor of C3aR signaling, SB290157. 23 Pre-incubating human macrophages with increasing concentrations of the C3aR inhibitor in the nanomolar range resulted in loss of the rat serum-mediated down-regulation of pro-inflammatory cytokine induction in response to N. gonorrhoeae (Figure S3A). Under similar conditions, the effect on LPS induced cytokine induction was only partial (Figure S3B). For reasons not fully understood, increasing the dose of the inhibitor beyond 200 nM had the opposite effect, suggesting a narrow therapeutic window for inhibitory activity (data not shown).
In a third approach, we asked whether it was possible to neutralize the inhibitory activity of rat serum with increasing doses of purified recombinant human C3a. Similar to observations with the human serum competition experiment (Figure 4a, b), we found that increasing doses of purified human C3a abolished the down-regulatory effect of rat serum on both N. gonorrhoeae, as well as LPS induced ΙL-1β and TNF-α in human macrophages (Figure 5a, b). Collectively, these data support our hypothesis that only rat, but not human C3a, interactions with C3aR trigger a signal that dampens pro-inflammatory cytokine induction in macrophages in response to pathogens and pathogenic ligands that signal via MyD88. Thus, there appears to be a species-specific signal that is determined by the species from which the C3a, but not its receptor, is derived.
Rat C3a inhibits pro-inflammatory cytokine production in macrophages. Human MDM were infected for 16 h with (a) N. gonorrhoeae (GC) strain MS11; MOI 5:1 or (b) E. coli LPS (100 ng/ml) with ATP; either alone (solid black bars), or with rat serum (20% v/v) alone (white bars) or with rat serum (20% v/v) mixed with increasing doses (0.75 and 1.5 μg/ml) of purified recombinant human C3a (gray bars) as indicated on the x-axis. The y-axis shows fold change in production of IL-1β (top panels, a, b) and TNF-α (top panels, a, b) by human macrophages upon each treatment relative to either N. gonorrhoeae or LPS treatment alone as calculated. Mean (SEM) of three independent experiments is shown; *P < 0.05; **P < 0.005.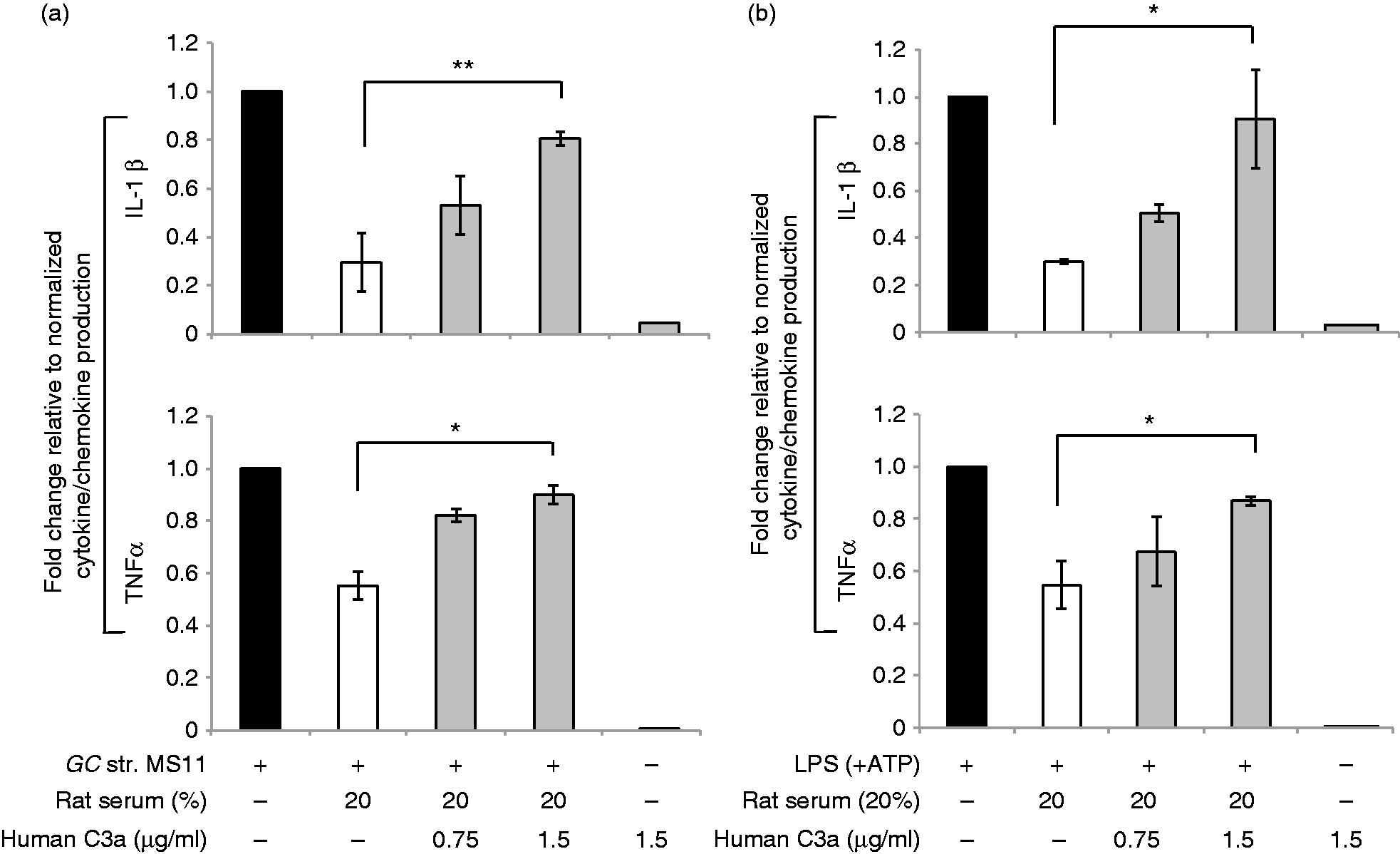
Rat serum down-regulates NF-κB activation in macrophages by N. gonorrhoeae and LPS through a C3a–C3aR-dependent mechanism
We next explored the mechanism by which rat serum dampens the inflammatory response in macrophages. The transcription factor NF-κB plays a central role in host defense and inflammation, and has been implicated in the expression of numerous pro-inflammatory cytokines and chemokines. NF-κB normally resides in the cytosol bound to the inhibitory protein, IκB, as an inactive complex. Upon activation, the IκB subunit is phosphorylated and degraded, followed by translocation of NF-κB to the nucleus and activation of NF-κB responsive genes, including pro-ΙL-1β and TNF-α. To determine if the inhibitory effects of rat serum occurred at the level of NF-κB activation, we examined the effect of rat serum on N. gonorrhoeae- and LPS-induced IκB degradation. We found that IκB degradation was blocked by rat serum, but not human serum, in response to N. gonorrhoeae or LPS (Figure 6a).
