
Abstract
Platelets are the main players in thrombosis and hemostasis; however they also play important roles during inflammation and infection. Through their surface receptors, platelets can directly interact with pathogens and immune cells. Platelets form complexes with neutrophils to modulate their capacities to produce reactive oxygen species or form neutrophil extracellular traps. Furthermore, they release microbicidal factors and cytokines that kill pathogens and influence the immune response, respectively. Platelets also maintain the vascular integrity during inflammation by a mechanism that is different from classical platelet activation. In this review we summarize the current knowledge about how platelets interact with the innate immune system during inflammation and infection and highlight recent advances in the field.
Introduction
Platelets are small anucleated cell fragments patrolling the vasculature, and immediately respond to vessel breaches and restore hemostasis. In recent decades it has become clear that they play roles beyond hemostasis and also contribute to (thrombo-)inflammatory processes like those unfolding after stroke. 1 Furthermore, they also play an important role during infection by either directly interacting with pathogens or by recruiting and stimulating immune cells. 2 More recently, we have come to understand that platelets also maintain vascular integrity in inflamed vessels in a process different from classical hemostasis.3,4 Acknowledging their multifaceted capabilities, platelets have lately been described as autonomous drones for hemostatic and immune surveillance. 5
The finding that platelets form aggregates around bacteria is not new. In fact, one of the first descriptions of this process dates back to 1901
6
when Levaditi showed that platelets aggregated upon incubation with Vibrio cholerae; however, more systematic investigations on how bacteria cause platelet aggregation were only performed in the 1970s.7,8 Platelet surface receptors enable direct platelet–bacteria or platelet–immune cell interactions.
9
Factors stored in platelet granules that are released upon activation include cytokines, inflammatory mediators and antimicrobial peptides.10,11 In this review we discuss how platelets, being among the first cells to respond to vessel injury, are at the front line of antimicrobial host defense which allows them to orchestrate the innate immune response (Figure 1).
Platelets interact with bacteria and cells of the innate immune system. Platelets interact with bacteria directly through their surface receptors or indirectly through plasma proteins. Platelets orchestrate the immune reaction to inflammation and infection by direct interactions with cells of the innate immune system (neutrophils and Kupffer cells) or through the secretion of mediators.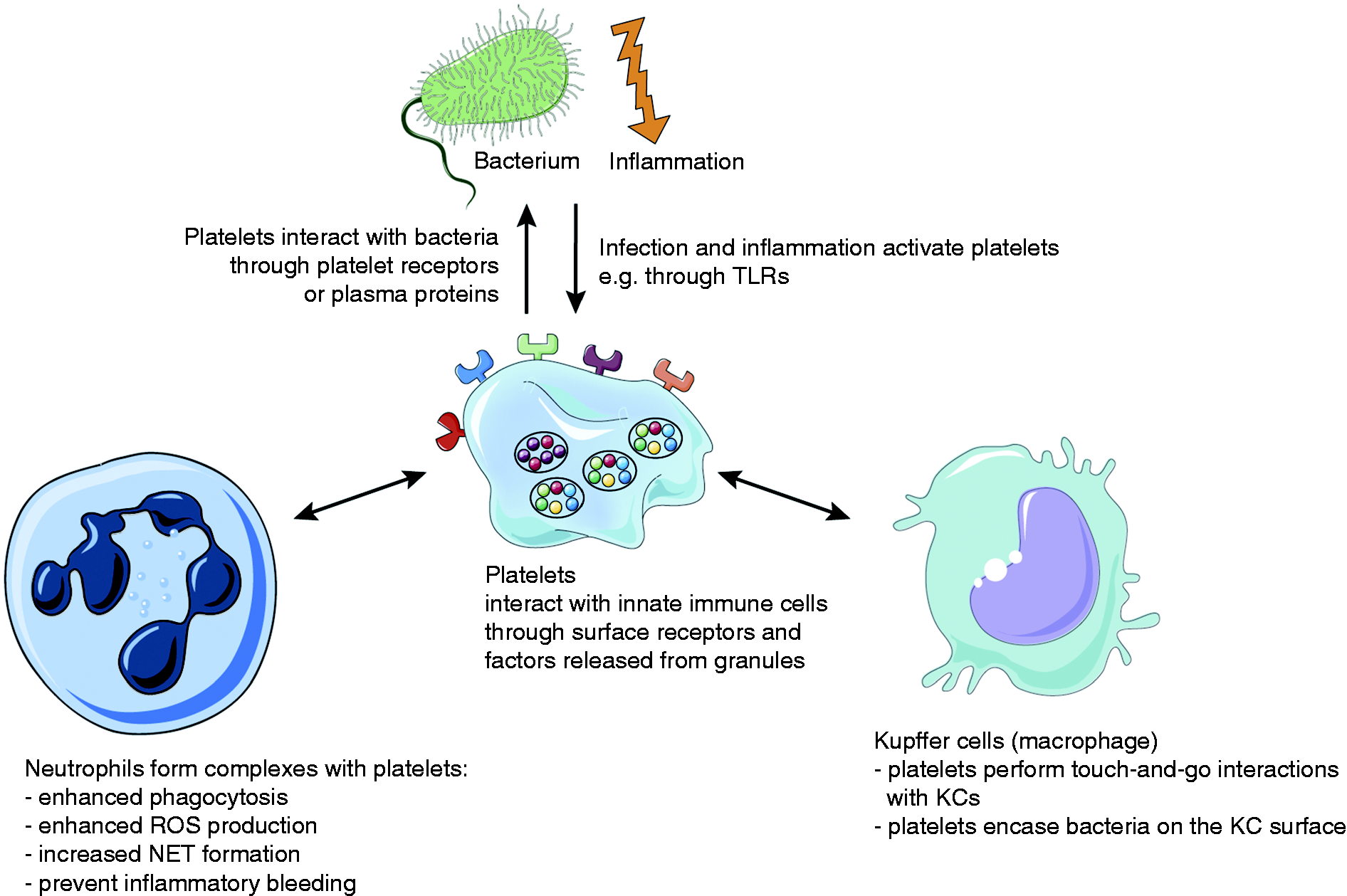
A lot of the findings presented here were discovered using experimental mouse models or knockout mouse lines. It is therefore worth to mention that there are some distinct characteristics between murine and human platelets such as their size, number and some histological features. However, they also share a lot of similarities, and mice offer an excellent model to study various aspects of platelet biology in vivo. 12
Platelets and sepsis
Severe sepsis—defined as infection in combination with acute organ dysfunction—is the leading cause of in-hospital death in the United States (US).13,14 Estimations suggest there are 750,000 cases of severe sepsis every year in the US, with most of them requiring intensive care. Case numbers for patients with severe sepsis admitted to a hospital have seen a significant rise in recent years, 14 with the highest incidence rates reported for newborns younger than 12 months (5.3 per 1000) and senior patients older than 85 years (26.2 per 1000). 15 In the 1960s, some studies reported mortality rates of up to 80% for septic shock patients. Thanks to improvement in monitoring and therapy, this number has decreased significantly; however, it still remains at around 25% today.13,16 In addition, total hospital costs for patients with severe sepsis have seen a steady increase, and it is now a major burden for the public health system with estimated annual costs of more than 24 billion USD in the US. 14
During severe sepsis both pro- and anti-inflammatory responses occur simultaneously. The two processes aim at eliminating the pathogen and at the same time try to restrict the immune reaction to prevent excessive damage. Both responses have to be delicately balanced to provide a response that is powerful enough to clear the pathogen and prevent secondary infections but also to minimize collateral tissue damage. 13
Severe sepsis is often accompanied by disseminated intravascular coagulation and thrombocytopenia; however, the underlying mechanisms are incompletely understood.13,17 It is well established, however, that there is extensive cross-talk between the inflammatory and coagulation pathways. Tissue factor (TF) is up-regulated by leukocytes, platelets, endothelial and smooth muscle cells, which triggers thrombin generation leading to a sustained pro-inflammatory and thrombotic response while at the same time dampening anticoagulant mechanisms and fibrinolysis.18,19
A recent study showed that thrombocytopenia led to severely impaired survival and enhanced bacterial growth in blood and lungs in a mouse model of pneumonia-derived sepsis. 20 Thrombocytopenia also caused hemorrhage at the site of infection, in line with previous results demonstrating that inflammation induces hemorrhage during thrombocytopenia.20,21 A clinical study including more than 900 sepsis patients grouped them according to their platelet counts upon admission to the intensive care unit. Remarkably, patients with very low or intermediate–low platelet counts showed significant increases in both 30-day mortality as well as cytokine levels and enhanced endothelial cell activation. 17 These results show that platelet count might be used as a prognostic marker during sepsis.
Platelet receptors enable interactions with bacteria
