
Abstract
Cyclooxygenase-2 (COX-2) and IL-8 are two inflammatory mediators induced by protein kinase C (PKC) via various stimuli. Both contribute significantly to cancer progression. Bufalin, a major active component of the traditional Chinese medicine Chan Su, is known to induce apoptosis in various cancer cells. This study clarifies the role and mechanism of bufalin action during PKC regulation of COX-2/IL-8 expression and investigates the associated impact on breast cancer. Using MB-231 breast cancer cells, bufalin augments PKC induction of COX-2/IL-8 at both the protein and mRNA levels, and the production of prostaglandin E2 (PGE2) and IL-8. The MAPK and NF-κB pathways are involved in both the PKC-mediated and bufalin-promoted PKC regulation of COX-2/IL-8 production. Bufalin increases PKC-induced MAPKs phosphorylation and NF-κB nuclear translocation. PGE2 stimulates the proliferation/migration of breast cancer cells. Furthermore, PKC-induced matrix metalloproteinase 3 expression is enhanced by bufalin. Bufalin significantly enhances breast cancer xenograft growth, which is accompanied by an elevation in COX-2/IL-8 expression. In conclusion, bufalin seems to promote the inflammatory response in vitro and in vivo, and this occurs, at least in part, by targeting the MAPK and NF-κB pathways, which then enhances the growth of breast cancer cells.
Introduction
Breast cancer is a frequently diagnosed cancer and the leading cause of cancer death among women. 1 The tumor microenvironment is largely orchestrated by inflammatory cells, and this often results in the proliferation, survival and migration of cancer cells during cancer progression. 2 Malignant progression during carcinogenesis is often associated with a persistent chronic inflammatory microenvironment that facilitates angiogenesis and promotes the growth, invasion and metastasis of tumor cells; these events occur via the involvement of various inflammatory mediators, as well as their downstream targets. 2 Accumulated evidence has indicated that inflammation contributes greatly to carcinogenesis by producing a variety of inflammatory mediators, such as cyclooxgenase-2 (COX-2) and IL-8, both of which are well known to affect an array of different signaling pathways that lead to cancer development. 3 An animal model has revealed that breast cancer cells present in an involuting mammary microenvironment can result in large tumors; this is accompanied by increased COX-2 expression and an invasive phenotype within the mammary gland. In such circumstances, the inhibition of COX-2 would seem to reduce collagen fibrillogenesis, tumor growth and tumor-cell infiltration. 4 A previous study measuring the levels of COX-2 expression in normal and breast cancer tissues has suggested that COX-2 levels can act as an indicator of breast cancer risk, supporting the hypothesis that an aberrant upregulation of COX-2 leading to an accumulation of prostaglandin E2 (PGE2), which then plays an important role in the progression of breast cancer.5,6 In addition, the inflammatory chemokine IL-8 and its cognate receptor show a high correlation with an increased risk of breast cancer and breast cancer progression and, in this context, IL-8 has been demonstrated to mediate breast cancer-cell invasion.7,8 Moreover, breast cancer stem-like cells (CSCs) have been recently recognized as a potential therapeutic target, and it has been found that the activation of the cognate receptors of IL-8, CXCR1 and CXCR2, by IL-8 is involved in the regulation of breast CSC activity, and in this way IL-8 signaling is a critical component in targeting breast CSCs, and thus it has been proposed that blocking IL-8 functions in combination with other existing chemotherapy and endocrine therapy drugs would potentially eradicate both the CSCs and non-CSCs in breast tumors. 9 Taken together, these findings suggest that IL-8 is a crucial molecule in breast cancer progression.
Several signaling pathways have been demonstrated to play a variety of roles in the regulation of COX-2/IL-8 production. NF-κB and the MAPKs, namely p38, c-Jun N-terminal kinase (JNK) and ERK, have all been shown to be involved in COX-2 regulation.10–13 Furthermore, NF-κB, p38, JNK and ERK have also been demonstrated to be involved in IL-8 regulation.14–16 The protein kinase C (PKC) family contains a number of protein kinases that have distinct tissue distribution patterns and functionalities. Previously, studies have revealed that PKC is one of the main signal transduction systems involved in a variety of inflammatory cascades. 17 In fact, PKC has been shown to regulate the activation of both NF-κB and the various MAPKs. 18 In addition, PKC and its substrates are known to be associated with a range of human breast cancer-related phenotypes, and PKC has been reported to promote the motility of human breast cells. These findings, when taken together, suggest that the PKC-mediated pathways play critical roles in breast cancer progression.19–21
Chan Su, a popular traditional Chinese medicine, is extracted from the skin or parotid venom glands of Bufo gargarizans Cantor, the Asiatic toad. 22 The bufadienolides, including bufalin, cinobufagin and resibufogenin, are the major cardiotonic steroids isolated from Chan Su. These chemicals are well known as Na+/K+ ATPase inhibitors and are able to regulate renal sodium transport, and have cardiotonic, anesthetic and blood pressure stimulation effects. 23 Previous studies have revealed a valuable role for bufalin as an anti-tumor agent that targets multiple types of cancer cells; these include cells from leukemia, prostate, lung, liver, ovarian, colon and liver cancers.24–30 Thus, whether bufalin acts as a pro-inflammatory or an anti-inflammatory molecule in relation to cancer progression remains a puzzle. In fact, the impacts of bufalin in relation to inflammation on breast cancer have not been well characterized in either cell culture or any animal model. Therefore, the aim of this study was to investigate whether and how bufalin is able to act as an inflammation-modulating player in vitro and in vivo by exploring the effects of this chemical on various inflammation-associated signaling pathways and to determine whether bufalin might be involved in promoting the development of breast cancer.
Materials and methods
Chemicals and reagents
Bufalin was purchased from Sigma (St. Louis, MO, USA). The PKC activator 12-O-tetradecanoylphorbol-13-acetate (TPA) was purchased from Enzo Life Sciences (Farmingdale, NY, USA). Bisindolylmaleimide I (BIM I; a PKC inhibitor) and PGE2 were purchased from Cayman Chemical (Ann Arbor, MI, USA). FBS was obtained from HyClone (Logan, UT, USA). Reverse transcriptase and Taq polymerase were purchased from Promega (Madison, WI, USA). The following Abs were purchased: mouse monoclonal Ab against human IL-8 (R&D Systems; Minneapolis, MN, USA); mouse monoclonal Ab against COX-2 (Cayman Chemical); rabbit polyclonal Abs against p65(Neomarkers, Fremont, CA, USA); mouse monoclonal Ab against histone H3 (Santa Cruz Biotechnology, Santa Cruz, CA, USA); mouse monoclonal Ab against β-actin f(Novus Biologicals, Littleton, CO, USA); mouse monoclonal Ab against α-tubulin and sheep monoclonal anti-mouse IgG secondary Ab (Sigma); HRP-conjugated donkey anti-rabbit IgG secondary Abs (Amersham Life Science, Arlington Heights, IL, USA). Unless otherwise specified, all other chemicals and reagents used in this project were obtained from Sigma.