Rat serum inhibits NF-κB activation and caspase-1 cleavage. Human MDM were stimulated with (a) N. gonorrhoeae (GC) strain MS11 (MOI 5:1) for 2 h (immunoblot and densitometry graph) or (b) LPS (100 ng/ml) with ATP for 30 min (immunoblot and densitometry graph); either alone or with pre-incubation with 20% (volume/volume) of rat serum or human serum as indicated. Macrophages incubated with either rat or human serum alone were used as a negative control. Following treatment, cells were lysed and immunoblotted for IκBα and GAPDH as indicated in the representative immunoblots. The corresponding densitometry graph indicates ratio of band density of iκBα:GAPDH on the y-axis for each treatment as indicated on the x-axis. Mean (SEM) of three independent experiments is shown; *P < 0.05; **P < 0.005 in graph next to each blot. (c) Human MDM were stimulated with N. gonorrhoeae (GC) strain MS11 (MOI 5:1) for 5 h, in presence or absence of 20% human or rat serum. Following stimulation, cells were lysed and immunoblotted for pro-caspase-1, caspase-1 p20 fragment and GAPDH as indicated. (d) Cells were stimulated as in (c), and caspase-1 activation was measured using the FAM-FLICA caspase-1 assay. A representative histogram plot of the effect of rat and human serum on the level of caspase-1 activation by N. gonorrhoeae is shown on the right. Shaded gray histogram: no treatment; solid black line: N. gonorrhoeae, MS11 MOI 5:1; dotted line: N. gonorrhoeae plus 20% human serum; dashed line plot: N. gonorrhoeae plus 20% rat serum. The x-axis represents relative fluorescence for the FAM-FLICA staining while the y-axis represents relative cell number. The inset graph depicts the mean fluorescence intensity of FAM-FLICA staining for two independent experiments. Error bars indicate standard error with *P < 0.05 and **P < 0.005. n.s.: non-significant.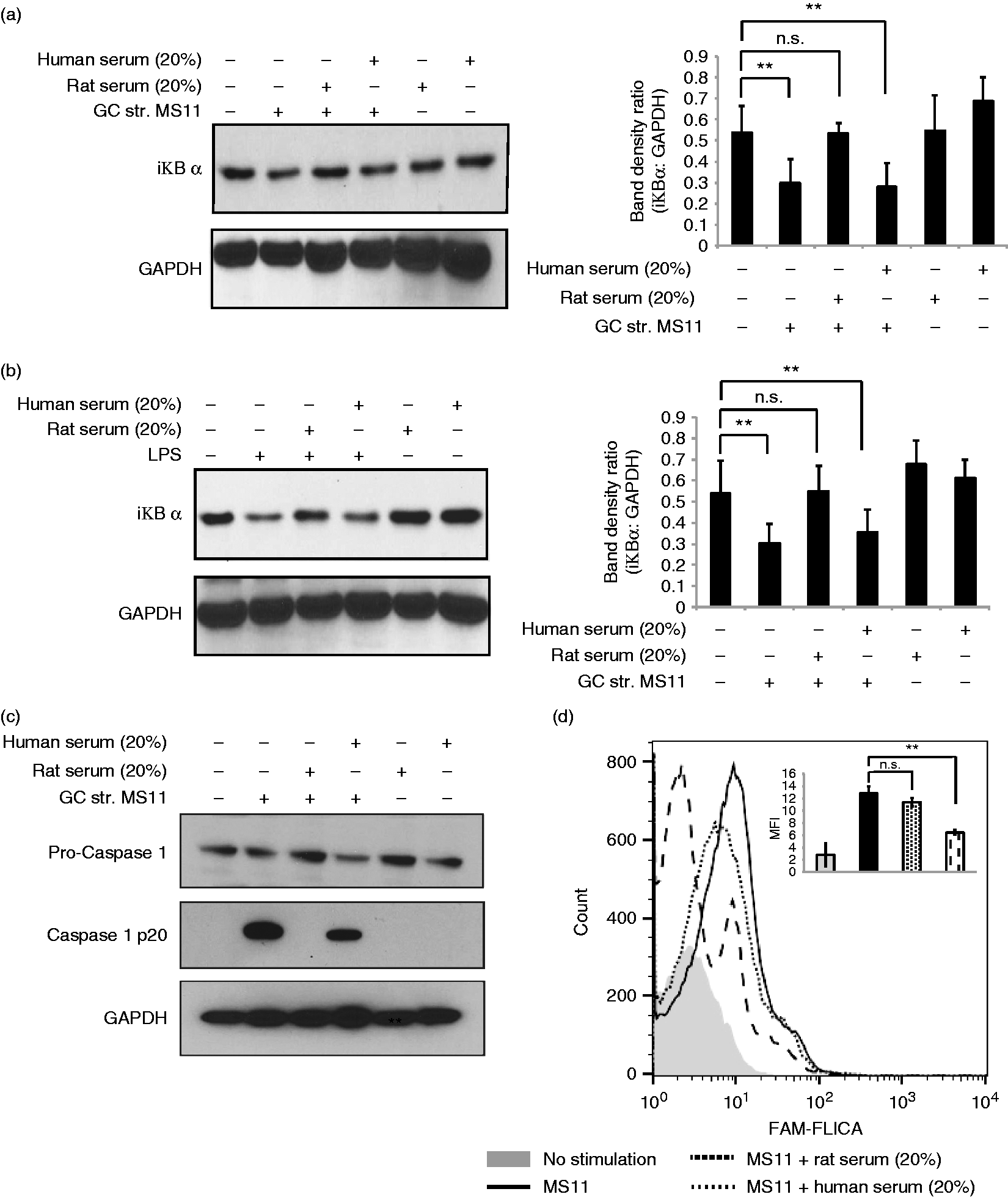
In addition to activating TLRs, N. gonorrhoeae has been shown to activate the NLRP3–Asc inflammasome, leading to cleavage of caspase-1 and the subsequent cleavage of pro-IL-β to its mature form.