Several platelet surface receptors are also involved in inflammation and infection in addition to thrombosis and hemostasis. Platelet–bacteria interactions can be either direct (bacterial surface proteins binding to a platelet receptor) or indirect (bacteria binding to a plasma protein, for example von Willebrand factor (vWF) or fibrinogen, which then binds to the respective platelet receptor).
Glycoprotein Ib (GPIb)
GPIb is exclusively expressed on the surface of platelets and precursor megakaryocytes. The interaction with its main ligand vWF is especially important for platelet adhesion under high shear conditions, for example in stenosed vessels or capillaries.22,23 GPIb can also bind to serine-rich repeat proteins like the Streptococcus sanguis platelet adhesin called serine-rich protein A (SrpA) which enables binding to GPIb in a sialic acid-dependent manner. 24 Staphylococcus aureus protein A (Spa) facilitates indirect interaction with platelets through both soluble and immobilized vWF which then binds to platelets via GPIb.25,26
Integrin αIIbβ3 (GPIIb/IIIa)
The platelet-specific integrin αIIbβ3 is the most abundant glycoprotein on the platelet surface and binds ligands that contain an arginine-glycine-aspartic acid (RGD) sequence such as fibrinogen, vWF, fibronectin and vitronectin. αIIbβ3 enables stable platelet–platelet interactions and adhesion to the extracellular matrix (ECM). Firm binding only occurs after a conformational change that puts αIIbβ3 in an activated or high-affinity state in which the RGD binding site is uncovered. 27 Binding of Borrelia burgdorferi to human platelets was shown to be mediated by αIIbβ3 and could be blocked by a synthetic RGD peptide. 28 Another bacterial protein that contains a RGD-like motif to interact with platelet αIIbβ3 is SdrG (Fbe) from Staphylococcus epidermis, which is present in many clinical strains and causes platelet aggregation which can be blocked using the αIIbβ3 antagonists abciximab or tirofiban, but also aspirin. SdrG also mediates indirect interactions through fibrinogen which in turn binds αIIbβ3 and the IgG receptor FcγRIIa.29,30 Another well-characterized indirect interaction between platelets and bacteria is through fibrinogen and clumping factors (Clf) on S. aureus. Like the Sdr proteins, ClfA and B both contain serine and aspartic acid dipeptide repeats (SD repeats) and bind fibrinogen to induce platelet aggregation via αIIbβ3.31,32
TLR2 / TLR4
TLRs recognize PAMPs, for example the bacterial cell wall components lipoteichoic acid (LTA) and LPS. 33 Platelets express TLR2 and TLR4 on their surface.34,35 Streptococcus pneumoniae triggers platelet aggregation through TLR2, and αIIbβ3, independent of pneumolysin toxin, causes activation of the phosphoinositide 3-kinase (PI3-K) pathway and provokes dense-granule release. 36 In mice, LPS injection was shown to induce an increase in platelet binding to fibrinogen under flow. Furthermore, LPS administration caused thrombocytopenia through P-Selectin-independent neutrophil-mediated pulmonary platelet sequestration in wildtype but not in TLR4-deficient mice. 37 Another study demonstrated that human platelets responded to LPS stimulation with release of soluble CD62p, epidermal growth factor, TGFb, IL-8, platelet activating factor 4 (PAF4) and platelet-derived growth factor (PDGF) α and β. 37 During hemolytic-uremic syndrome (HUS) caused by infection with enterohemorrhagic Escherichia coli (EHEC), platelets were activated by EHEC-LPS binding to TLR4. 38 Platelet TLR4 detects TLR4 ligands (LPS, but also others like high mobility group B1 (HMGB1) and heat-shock proteins) in the blood and causes platelets to bind to adherent neutrophils and the formation of neutrophil extracellular traps (NETs) in liver sinusoids and pulmonary capillaries, which facilitate bacterial capture during sepsis 39 but can also have detrimental effects such as causing vascular occlusion.40,41
(hem)ITAM receptors GPVI and CLEC-2
Platelets are activated by collagen through their main collagen receptor GPVI, which signals through an immunoreceptor tyrosine-based activation motif (ITAM) in the Fc receptor (FcR) γ-chain it forms a complex with.42,43 The platelet C-type lectin receptor 2 (CLEC-2) binds podoplanin, which is highly expressed in type 1 lung alveolar cells, lymphatic endothelial cells and kidney podocytes, but absent from endothelial cells and platelets. CLEC-2 contains a single cytosolic YXXL motif known as a hemITAM that becomes phosphorylated upon receptor multimerization to enable signaling.44,45 GPVI and CLEC-2 play important roles in thrombosis and hemostasis;42,46–48 however, they are also involved in maintaining vascular integrity in inflamed vessels thus preventing inflammatory bleeding,3,49,50 formation of cerebral blood vessels 51 and mediating blood/lymphatic vessel separation. 52 A recent report showed that CLEC-2 mediates inflammation-triggered thrombosis after salmonella infection in the liver 53 through platelets interacting with podoplanin-expressing monocytes and Kupffer cells. Lately, a thrombosis-independent role for CLEC-2 during sepsis was described: Platelet CLEC-2 interacts with podoplanin expressed on inflammatory macrophages to regulate immune cell recruitment as well as the cytokine/chemokine storm following infection to limit organ damage. 54 In another study, S. aureus α-toxin bound ADAM10 on the platelet surface to trigger platelet activation and platelet–neutrophil complex formation that enhanced neutrophil activity during sepsis. 55 ADAM10 is a metalloproteinase that cleaves the ectodomain of GPVI, 56 and in fact soluble GPVI (sGPVI) was released from platelets following incubation with S. aureus α-toxin. Whether this demonstrates a direct interaction of ADAM10 with S. aureus or a potential role for sGPVI needs further investigation. Moreover, GPVI-deficient mice showed increased bacterial growth in lungs and distant body sites after pneumonia-derived sepsis as well as reduced platelet activation and platelet–leukocyte complex formation in the bronchoalveolar space. 57 Interestingly, in a recent clinical study, sGPVI was identified as a marker for platelet activation and predictive for the occurrence of sepsis and overall survival in patients with thermal injury. 58 GPVI is also critically involved in the formation of platelet microparticles—submicrometer vesicles shed from activated platelets—that can have pro-inflammatory effects, for example in patients with rheumatoid arthritis. 59 Recently it was shown that microparticles shed from activated platelets lose GPVI expression while maintaining CLEC-2, 60 contributing to sGPVI levels. Indeed, sGPVI levels in plasma from patients with rheumatoid arthritis were significantly increased compared with healthy controls.