Cell culture
A non-tumorigenic mammary epithelial cell line, H184B5F5/M10, and three human breast cancer cell lines, MCF-7, MDA-MB-231 and Hs578T, were maintained at 37℃ in high-Glc DMEM (Biological industries, Bet-Haemek, Israel) containing 10% FBS. The various cells were plated and allowed to grow overnight (16–18 h) prior to the various treatments, which were carried out on the following day.
Animals
Female athymic nude mice at the age of 5 wk were allowed to acclimatize for 1 wk while housed at 25℃ on a 12-h light/12-h dark cycle; all animals were allowed free access to water and standard laboratory chow. All animal care and experiments were approved by the Institutional Animal Care and Use Committee of National Yang-Ming University.
Western blotting analysis
Protein extracts from the tumor xenografts, total cellular proteins and nuclear proteins were collected and processed as required, and their protein concentrations were determined using Bio-Rad protein assay reagent (Bio-Rad, Hercules, CA, USA). Protein samples were then subjected to a normal Western blotting assay in order to determine the expression profiles of the various target proteins.
ELISA
The concentrations of IL-8 and PGE2 in the culture medium were determined using ELISA kits; for IL-8 these were obtained from R&D systems and for PGE2 these were obtained from Assay Designs (Ann Arbor, MI, USA). The ELISAs were performed according to the manufacturers’ instructions.
Semi-quantitative RT-PCR
Primer sequences for RT-PCR.

Cell viability assay
MTT assay was used to monitor MB-231 cell viability. Briefly, MB-231 cells were plated at 1 × 104 cells/well overnight into a 96-well plate format. The plated cells were then washed and this was followed by the various treatments as indicated. The treated cells were then mixed with 0.5 mg/ml MTT (Sigma) at a dilution of 1:10 from a stock solution (5 mg MTT/ml); this was adjusted based on the volume of culture medium. The cells were then incubated for 4 h at 37℃. At the end of incubation, the MTT solution was removed and 200 μl isopropanol was added to dissolve the dark-blue formazon crystals that had formed. The proportions of viable cells were determined by examining the OD at a test wavelength of 570 nm and a reference wavelength of 630 nm using an ELISA reader (Power Wave X340; Bio-Tek Instrument Incorporation, Winoski, VT, USA). Cell proliferation was also measured by the alamarBlue assay (Invitrogen, Carlsbad, CA, USA), which involves the addition of a fluorogenic redox indicator to the cell culture. The reduction reactions of the metabolically active cells then bring about the reduction of non-fluorescent resazurin to bright-red fluorescent resorufin. 19 Four h before termination of the various treatments, alamarBlue dye (Invitrogen) was added and the mixture was allowed to form during the incubation for 4 h at 37℃. The culture medium containing the alamarBlue dye was then removed from wells and transferred into a black 96-well plate (OptiPlate-96F; PerkinElmer, Waltham, MA, USA) and this plate was then subjected to spectrofluorometry (Varioskan Flash Spectral Scanning Multimode Reader; Thermo Electron, Waltham, MA, USA) with excitation at 550 nm and emission at 590 nm.
Wound-healing assay
Culture inserts (ibidi GmbH, Martinsried, Germany) were placed on the surface of each well in a 24-well plate. Next, 70 μl cell suspension was added to either side of reservoirs and after overnight incubation the inserts were removed to form a wound-like gap. The migration profiles of the MB-231 cells were then determined by microscopy at 100 × magnification, and quantification of migration ability was calculated by measuring the surface area of wound at the 24 and 48 h after the removal of the insert. The new reduced surface area was deducted from the surface area of wound at the 0 h time point, and then divided by the surface area of wound at the 0 h time point.
The MB-231 human breast tumor xenograft model
Female athymic nude mice were injected s.c. in both dorsal flank regions with 5 × 106 MB-231 cells. The tumor volumes were estimated weekly by measuring the length, width and depth of the tumor using calipers and calculated using the formula L × W × D × 103 (mm3). Once an approximate tumor volume of ∼20 mm3 was established, which was at around 4 wk, the tumor xenograft on the left flank was injected intratumorally with 10 μl bufalin (1 μM, in 0.9% normal saline). The tumor xenograft on the right flank was injected with an equal 10-µl volume of normal saline as a control. The tumors were excised as pairs from each mouse after an additional 4 wk of growth. The size and mass of each tumor was then measured and this was then followed by Western blotting analysis of the proteins expressed by the tumor.
Statistical analysis
Experimental data are expressed as the mean ± the SEM, in order to compare the difference in mean between each treatment group and the control group. The results were analyzed by one-way ANOVA, which was followed by a least-significant difference test. Differences with a value of P < 0.05 were considered statistically significant.
Results
Bufalin in MB-231 cells augments PKC-induced COX-2/IL-8 mRNA and protein expression levels, as well as the PGE2/IL-8 secretion
To define the inflammation-modulating effect of bufalin on COX-2 and IL-8 expression that has been triggered by PKC activation, MB-231 cells were pretreated with different concentrations of bufalin (0.001, 0.01, 0.1, 1 μM) for 2 h, followed by the exposure to TPA (20 ng/ml) for 24 h. Indeed, PKC activation increased the protein levels of COX-2 and IL-8, as well as PGE2 and IL-8 secretion (Figure 1a, b). Bufalin at 1 µM significantly induced COX-2 and IL-8 expression, and at 0.1 or 1 μM also significantly further increased the PKC-induced COX-2 and IL-8 protein expression, as well as the resulting PGE2 and IL-8 secretion (Figure 1a, b). Bufalin (1 μM) alone treatment was able to increase PGE2 secretion (Figure 1a). To confirm the specific PKC-activation effect by TPA, MB-231 cells were pretreated with a selective PKC inhibitor, BIM I (1, 5, 10 µM), for 2 h, and then the PKC activator TPA (20 ng/ml) was included for an additional 24 h. BIM I at 5 or 10 µM significantly attenuated TPA-induced COX-2 and IL-8 expression (Figure 1c). In addition, to rule out the possibility that the effect of bufalin was due to a promotion of cell proliferation, the viability index of the MB-231 cells with the same treatments as shown in Figure 1a was determined by alamarBlue and MTT assays. There was no apparent difference in the readouts across all the treatments for either assay (data not shown). Next, we assessed whether bufalin has a similar promoting effect on COX-2 and IL-8 protein levels under PKC activation in another breast cancer cell line, MCF-7. Similar to our findings with MB-231 cells, the COX-2 and IL-8 protein expression levels were induced by TPA and further enhanced by bufalin (Supplementary Figure S1A); however, a lower dose at 0.01 µM was able to mediate the most profound promotion effect on TPA-induced COX-2 and IL-8 expression (Supplementary Figure S1A). Meanwhile, we also tested the bufalin effect on TPA-induced inflammation in the breast cancer cell line Hs578T. TPA clearly induced both COX-2 and IL-8 protein expression, and PGE2/IL-8 secretion and bufalin alone did not have any effect (Supplementary Figure S2); however, surprisingly, bufalin in combination with TPA appeared to suppress the inducing effect of TPA on COX-2 expression and PGE2 secretion (Supplementary Figure S2A); bufalin at 1 or 0.1 µM was able to attenuate TPA-induced IL-8 expression, but lower doses of bufalin further enhanced TPA-mediated IL-8 expression (Supplementary Figure S2B) and bufalin at all doses did not affect TPA-mediated IL-8 secretion (Supplementary Figure S2B). In addition, we also examined the effect of bufalin on a human mammary epithelial cell line H184. The level of COX-2 protein expression was low in this cell line and was not affected by either TPA or bufalin (Supplementary Figure S1B); a low level of IL-8 protein was detected, but this was not affected by either TPA or bufalin (Supplementary Figure S1B).