11
To determine if rat serum could also affect this signaling pathway, we assayed for caspase-1 activation using two approaches: immunoblot analysis for the cleaved p10 subunit of caspase-1 and flow cytometric analysis using the active caspase-1 staining dye FAM-FLICA. As shown in Figure 6(b), N. gonorrhoeae-induced generation of the p10 caspase-1 fragment was blocked in the presence of rat serum but not human serum. Similarly, FLICA staining induced by N. gonorrhoeae was also blocked by rat serum but not human serum (Figure 6c). We further validated that the opposing effects on caspase 1 activation is not because of difference in uptake of N. gonorrhoeae by human macropages in presence of rat v/v human serum (Figure S5). Thus, the anti-inflammatory signaling induced by rat serum also effects caspase-1 activation. Based on these data, we propose a model where the interaction of rat C3a with C3aR (either human or rodent) blocks pathogen-induced activation of the transcription factor, NF-κB, as well as inflammasome-mediated caspase-1 activation (Figure 7).
Model for the site of C3a activity in the TLR and inflammasome signaling cascades. Hypothetical mechanism showing rat serum mediated down-regulation of pro-inflammatory cytokine response in macrophages in response to TLR activation. Neisseria gonorrhoeae or pathogenic ligands such as LPS activate complement in rat serum generating the C3a fragment following cleavage of C3. Rat C3a binds to C3aR on macrophages and in two distinct pathways engages in a two-way cross talk with the TLR–MyD88 and NLRP3–caspase 1 pathways. Through unknown signaling mechanisms, the C3a–C3a receptor (1) down-regulates NF-κB activation by preventing IκB degradation and thereby down-regulating downstream activation of NF-κB responsive genes to induce inflammatory cytokines like IL-1β and TNF-α and (2) down-regulates the NLRP3–inflammasome-mediated activation of caspase 1 activation, thereby down-regulating pro-ΙL-1β cleavage to ΙL-1β and release.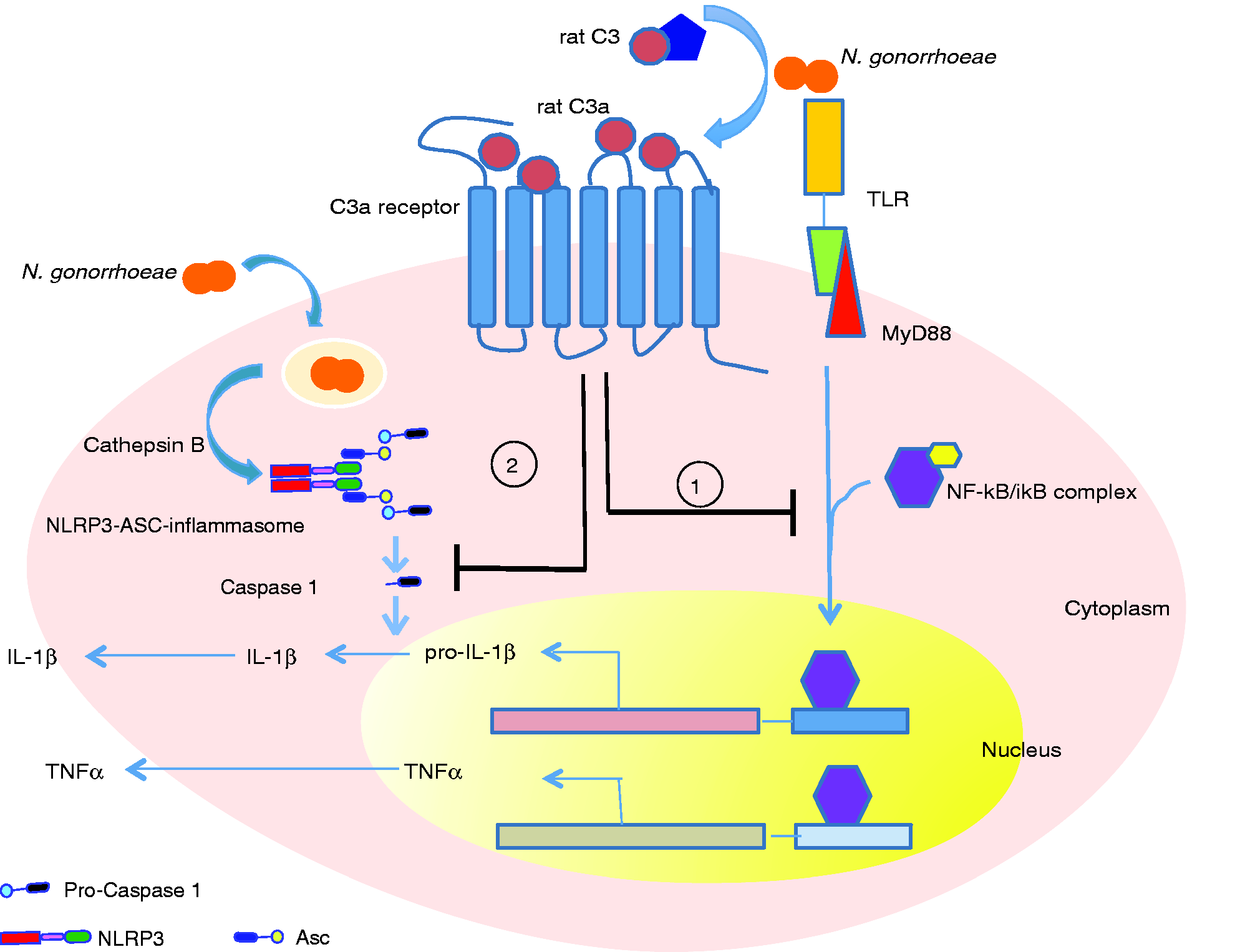