Platelet granule-derived factors and their impact on innate immunity
Upon activation, platelets release a plethora of different factors stored in two major types of granules: α-granules and dense granules. α-Granules are highly abundant, with 50–80 granules per mouse platelet,61,62 while dense granules are considerably less abundant with 5–6 granules per platelet. α-Granules contain more than 300 different membrane and soluble proteins, which are recruited to the plasma membrane or secreted upon platelet activation, respectively, and are involved in processes such as platelet adhesion, coagulation, thrombo-inflammation, wound healing, tumor growth, angiogenesis and antimicrobial host defense.61,63,64
There is increasing evidence that platelets contribute to the onset and spread of inflammation. 65 Platelets adhere to the activated endothelium or form complexes with immune cells to activate, attract or differentiate other immune cells by several mechanisms. Many platelet-derived factors contribute to shaping the inflammatory response, and one of the most important ones is P-Selectin, which is exposed on the platelet surface upon activation and mediates interactions of platelets with immune cells and the endothelium. Platelet P-Selectin binds to P-Selectin glycoprotein ligand-1 (PSGL1) on endothelial or immune cells, thereby enabling platelets to bind to the inflamed endothelium, to recruit circulating monocytes, neutrophils and lymphocytes and to initiate an inflammatory response at the site of injury. Importantly, blocking P-Selectin or PSGL-1 using antibodies almost completely abolished platelet tethering, rolling and adhesion on activated endothelium. 66 In a mouse model of acute lung injury, blocking platelet P-Selectin reduced the number of platelet–neutrophil complexes, improved gas exchange, reduced neutrophil recruitment and permeability and prolonged survival of the animals. 67
Upon activation, platelets secrete numerous chemokines, including CXCL1, CXCL4, CXCL5, CXCL7, CXCL8, CXCL12, CCL2, CCL3 and CCL5. 10 The most abundant platelet chemokine CXCL7 is present in several variants: platelet basic protein, connective tissue-activating peptide III (CTAP-III), β-thromboglobulin (β-TG) and neutrophil-activating peptide-2 (NAP-2), and all of them are generated by proteolytic cleavage from a precursor protein.10,61 However, the only variant that possesses chemotactic activity is NAP-2. 68 It was shown that both CTAP-III and NAP-2 induce neutrophil adhesion to human umbilical vein endothelial cells (HUVECs); however, only NAP-2 triggered neutrophil transendothelial migration. 69 Interestingly, CXCL7 is also involved in the recruitment of circulating endothelial progenitor cells after arterial injury through its receptor CXCR2, indicating that CXCL7 secreted by platelets may contribute to revascularization after vessel injury. 70
Platelets are a major source of CCL5 (RANTES) in the circulation, as its concentration was highly correlated to platelet counts in a study of patients with hematological malignancy undergoing chemotherapy. 71 Platelet-derived CCL5 contributes to recruiting monocytes to the vessel wall. In a mouse model of atherosclerosis, activated platelets were shown to deliver CCL5 and CXCL4 (platelet factor 4, PF4) to the surface of both monocytes and endothelial cells in atherosclerotic lesions in a P-Selectin-dependent manner. 72 It was further shown that CXCL4 facilitates CCL5 oligomerization and amplifies its effect on monocyte recruitment. 73
Platelets (and their precursors megakaryocytes) are the exclusive source of CXCL4 (PF4). That platelets have an abundance of CXCL4 is strikingly demonstrated by a 1000-fold increase in the serum concentration after thrombin stimulation.61,74 Although CXCL4 lacks chemotactic activity, 75 it causes firm neutrophil adhesion to endothelial cells and degranulation. While the first process is a direct effect of Src kinase activation, the latter requires costimulation, for example by TNF through p38 MAP kinase and PI3 kinase. 76 Furthermore, CXCL4 triggers several functions in monocytes, including phagocytosis, respiratory burst, survival and cytokine secretion. CXCL4-initiated respiratory burst was shown to depend on rapid activation of the PI3 kinase, Syk and p38 MAP kinase. By contrast, monocyte differentiation and survival is mediated by CXCL4-mediated delayed Erk activation approximately 6 h after stimulation.77,78 CXCL4 released from platelets is also capable of inducing differentiation of monocytes into macrophages and prevents them from undergoing spontaneous apoptosis in culture. 79 In combination with IL-4, CXCL4 induces a rapid differentiation of monocytes into specialized APCs that stimulates lymphocyte proliferation and lytic NK activity while inducing only moderate cytokine release. 80
CXCL4 also acts on other cell types such as endothelial cells. Platelet-derived CXCL4 inhibited fibroblast growth factor-2 (FGF-2) and vascular endothelial growth factor function by blocking the binding to their respective receptors. Furthermore, both CXCL4 and its variant CXCL4L1 potently inhibit chemotaxis and proliferation of endothelial cells as well as in vitro and in vivo angiogenesis.81,82 CXCR3-B, a splice variant of CXCR3, was shown to be a receptor for CXCL4 and might be involved in the angiostatic activity of CXCL4 released by platelets 83 that causes inhibition of endothelial cell growth.