Enhancement of PKC-induced COX-2 and IL-8 protein levels, as well as PGE2 and IL-8 secretion. (a, b) Plated MB-231 cells were untreated (0.1% DMSO; Control) or pretreated with various doses of bufalin for 2 h, followed by TPA (20 ng/ml) administration for additional 24 h. Alternatively, cells were pretreated with PKC inhibitor BIM I (1, 5, 10 µM) for 2 h and then (c) TPA (20 ng/ml) was included for an additional 24 h. Total cell lysates were harvested to evaluate the protein levels of (a, c) COX-2 and (b, c) IL-8 by Western blotting using α-tubulin as an internal control. The concentrations of (a) PGE2 and (b) IL-8 secreted into the cultured medium were measured by ELISA. The results represent the mean ± SEM from (a) six or (b, c) four independent experiments. *P < 0.05 compared with the control; #P < 0.05 compared with the TPA treatment; &P < 0.05 compared with the bufalin treatment.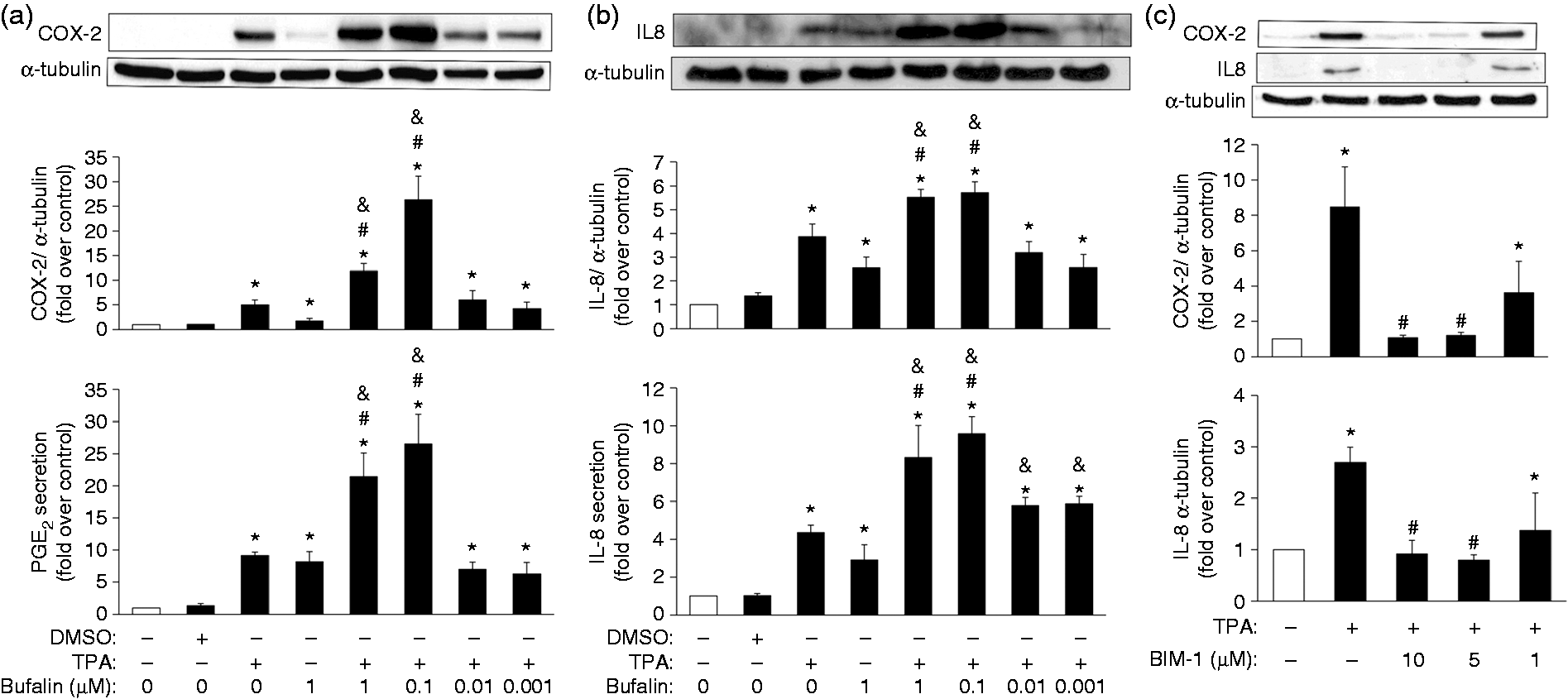
In addition to the above, in order to clarify the effect of bufalin on the COX-2 and IL-8 mRNA expression after PKC stimulation, MB-231 cells were pretreated with bufalin for 1 h and then TPA was included for an additional 12 h. The mRNA levels of COX-2 and IL-8 were, indeed, increased by TPA or bufalin treatment alone (1 μM). Furthermore, when bufalin at 0.1 or 1 μM was combined with TPA, this resulted in a moderate increase in mRNA levels of COX-2 and IL-8 (Figure 2a, b).