Discussion
Complement is a critical arm of the innate immune system and forms one of the first lines of defense against invading pathogens. The balance between activation and regulation of complement is a critical factor that determines the outcome of many infectious, autoimmune and neurodegenerative diseases, as well as the efficacy of the adaptive immune system.24,25 Previously, complement activation was largely seen as a mechanism to attack and lyse invading pathogens, or eliminate apoptotic and necrotic cells. While several groups have highlighted a novel role of the complement system in regulating several aspects of the immune system, the reported effects are sometimes contradictory.5,22,26,27
The novelty of our study lies with the observed differences in the biological effects of human and rat complement proteins on the induction of pro-inflammatory signals in response to N. gonorrhoeae and the MyD88-dependent TLR ligands, LPS and Pam3CysSK4
On the evolutionary time line, humans and rodents split over 70 million yr ago. Although genomic sequence comparison shows that < 300 genes are unique to either species, there are considerable differences in gene expression and function, particularly evident in the immune system. 33 A recent study highlights these differences by comparing the expression profile of about 5000 genes between human macrophages and mouse macrophages in three distinct acute inflammatory conditions: trauma, burns and endotoxemia. 34 The authors observed a high correlation in gene expression between all three conditions in humans but not in mice. Further, there was very poor correlation in the directionality of the change in gene expression between humans and mice in all three conditions. These differences may be responsible for the discrepancies observed between studies in rodent disease models and humans,35,36 and has led to exercising caution while extrapolating data obtained from studies in mice to humans.