Platelets as phagocytes and platelet-derived antimicrobial peptides
We know that platelets express TLR436 and other receptors which enable them to detect and bind to bacteria, which raises the question of their capability of phagocyting bacteria. Indeed, electron microscopic studies demonstrated that activated platelets engulf S. aureus in vacuoles and appear to secrete granule content into the vacuole. 84 However, whether platelets actually killed the bacteria or transferred them to professional phagocytes was not clarified in this study. Interestingly, in an earlier report, Yeaman and colleagues isolated and characterized cationic proteins from rabbit platelets that displayed in vitro microbiostatic or microbicidal activity against S. aureus, Escherichia coli and Candida albicans. 85 In another study, thrombin-induced platelet microbicidal protein (tPMP-1) potently lysed S. aureus during logarithmic-phase growth. 86 Releasate from thrombin-activated platelets reduced the number of adherent bacteria in a rabbit model of infective endocarditis using Streptococcus sanguis. 87 Using the same model, experimentally induced thrombocytopenia led to higher densities of streptococci within vegetations as well as higher total number of bacteria per valve. 88 Another group of antibacterial proteins found in platelets is called thrombocidins (TCs). TC-1 and TC-2 were able to kill Bacillus subtilis, E. coli, S. aureus and Lactococcus lactis 89 using a mechanism that did not lyse the bacterial cell wall, indicating that they act differently than tPMP-1. Platelets were also shown to bind Plasmodium falciparum-infected erythrocytes and kill the parasite inside the cell. 90 Treating the platelets with aspirin or a P2Y1 antagonist rendered them incapable of killing the parasite. Accordingly, treating mice with antiplatelet Abs or aspirin resulted in lower overall survival after infection with Plasmodium chabaudi. Lately, E. coli were found to be killed by human platelets through a process involving CXCL4 and FcγRIIA: Anti-CXCL4/polyanion Abs opsonized E. coli coated with platelet-derived CXCL4. The Ab complex was then detected by platelet FcγRIIA, 91 causing platelets to cover the opsonized bacteria and release antimicrobial factors in a concerted way to effectively kill them.
Platelet interactions with innate immune cells in infection and inflammation
Platelets interact with a number of key players during infection and inflammation, namely macrophages (e.g. Kupffer cells in the liver), neutrophils, monocytes, NK cells, dendritic cells and components of the complement system. Here, we focus on reviewing their interactions with Kupffer cells, neutrophils, the complement system and how they maintain vascular integrity during inflammation and infection.
Kupffer cells
The liver is not only the largest internal organ with important roles in detoxification and metabolism, it is also the first line of defense against pathogens present in the blood stream. It filters about a third of the body’s total blood volume each minute and contains the largest population of phagocytes in the body. 92 The liver-resident macrophage population known as Kupffer cells (KCs) are very large, immobile cells that reside in the sinusoidal space where they scan for foreign objects in the blood. More than 25 years ago, Endo and colleagues discovered that LPS injection into mice caused an increase in hepatic serotonin levels which was independent of mast cells and correlated with a drop in the number of circulating platelets. They found that LPS application caused accumulation of non-activated platelets in the sinusoidal space where they frequently interacted with KCs. 93 Subsequent studies showed that while aspirin or heparin did not affect serotonin accumulation in the liver, KC depletion using clodronate liposomes almost completely abolished platelet and serotonin accumulation. 94 This suggested that LPS stimulation causes platelet retention in the liver through a process that is different from classical platelet aggregation and that involves KCs.
We now know that KCs rapidly capture a striking quantity of pathogens from the bloodstream in a process that often involves platelets. KCs were shown to bind B. burgdorferi and prevent them from using sinusoidal endothelial cells to gain access to the extravascular space. 95 It was also demonstrated that methicillin-resistant S. aureus (MRSA) is primarily sequestered and killed by KCs through an interaction of the complement receptor of the immunoglobulin superfamily (CRIg) with LTA on the bacterial surface. 96 A minority of staphylococci, however, can overcome the antimicrobial activity of the KCs to survive and proliferate inside this intracellular niche. 97 Importantly, KCs were found to collaborate with platelets to eradicate blood-borne infections with Bacillus cereus and MRSA: 98 In the naïve mouse liver, platelets performed touch-and-go interactions through GPIb with vWF constitutively expressed on KCs. Upon infection, KCs captured bacteria and platelets rapidly adhered and formed aggregates around them in an integrin αIIbβ3-dependent way to contain the bacterium. This suggested an important role for platelets in KC-mediated bacterial clearance. Indeed, platelet depletion or lack of GPIb resulted in severely increased mortality in mice following infection. Importantly, opsonization with complement factor C3 was necessary for successful bacterial clearance, indicating a complex interplay between KCs, platelets and the complement system during bacterial clearance in the liver. Another group investigating the clearance of Listeria monocytogenes bloodstream infections by KCs discovered a dual-track mechanism consisting of a slow clearance of bacteria–platelet complexes that requires platelet GPIb, CRIg and C3 opsonization as well as a fast clearance of free bacteria independent of complement and platelets that requires scavenger receptors.99,100 The authors hypothesized that the slow clearance allows a small number of platelet–bacteria complexes to remain in the circulation long enough to be detected by splenic CD8α+ dendritic cells to launch an antibacterial cytotoxic T cell response.