Promotion of PKC-induced COX-2 and IL-8 mRNA expression. Plated MB-231 cells were treated with various doses of bufalin for 1 h, followed by exposure to TPA (20 ng/ml) for an additional 12 h. Total cell lysates were harvested to measure (a) COX-2 and (b) IL-8 mRNA expression by semi-quantitative RT-PCR assay using (a) human β-actin or (b) GAPDH as an internal control. The results represent the mean ± SEM from (a) five or (b) four independent experiments. *P < 0.05 compared with the control; #P < 0.05 compared with the TPA treatment; &P < 0.05 compared with the bufalin treatment.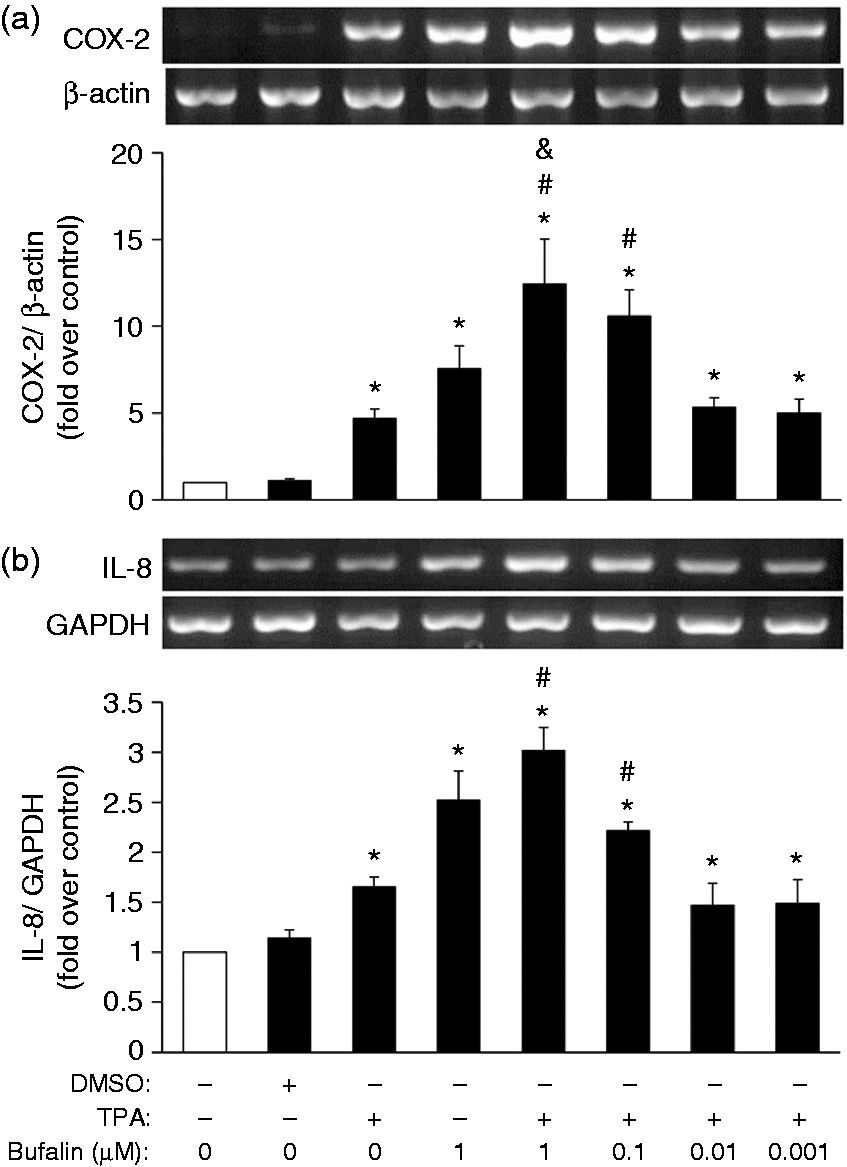
PKC activation modulates COX-2 and IL-8 expression via the JNK and ERK pathways, while bufalin targets all the MAPK and NF-κB pathways
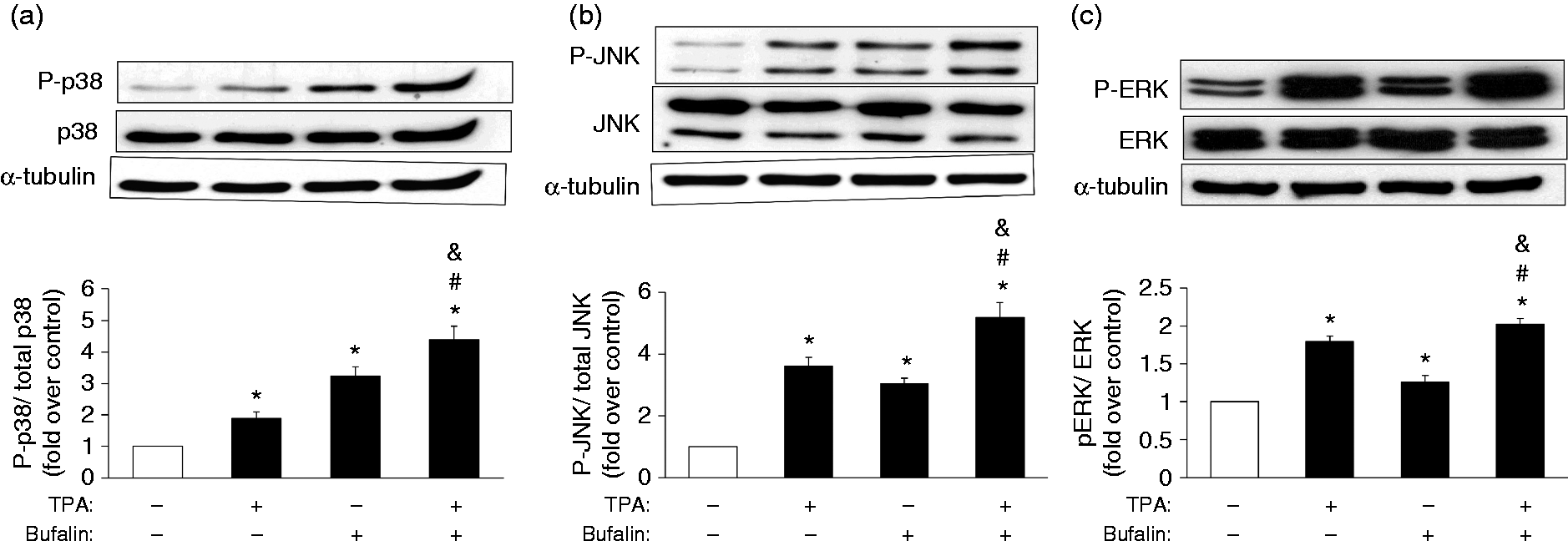
After confirming the impact of bufalin on PKC-induced inflammation (Figure 1), we then went on to clarify what signaling pathways may be the target of bufalin in MB-231 cells. Initially, PKC activity was examined and it was found that TPA significantly induced PKC activation, while bufalin did not seem to affect PKC activity (Supplementary Figure S3). Consequently, to pinpoint other potential targets of bufalin by which it might exert its inflammation-promoting properties via PKC-mediated COX-2 and IL-8 expression, a panel of inhibitors targeting to various signaling cascades, namely the MAPK cascades (SB2030580 for p38, SP600125 for JNK and PD98059 for ERK) and the NF-κB cascade (APDC), were used to identify the critical signaling components involved in mediating PKC- and bufalin-regulated COX-2 and IL-8 expression. The results show that JNK and ERK are involved in the PKC induction of COX-2 and IL-8 expression (Figure 3), while all the MAPKs, as well as NF-κB, were found to be involved in the combined bufalin-induced and PKC-induced COX-2 and IL-8 expression (Figure 3). We then further explored whether bufalin affected the activation of the MAPK and NF-κB pathways via phosphorylation and nuclear translocation, respectively. Phosphorylation of p38 was noted to be significantly induced by TPA at 15, 30 and 60 min, and by bufalin at 120 min (Supplementary Figure S4A), while JNK phosphorylation was significantly induced by TPA at 5 and 15 min, and by bufalin at 5 and 120 min (Supplementary Figure S4B). Finally, ERK phosphorylation was significantly induced by TPA at 5, 15, 30, 60 and 120 min, and by bufalin at 15, 30 and 60 min (Supplementary Figure S4C). In order to evaluate the combined effect of TPA and bufalin, and owing to the variations in the induction profiles of each MAPK by TPA or bufalin, cells were treated with bufalin for 90 min, followed by the inclusion of TPA for additional 30 min for p38 phosphorylation. For JNK phosphorylation, the cells were treated with bufalin for 105 min, followed by the inclusion of TPA for an additional 15 min. For ERK phosphorylation, cells were treated with TPA or bufalin alone, or with a combination of TPA and bufalin for 60 min. Intriguingly, under the last condition involving the combined TPA and bufalin treatment, the result was a strikingly higher degree of phosphorylation of p38 and JNK, but only a further minor increase of ERK phosphorylation, than TPA or bufalin treatment alone (Figure 4a–c).