Previous studies have demonstrated a protective anti-inflammatory role of the C3a–C3aR interactions in both endotoxemia and bacteremia in mouse models, 32 as well the ischemia–reperfusion model in mice. 22 However, several studies in humans have shown that elevated plasma levels of C3a and other anaphylatoxins are associated with severity of acute inflammatory conditions, such as septic shock.37,38 Our data may partially explain these conflicting observations. While the mechanism behind these species-specific effects remains unclear, we speculate that differences in rat and human C3a may determine the observed differences in function. It should be noted that an inherent regulatory mechanism to prevent unchecked activation of C3a and C5a is the rapid proteolytic cleavage of the terminal arginine residue by a carboxypeptidase to generate C3a-desArg and C5a-desArg in vivo, which leads to significant loss in binding affinity to their corresponding receptors.39,40 So most studies, including ours, that investigate C3a/C5a activity in serum are also studying C3a-desArg/C5a-desArg. Thus, an alternative explanation for the difference between the activity of rat and human serum could be that the rapidly generated rat C3a-desArg may retain relatively more ‘C3a-like’ activity than the human C3a-desArg generated in human serum. C5a-desArg has reduced activity and binding affinity for C5a receptor compared with C5a. However, when compared with their C5a parent molecules, the spasmogenic potency of rat C5a-desArg on guinea pig ileum was only fourfold less than the parent rat C5a. In contrast, human C5a-desArg was 3000-fold less potent than human C5a in the same assay. 41 Although we are not aware of any experimental evidence for this, we speculate that rat and human C3a-desArg may also differ in their affinity for C3a receptor and functional potency. The primary amino-acid sequence of human and rat C3a exhibit only 63% identity; the length of rat C3a also exceeds human C3a by four amino acids. In contrast, mouse and rat C3a show > 80% primary sequence identity (shown in Figure S4). This heterology between rat, mouse and human C3a and C5a is the same. The higher affinity of rat and human C3a-desArg for the C3a receptor may result in differential activation of the receptor leading to inhibitory/dampening effect on inflammatory response. Another explanation could be that rat C3a-desArg binds to C3a receptor at a site distinct from that recognized by human C3a-desArg, which could lead to opposing effects on macrophage inflammatory response. However, experimental proof for this would require purification or synthesis of rat and human C3a and C3adesArg, receptor binding studies, and functional assays, which are beyond the scope of this current manuscript.
As therapeutic strategies that target complement for various diseases are becoming increasingly common, it is important to consider cross regulation of different arms of innate immune system by complement.42–44 The activation of complement C3 is the point of convergence of all the three complement pathways. Targeting C3 activation and/or inhibiting C3a–C3aR interactions using Abs, complement regulators, and synthetic inhibitors are potential therapeutic approaches for several inflammatory disorders.45,46 This study raises the possibility of developing synthetic or small molecule mimics of rat C3a as novel inhibitors of inflammation.
Footnotes
Acknowledgments
The authors acknowledge support from the Boston University Flow Cytometry Core Facility and thank Nancy Nowak for technical assistance.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported, in part, by the National Institutes of Health (NIH) Grant AI084048 (RRI and SR) and AI114790 and AI118161 (SR); the Ruth L. Kirchstein NIH T32 HL007224 (TDR); and a 2016 developmental grant from the Providence/Boston Center for AIDS Research (CFAR) AI042853 (RRI).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.