Neutrophils
It is well established that platelets interact with neutrophils during inflammation and infection. Indeed, circulating platelet–neutrophil complexes (PNC) have been found in a variety of diseases such as asthma, rheumatoid arthritis, inflammatory bowel disease, multiple sclerosis, stroke and severe sepsis.101–102 The effect of platelets binding to neutrophils includes increased neutrophil adhesion to the endothelium, increased reactive oxygen (ROS) production and NET formation (Figure 2). P-Selectin, which upon activation is recruited to the platelet surface from α-granular stores, seems to be the most important platelet surface receptor for platelet–neutrophil interactions. Platelet P-Selectin binds to the high-affinity counter-ligand PSGL-1 on neutrophils.104,105 P-Selectin-deficient mice show severe leukocyte defects, for example abrogated leukocyte rolling as well as delayed neutrophil recruitment and reduced neutrophil extravasation.106,107 Using P-Selectin-deficient mice or treating mice with an anti-P-Selectin Ab and subjecting them to different models of lung inflammation resulted in reduced neutrophil recruitment and less lung damage.108,109 However, all those studies looked at global P-Selectin deficiency in which the individual contribution of endothelial and platelet P-Selectin are difficult to isolate. In later studies it was shown that platelet depletion significantly inhibits neutrophil recruitment to the site of inflammation in a zymosan-induced peritonitis and LPS-induced lung inflammation model.
110
In inflamed glomerular capillaries, platelets are essential for leukocyte adhesion via a non-classical cascade that involves platelet P-Selectin binding to endothelial PSGL-1 as well as β2 integrin/ICAM-1 and nonrolling interactions.
111
Depletion of neutrophils and platelets reduced urinary protein excretion induced by anti-glomerular basement membrane Abs, underlining their importance for the development of renal injury. In addition, it was shown that platelet P-Selectin is important for neutrophil recruitment into the outer and inner medulla during acute post-ischemic renal failure.
112
In another paper, Sreeramkumar et al. showed that in inflamed vessels, neutrophils scan for the presence of activated platelets using PSGL-1 clusters. Migration and NET formation only occurred once activated platelets had bound.
113
In a mouse model of multiple sclerosis called experimental autoimmune encephalomyelitis (EAE), platelet depletion significantly improved the disease state and slowed its progression through reduced recruitment of leukocytes to the inflamed central nervous system and attenuated inflammation. More specifically, targeting GPIb or αIIbβ3 led to a pronounced improvement of EAE outcome.
114
Platelets form complexes with neutrophils to potentiate their activity. Platelets interact with neutrophils through multiple receptors: Activated platelets express P-Selectin on their surface which binds PSGL-1 on neutrophils and endothelial cells, CD40L is expressed on the platelet surface upon activation and binds CD40, Neutrophil Mac-1 binds platelet GPIb as well as αIIbβ3. Together these interactions promote ROS production, NET formation, adhesion, transmigration and degranulation of neutrophils. Upon activation (e.g. through PAMPS binding to TLRs), platelets release microbicidal proteins like TC1+2 which can kill bacteria. They also secrete large quantities of CXCL4 and CXCL7 that promote neutrophil adhesion, degranulation and transmigration.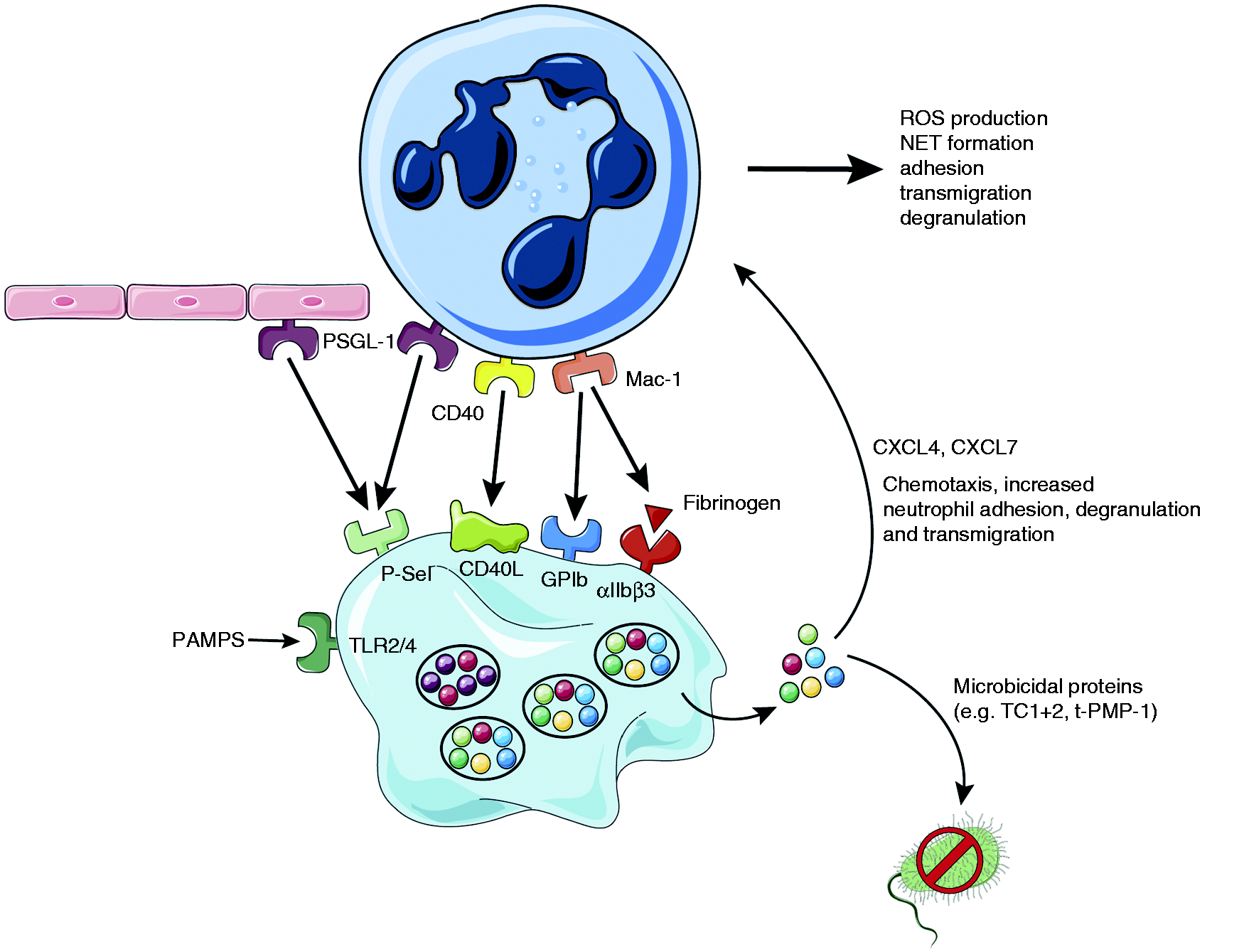
Interestingly, platelets not only express P-Selectin, but also its counter-receptor PSGL-1 which mediates platelet–endothelium interaction.115,116 Furthermore, platelet GPIb was also shown to bind to (endothelial) P-Selectin, 117 thereby allowing platelets to scan for the activation status of the vasculature. In addition, platelets can stimulate the secretion of Weibel–Palade bodies from endothelial cells and leukocyte rolling through P-Selectin. 118 Therefore, it seems that platelets can interact with the endothelium to pave the way for leukocytes to bind to both platelets and the endothelium.
P-Selectin/PSGL-1 interactions between activated platelets and neutrophils in the inflamed vasculature promote further interactions through β2 integrins (CD18), especially Mac-1 (αMβ2, CD11b/CD18) which follows a three-step process that involves binding to fibrinogen/GPIb and outside-in-signaling through Src family kinases.119,120 In addition, activated platelets express intercellular adhesion molecular-2 (ICAM-2, CD102) which enables the formation of firm and shear resistant platelet-neutrophil-complexes under flow conditions by binding to β2 integrins.121,122