Involvement of the MAPK and NF-κB signaling pathways in the mediation of bufalin-regulated and PKC-regulated COX-2 and IL-8 expression. Plated MB-231 cells were pretreated with various inhibitors of the different signaling pathways, namely SB2030580 (SB; 20 µM) for p38, SP600125 (SP; 40 µM) for JNK, PD98059 (PD; 20 µM) for ERK and APDC (50 μM) for NF-κB for 1 h, followed by treatment with TPA (20 ng/ml) for an additional 24 h. Finally, the (a) COX-2 and (b) IL-8 protein levels were measured by Western blotting. The results represent the mean ± SEM from four independent experiments. *P < 0.05 compared with the control; #P < 0.05 compared with the TPA treatment; &P < 0.05 compared with the combined bufalin and TPA treatment. The promoting effects of bufalin on PKC-induced MAPKs activation. To examine the combined effects of bufalin and TPA, (a) overnight plated MB-231 cells were pretreated with bufalin (100 nM) for 90 min, and then TPA (20 ng/ml) was included for an additional 30 min (bufalin for a total of 120 min) when assessing p38 phosphorylation; (b) pretreated with bufalin (100 nM) for 105 min and then TPA (20 ng/ml) was included for additional 15 min (bufalin for a total 120 min) when assessing (b) JNK phosphorylation; (c) or treated with bufalin (100 nM) or TPA (20 ng/ml) alone, or in combination for 1 h when assessing ERK phosphorylation. The results are presented as the mean ± SEM from five individual experiments. *P < 0.05 compared with control; #P < 0.05 compared with TPA treatment; &P < 0.05 compared with bufalin treatment.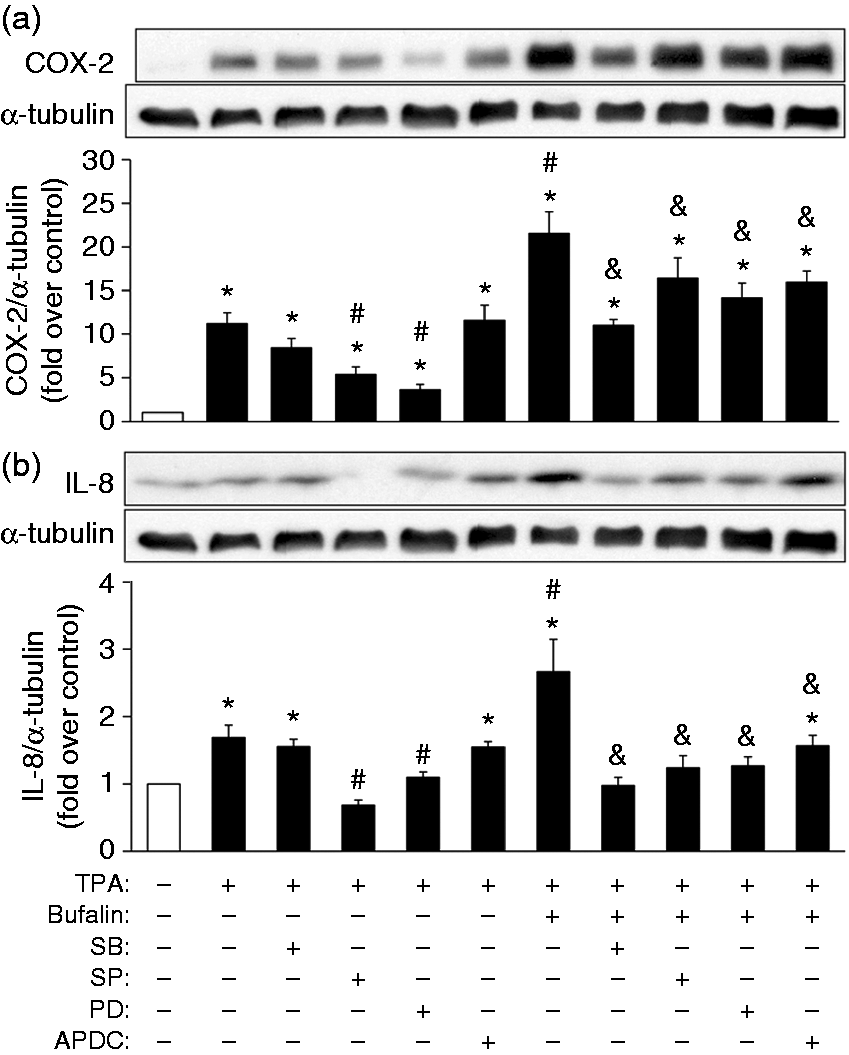
The NF-κB inhibitor APDC was found to attenuate significantly the combined effects of bufalin and TPA on COX-2 and IL-8 expression (Figure 3). On stimulation by inflammation, NF-κB is well known to translocate from the cytoplasm to the nucleus in order to upregulate the transcription of various NF-κB-dependent genes.
31
Thus, we investigated the effects of bufalin and TPA on the translocation of the NF-κB p65 subunit from the cytosol into the nucleus. Notably, TPA increased p65 nuclear translocation at 30 and 60 min (Figure 5a), while bufalin induced p65 nuclear translocation at 15, 30 and 60 min (Figure 5b). To evaluate the combined impact of bufalin and TPA, MB-231 cells were treated with bufalin for 30 min, and then TPA was included for additional 30 min (bufalin for 60 min and TPA for 30 min in total). The combined TPA and bufalin treatment appeared to result in a minor increase in induction of p65 nuclear translocation than either TPA or bufalin alone (Figure 5c).