Neutrophil accumulation on activated platelets under flow conditions also involves interactions of Mac-1 with fibrinogen bound to platelet αIIbβ3, as found by using blocking Abs against the major platelet integrin. Furthermore, platelets from patients with Glanzmann thrombasthenia harboring genetic defects in their ITGA2B or ITGB3 genes that cause impaired αIIbβ3 function or expression 123 demonstrated significantly reduced neutrophil adhesion to platelets under flow compared with healthy controls. 124
Platelets are a major source of CD40 ligand (CD40L), which they express on their surface upon activation. 125 Patients with diabetes, ischemic stroke or acute coronary syndromes often show elevated levels of circulating soluble CD40L. Stimulation of endothelial cells through CD40 by platelet CD40L induces recruitment of neutrophils, likely via platelet P-Selectin and neutrophil Mac-1 expression. Furthermore, CD40L increased neutrophil oxidative burst and degranulation.126,127
Indeed, neutrophils in PNCs show increased activation, CD11b expression, phagocytosis and ROS production compared with free neutrophils. 128 Upon stimulation of platelets, the activation state of neutrophils in PNCs was even more pronounced.128,129 Interestingly, incubation with resting platelets was able to restrict neutrophil activation, indicating that there is a strong interconnection between the activation state of platelets and neutrophils in PNCs, which is most likely mediated through P-Selectin.129,130
Besides phagocytosis, neutrophils possess another clever way to capture bacteria through the formation of NETs. During NET formation, neutrophils expel large amounts of chromatin and granular proteins (e.g. elastase and myeloperoxidase (MPO)), thereby forming extracellular fibers to immobilize and kill bacteria.131,132 Initially, it was thought that all neutrophils die during NETosis; however, early in infection, live neutrophils in fact release NETs to prevent bacterial dissemination. 133 While this may sound counter-intuitive at first, we know that red blood cells or platelets live without a nucleus for several days and that neutrophils whose nucleus has been removed retain their ability to crawl, transmigrate, phagocytose and kill bacteria at least for a short time. 134
Platelets are critically involved in NET formation through their TLR4 receptor that can detect PAMPs in the bloodstream. Platelet activation through TLR4 causes them to bind to neutrophils adhering primarily to liver sinusoids and pulmonary capillaries, causing neutrophil activation and NET formation. 39 Significantly, adding plasma from patients with severe sepsis to platelets and neutrophils from healthy donors triggered TLR4-dependent PNC formation. Moreover, neutrophils migrate to liver sinusoids during sepsis to release NETs and prevent bacterial dissemination to other organs by a mechanism that requires PNC formation through LFA-1 (CD11a/CD18). 135 Platelets were shown to form aggregates around S. aureus thereby limiting their growth. In addition, human platelets also release an antimicrobial peptide called human β-defensin-1 (hBD-1) after stimulation with S. aureus α-toxin. hBD-1 significantly impairs bacterial growth but was also shown to induce robust NET formation. 136
Interestingly, NETs can also be found under sterile inflammation conditions in the lungs and plasma of patients with transfusion-related acute lung injury (TRALI). Targeting platelet activation using aspirin or an αIIbβ3 inhibitor decreased NET formation and lung injury in a mouse model of TRALI. 137 Blocking Mac-1 but not LFA-1 during acute lung injury also significantly reduced the amount of NET formation and lung injury. 138 Platelets are critically involved in the propagation of deep vein thrombosis (DVT) by promoting leukocyte accumulation and NET formation, which provides a prothrombotic surface through its decoration with TF. 40 Importantly, treating mice with DNase significantly reduced NET formation and DVT growth. A recent publication showed that both mice and humans harbor endogenous DNases that help to contain NET formation in the host. 41 Serum from mice deficient in DNase1 or DNase1L3 was able to degrade NETs; however, deficiency in both DNases completely abrogated NET-degrading capacity. Double-deficient animals subjected to chronic neutrophilia or septicemia showed a high mortality due to vascular occlusion through “pure” NET clots independent of classical hemostasis or thrombus formation.
Platelets were recently found to be capable of actively probing their local environment and migrating using actomyosin-generated forces. 139 While migrating they act as cellular scavengers that collect bacteria both in vitro and in vivo. Platelet–bacteria bundles generated this way facilitate phagocytosis by neutrophils as well as NET formation.
Complement
The complement system facilitates lysis of pathogens and damaged cells by forming a pore in the target cell membrane through the membrane attack complex. Platelets are capable of activating both the classical and alternative complement pathway; however, the mechanism is still incompletely understood. 140 It is known that activated human platelets express gC1qR—a receptor for C1q and the first factor of the classical complement pathway141,142 which also binds to other ligands such as SpA expressed on the surface of S. aureus.143,144 Platelet granules store complement C3 and C4 precursor but also C1 inhibitor, which indicates that platelets might in fact regulate the complement response. 61 Chondroitin sulfate released from activated platelets causes complement activation through interactions with C1q. 145 In addition, platelet P-Selectin can bind C3b and trigger the alternative complement pathway. 140
S. sanguis induces platelet aggregation in a complement-dependent way with a lag time that can be explained by the time needed for the assembly of the C5b-9 complex on the bacterial surface. 146 S. aureus ClfA exhibits an alternative route to bind platelets that is fibrinogen-independent and involves FcγRIIa and the assembly of complement proteins as well as a complement receptor. 147
The complement factors C1q, C4, C3, and C9 bind TRAP (thrombin receptor-activating peptide)-activated platelets without, however, activating the complement cascade, indicating that under physiological conditions there is no activation of the complement system on the platelet surface. 148 Assembly of the lytic terminal complement complex C5b-9 on the platelet plasma membrane can activate platelets and induce platelet procoagulant activity. 149 Patients suffering from HUS display hemolytic anemia, acute kidney failure, complement system activation and microvascular thrombosis leading to thrombocytopenia. 150 During HUS, endothelial and complement system activation lead to vWF release from endothelial Weibel–Palade bodies as well as P-Selectin and TF recruitment in a C3a or C5a-dependent way that leads to platelet adhesion and establish the prothrombotic state.150–152
Platelets maintain vascular integrity during inflammation
In patients with immune thrombocytopenia (ITP), the immune system wreaks havoc on endogenous platelets through antiplatelet Abs against major platelet receptors, in most cases GPIb and αIIbβ3. 153 This results in a significantly reduced number of circulating platelets (<100 × 109/L), causing varying degrees of bleeding culminating—in some cases—in intracranial hemorrhages. 154 Often the degree of thrombocytopenia observed in ITP patients does not predict the severity of bleeding, 155 indicating that thrombocytopenia alone is not sufficient to cause bleeding and that an additional trigger—such as inflammation—is needed.
In 2008, Goerge et al. showed that inflammation induces hemorrhage in thrombocytopenic mice.
21
Using multiple models to induce local inflammation in the skin, brain and lung, they observed no signs of bleeding in mice with normal platelet counts. In striking contrast, when using thrombocytopenic mice, they observed massive hemorrhage at the site of inflammation. Remarkably, as little as 5% of the baseline platelet count was sufficient to significantly reduce bleeding. Interestingly, similar results were obtained in a model of pneumonia-derived sepsis using Klebsiella pneumoniae: Thrombocytopenic mice displayed increased bacterial growth and hemorrhage in the lung.
20
Later, the importance of platelet ITAM signaling downstream of GPVI and CLEC-2 in maintaining vascular integrity during skin and lung inflammation was demonstrated.
49
Recent findings show that the CLEC-2 ligand podoplanin is upregulated on macrophages and other extravascular cells during skin inflammation, indicating that in the absence of GPVI, binding of platelet CLEC-2 to podoplanin-expressing cells contributes to limiting bleeding in the inflamed skin (Figure 3).156,157 The study also showed that vascular integrity during lung inflammation was partially dependent on GPIb.
Platelets maintain vascular integrity during inflammation. During local inflammation (e.g. in the skin), platelet GPVI plays a dual role: On the one hand it promotes neutrophil infiltration and ROS production which causes tissue damage (a) and on the other hand it binds to the extracellular matrix protein collagen which gets exposed and facilitates platelet adhesion to restore vascular integrity (b). In the ischemic brain αIIbβ3 facilitates platelet–platelet interactions to prevent intracranial hemorrhage (c). Platelets can also bind to podoplanin, expressed on inflammatory macrophages via CLEC-2 (d). Factors secreted from platelet granules support cerebral hemostasis after stroke, for example by acting on endothelial cell receptors that stabilize cellular junctions (e).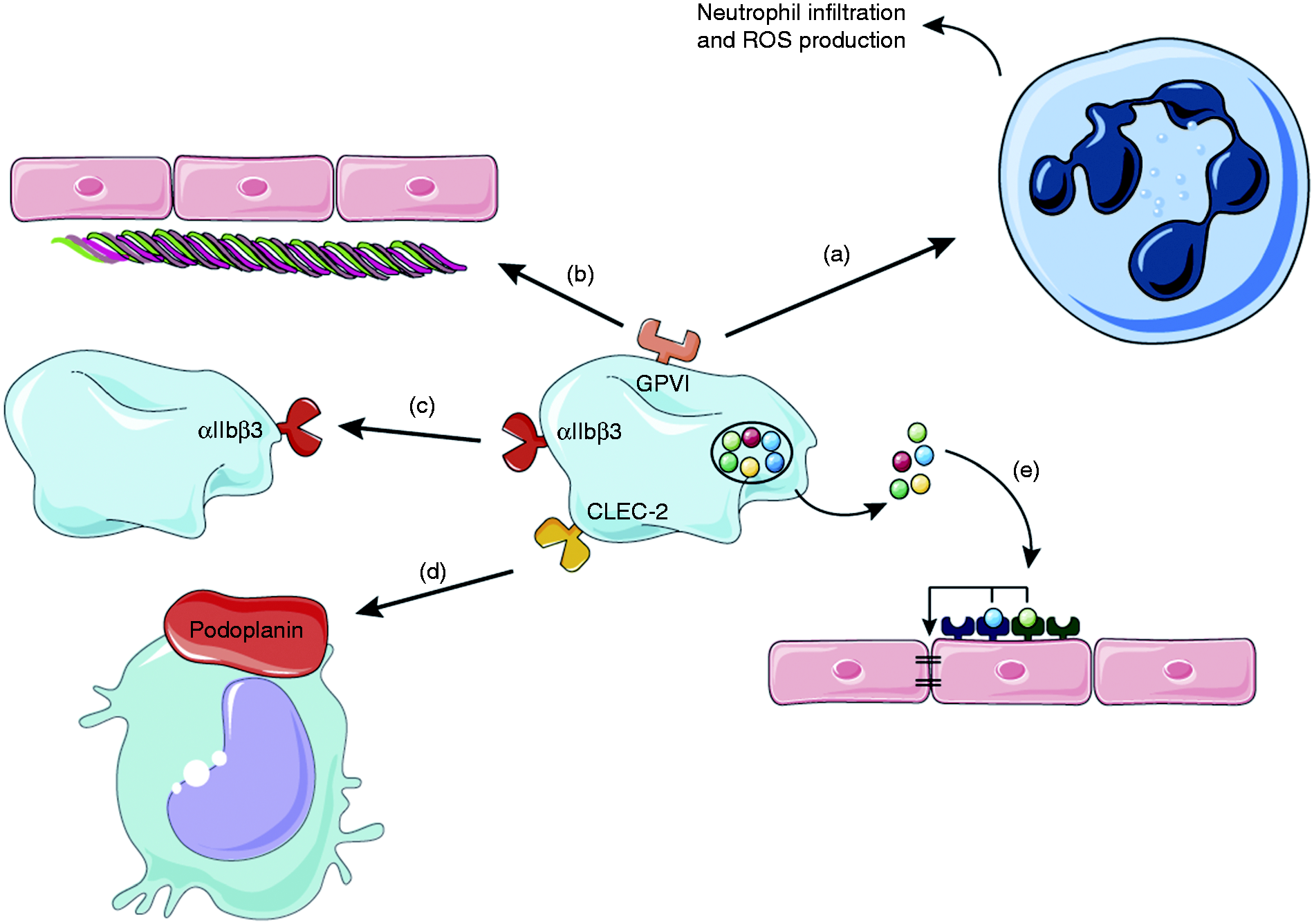