Promotion of TPA-induced NF-κB translocation by bufalin. Overnight plated MB-231 cells were treated with (a, b) TPA (20 ng/ml) or bufalin (100 nM) for 5, 15, 30, 60 or 180 min, or (c) pretreated with bufalin (100 nM) for 30 min and then TPA (20 ng/ml) was included for additional 30 min. The cells were then monitored to measure the bufalin-mediated and PKC-mediated accumulation of nuclear NF-κB p65 subunit by Western blotting with histone H3 as an internal control. The results are presented as the mean ± SEM from (a, c) five or (b) eight independent experiments. *P < 0.05 compared with the control; #P < 0.05 compared with the TPA treatment; &P < 0.05 compared with the bufalin treatment.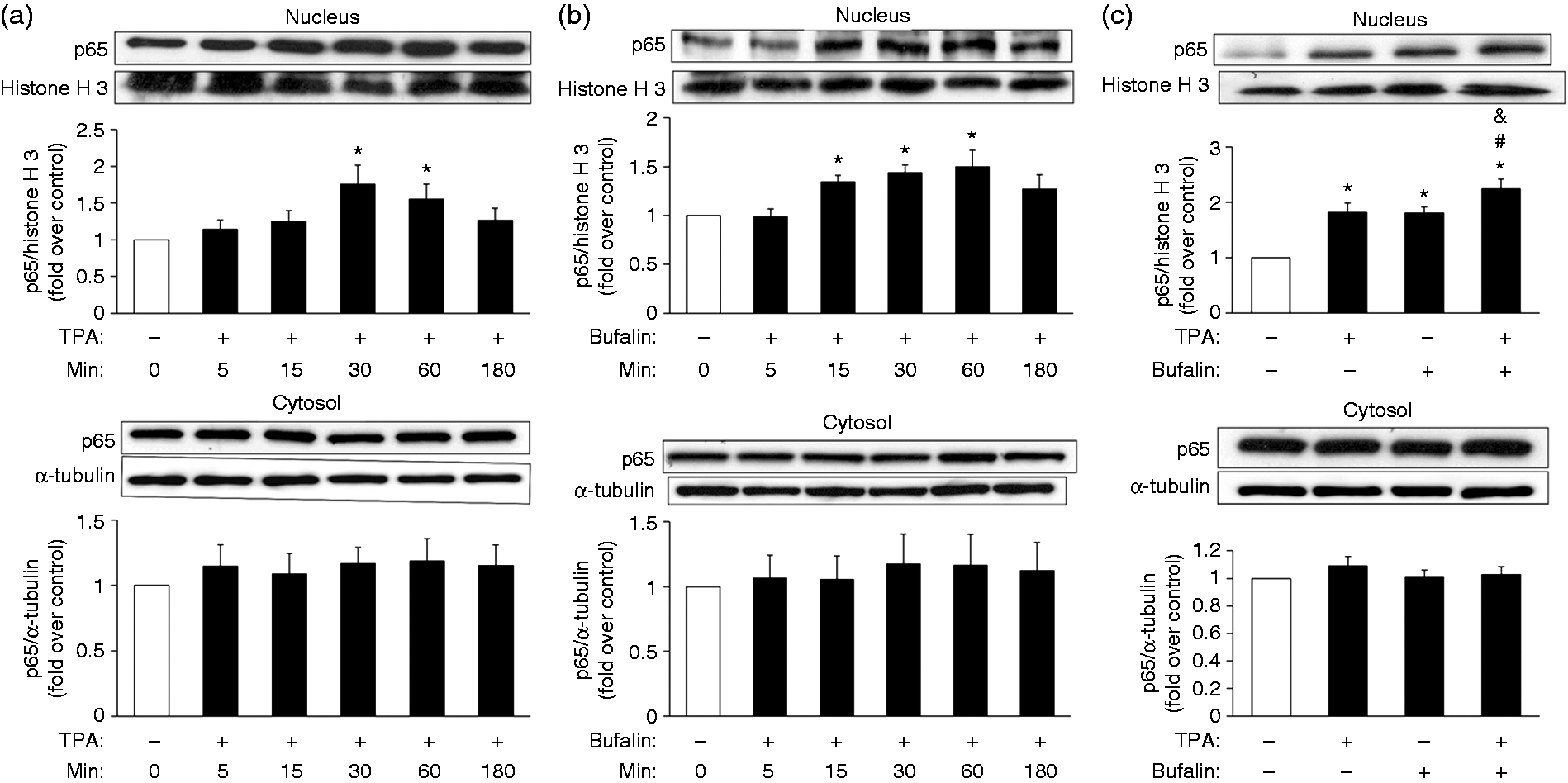
PGE2 stimulates cell proliferation and migration in MB-231 cells
Bufalin has been shown here to further enhance the PKC-mediated COX-2 expression and PGE2 production (Figure 1). Numerous previous investigations have demonstrated that PGE2 plays a role in cancer progression by promoting cell proliferation, metastasis and angiogenesis.
32
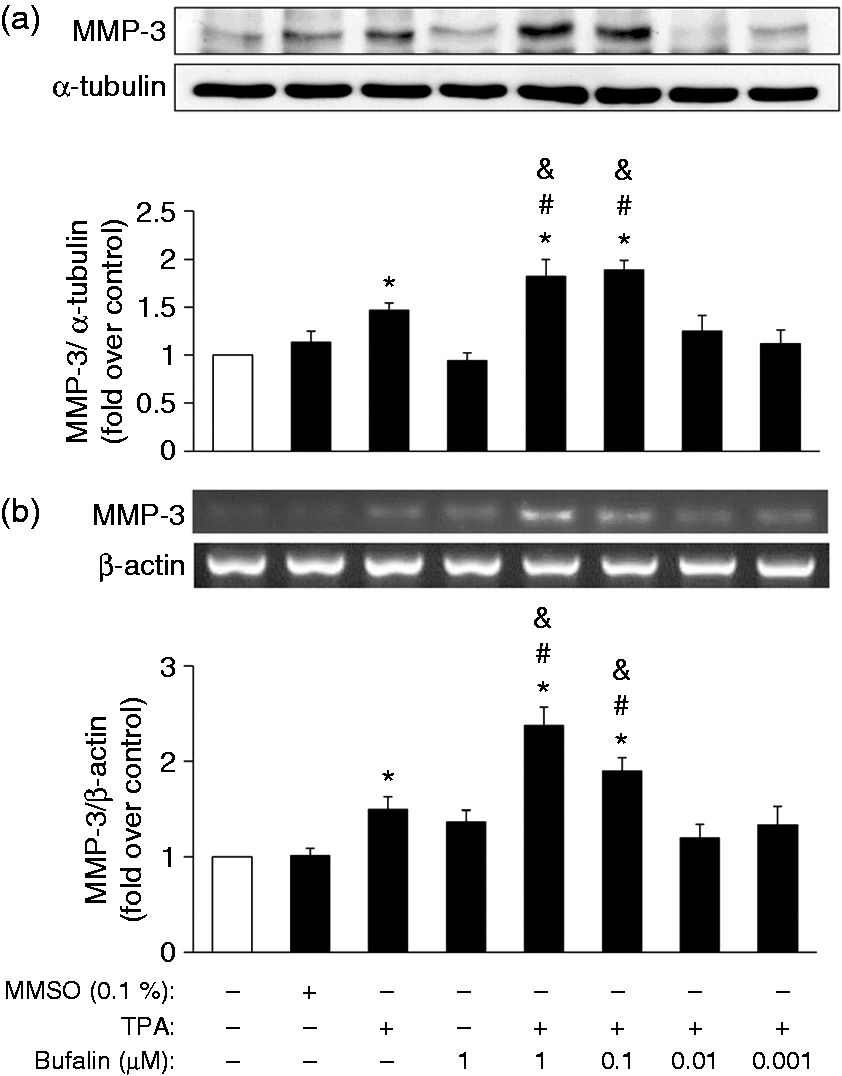
Based on these, we went on to examine the role of PGE2 in cell proliferation and migration in MB-231 cells. Our findings showed that PGE2 at 50 or 100 μM was able to increase MB-231 cell proliferation (Figure 6a), while PGE2 at 50 μM apparently induced cell migration at 24 and 48 h (Figure 6b, c). In addition, it has been previously reported that matrix metalloproteinase (MMP)-3 is involved in tumor invasion in MB-231 cells and it also has been proposed that PGE2 is able to induce cell migration/invasion via MMP-3 during endometriosis.33,34 Based on our gene microarray assay of MB-231 cells, we discovered that MMP-3 is not only upregulated by TPA, but this up-regulation is further enhanced by bufalin (data not shown). Thus, we evaluated the impact of bufalin and TPA on MMP-3 expression. Notably, both the protein and mRNA expression levels of MMP-3 were increased by TPA, and this induction manner was further elevated by bufalin at 0.1 or 1 μM (Figure 7).