It was suggested that platelet granule secretion following platelet activation through the aforementioned pathways could be part of the mechanism that maintains vascular integrity.21,49,50,158 However, recent findings demonstrate that platelet α- and dense-granule contents are dispensable for maintaining vascular integrity during acute inflammation in the skin and lung. 159 Strikingly, when mice lacking platelet α- and dense-granule secretion were subjected to transient middle cerebral artery occlusion (tMCAO), this resulted in significantly impaired hemostasis in the ischemic brain, causing increased intracranial hemorrhage and 50% mortality comparable to that observed in mice treated with αIIbβ3-blocking Abs. 160 This is especially interesting, since mice with a single deficiency in either platelet α- or dense-granule secretion did not show signs of spontaneous hemorrhage during thrombosis,64,161 thrombo-inflammation after stroke63,64 or tumor metastasis.162,163 This indicates that factors from both α- and dense granules are necessary to maintain cerebral hemostasis after tMCAO; for example platelet-derived angiopoietin-1 or serotonin—which were previously shown to prevent intra-tumor hemorrhage165—might play a role in this setting. Of note, angiopoietin-1 was also shown to be critical for the maintenance of vascular integrity and survival in a mouse model of cerebral malaria. 165 In addition, brain endothelial cells express the P2Y2 receptor that binds nucleotides such as ADP and ATP, which could also contribute to permeability. 166 Indeed, platelet-derived ATP was shown to enable tumor cell transendothelial migration and metastasis via P2Y2. 163
During inflammatory bleeding in the skin, neutrophil extravasation and RBC loss colocalize, 167 and inhibiting neutrophil capturing, adhesion and crawling on the endothelial cell layer significantly reduced hemorrhage. Furthermore, neutrophil diapedesis opens endothelial junctions via dephosphorylation of VE-cadherin during skin inflammation. Interestingly, platelet GPVI on the one hand enhances neutrophil infiltration and ROS production, thereby causing more endothelial damage, 50 while on the other hand it enables platelets to adhere to binding sites exposed by neutrophils. GPVI therefore has a Janus face in this process: It contributes to the pro-inflammatory role of platelets while at the same time helps to repair the damage inflicted by neutrophils and thereby maintains vascular integrity. 168
Conclusion
The roles of platelets besides those in thrombosis and hemostasis have long been neglected, but today we know that they also contribute to inflammation during sepsis, thrombo-inflammation, atherosclerosis and stroke. In most of these cases, platelets present themselves as a most versatile actor: They can form complexes with neutrophils and enhance their phagocytosis, ROS production and NET formation capacity, encase bacteria on the surface of KCs to assist with their destruction or confront pathogens on their own by acting like a wannabe-phagocyte.
First reports of platelets aggregating around bacteria are more than 100 years old; however, only recently we have started to understand the complex interplay between platelets and the cells of the innate immune response during inflammation and infection. Platelets interact with bacteria by direct interactions between platelet receptors and proteins on the bacterial surface. Upon activation platelets release a plethora of factors, for example microbicidal agents but also factors that modulate the innate immune response.
NET formation by neutrophils is a powerful tool to capture and destroy bacteria and it has become clear that platelets critically contribute to this process. A very new concept is that platelets assist in bacterial clearance in the liver through KCs and the complement system, and we will probably see a lot of exciting new findings in this field in the near future.
Another emerging role for platelets is in maintaining vascular integrity during inflammation through an organ-specific process that is independent of classical activation and involves platelet receptors as well as the content of α- and dense granules.
Footnotes
Acknowledgements
We thank Servier for providing Servier Medical Art which was used for creation of figures.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: C.D. is supported by Deutsche Forschungsgemeinschaft (DFG) Research Fellowship DE 2654/1-1. P.K. is supported by grants from the Canadian Institute of Health Research (CIHR), Alberta Innovates Health Solutions (AIHS), the Heart and Stroke Foundation of Canada and the Canada Research Chairs programme.