Induction of cell proliferation and migration by PGE2. Plated MB-231 cells were (a) treated with different concentrations of PGE2 (5, 10, 25, 50, 100 µM) for 24 h to evaluate the cell viability by MTT assay, or (b, c) treated with PGE2 (25, 50 µM) for 24 or 48 h to determine their migration profiles by wound-healing assay. The results are presented as the mean ± SEM from (a) four or (b, c) six independent experiments. *P < 0.05 compared with the control group at the same time point. Elevation of TPA-induced MMP-3 protein and mRNA expression by bufalin. Plated MB-231 cells were pretreated with bufalin (0.001, 0.01, 0.1, 1 µM) for 2 h, and then TPA (20 ng/ml) was included for an additional (a) 24 h or (b) 12 h. The (a) protein and (b) mRNA levels of MMP-3 were detected by Western blotting with α-tubulin as the internal control and by RT-PCR with β-actin as an internal control, respectively. The results are presented as the mean ± SEM from five independent experiments. *P < 0.05 compared with control; #P < 0.05 compared with TPA treatment; &P < 0.05 compared with bufalin treatment.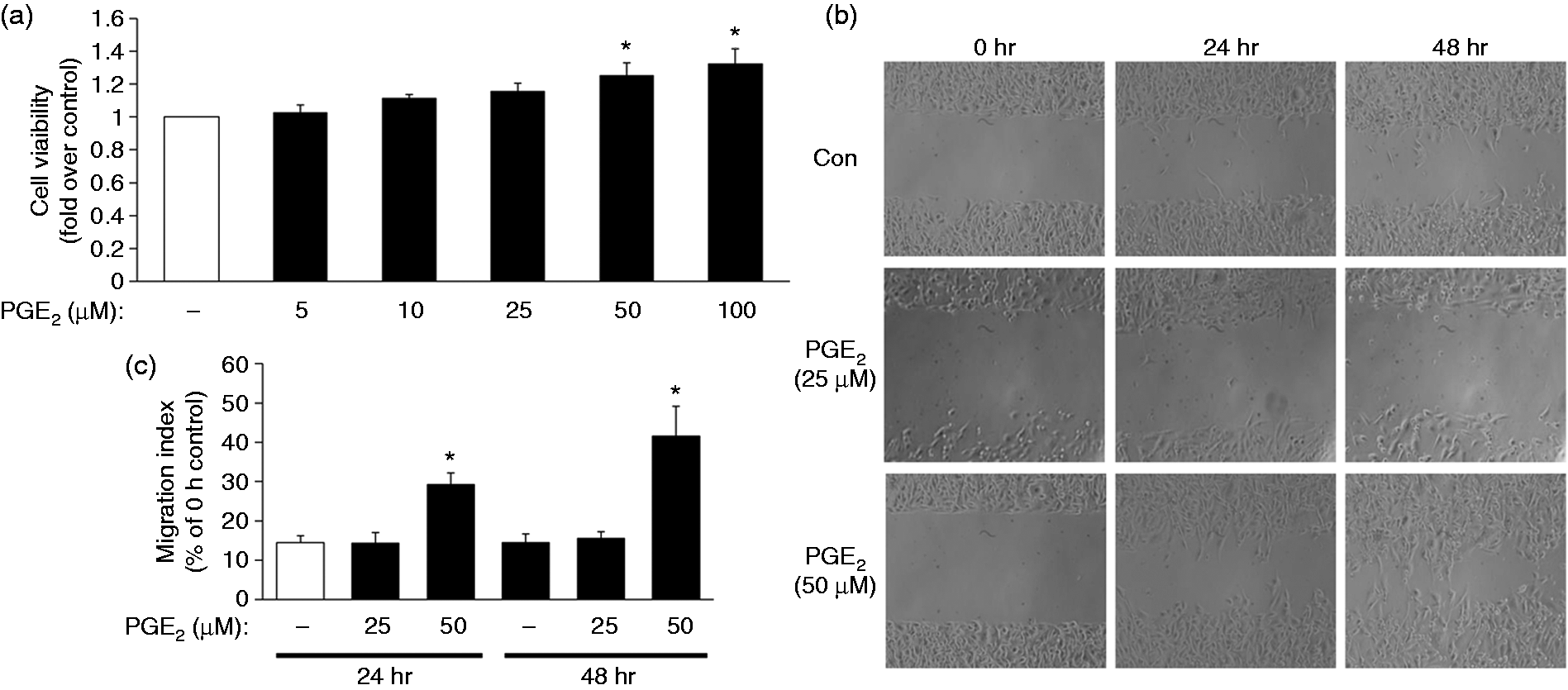
Bufalin increases tumor size and mass, as well as COX-2 and IL-8 protein levels, using a breast cancer xenograft model
In order to verify the effect of bufalin on breast tumors in vivo, we examined the effect of bufalin on the growth of MB-231 breast tumor xenografts in parallel with measuring COX-2 and IL-8 protein levels in the tumors. This was conducted by s.c. injection of MB-231 cells into the dorsal flank regions of nude mice followed by treatment with bufalin as described in the ‘Materials and methods’ section. Two wk after bufalin treatment had begun, a significant increase in tumor xenograft size was noted in the bufalin treatment group compared with the control tumors on the opposite flank that were injected with saline (Figure 8a, b). This was accompanied by an elevation in tumor mass of the bufalin treatment group compared to the control group (Figure 8b). Finally, it was verified that the administration of bufalin was also able to increase significantly COX-2 and IL-8 protein levels in the breast tumor xenografts (Figure 8c).
Bufalin increases the size and mass of xenographic breast tumors, as well as the expression of COX-2/IL-8 proteins in the tumors. Changes in volume of breast tumor xenografts were induced by s.c. injection of MB-231 into the dorsal flanks of mice with one of these tumors undergoing intratumoral administration of bufalin (bufalin, filled circle), and the other undergoing intratumoral injection with normal saline (Con, open circle). The tumors were measured each week for 8 wk; (a, b) the final tumor mass was measured after sacrifice. (c) The protein levels of COX-2 and IL-8 in xenograft tumor total lysates were evaluated by Western blotting with β-actin as an internal control. Each value represents mean ± SEM from eight animals. *P < 0.05 control group at each week compared with the control on wk 4 (the beginning of bufalin injection); #P < 0.05 bufalin group at each week compared with the control on wk 4; (a) &P < 0.05 compared between bufalin treatment and the control group within the same week; (b, c) *P < 0.05 compared with control group.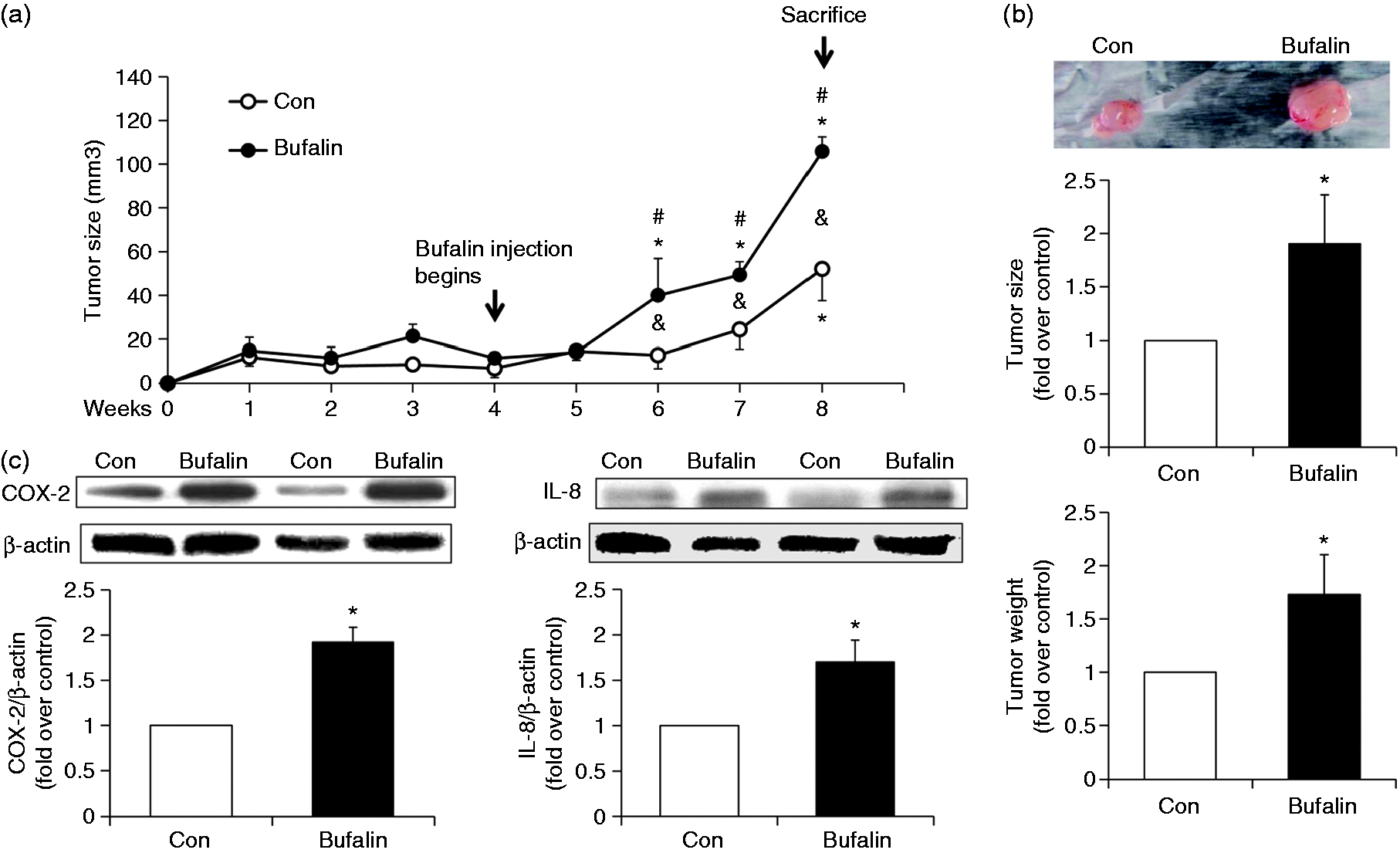
Discussion
Under inflammatory conditions, PKC signaling pathways are activated to regulate COX-2 and IL-8 expression.35,36 Previous studies have noted that PKC expression is elevated in human breast tumors compared with normal breast tissue, and many researchers have also shown that PKC participates in cancer proliferation, invasion, metastasis and angiogenesis.37,38 In the current study, TPA was used to activate the PKC signaling pathways and this system was then used as a model to investigate the significance of inflammatory events in breast cancer progression. The major finding of the present study is that bufalin seems to act as a pro-inflammatory molecule in a cell culture system by increasing COX-2 and IL-8 production in breast cancer cells, as well as in a xenograft animal model by increasing tumor size and mass, as well as increasing COX-2 and IL-8 expression. Furthermore, bufalin causes up-regulation of the PKC-induced COX-2 and IL-8 mRNA and protein expression, in parallel with an elevation in PGE2 and IL-8 secretion. Thus, bufalin appears to evoke pro-inflammation activity and this occurs, at least in part, by targeting to both the MAPK signaling pathways, including p38, JNK and ERK, as well as the NF-κB signaling pathway.
Numerous investigations have determined that IL-8 and COX-2, together with the PGE2 derived from it, are highly induced in the tumor microenvironment, which would promote cell proliferation, metastasis and angiogenesis; this, in turn, exacerbates breast cancer progression.39,40 COX-2 and its resulting product, PGE2, have been well recognized as important positive regulators in breast cancer tumorigenesis via a modulation of the immune response.41,42 In addition, the aberrant elevation of the expression of several cytokines, including IL-8, has been implicated in contributing to the development of breast cancer. 7 Along with the TPA-mediated PKC activation in our study, PGE2 also increased the proliferation and migration of MB-231 cells (Figure 6). During the process of cancer metastasis, cancer cells may produce various MMPs that cause disruption of the extracellular matrix, allowing invasion into blood vessels. As a result of this invasion, cancer cells are able to metastasize to other organs through the blood circulation. 43 During the development of breast tumors, MMPs seem to act as key modulators of cancer cell invasion and migration activity. 44 Among such MMPs, MMP-3, in particular, has been reported widely to participate in tumor invasion and mammary carcinogenesis. 33 PKC-induced MMP-3 expression, both at the protein and at the mRNA level, were found to be further enhanced by bufalin (Figure 7). This finding is consistent with previous reports that MMP-3 is important to MB-231 cell invasion and that PGE2 is able to mediate cell migration/invasion via MMP-3 during endometriosis.33,34 This suggests that bufalin may act via its up-regulation of PKC-mediated COX-2/PGE2 production, which, in turn, increases MMP-3 expression (Figure 7), which then promotes cell migration of MB-231 cells (Figure 6). These findings further support the critical role of PKC-mediated COX-2 and PGE2 production in breast cancer cell proliferation and tumor development.
In our study, bufalin was shown to further elevate TPA-induced PGE2 secretion (Figure 1). A previous study has demonstrated that the serum PGE2 level is significantly higher in patients with breast cancer (1499.0 ± 283.3 pg/ml) than in healthy women (587.0 ± 82.28 pg/ml). 45 It is our understanding that the concentration of PGE2 secreted (around 120 nM) during the bufalin-enhanced TPA-induction was relatively low compared with the amount that is exogenously administered (50–100 μM) to bring about cell proliferation and migration (Figure 6), and the concentration of the exogenous PGE2 treatment we used in his study was much higher (μM level) than the measurement from this study. 45 Besides, the conditioned medium from TPA- and bufalin-treated MB231 cells was tested in the proliferation and migration experiments, but we did not notice an effect after for up to 48 h incubation (data not shown). However, it seems likely that the level of PGE2 will be much higher within the tumor microenvironment in vivo, as it can be produced not only by cancer cells, but also by nearby fibroblasts, various immune cells and endothelial cells. 46 Furthermore, production of the COX-2-derived PGE2 is likely to be further promoted via the positive feedback created in the tumor microenvironment by breast cancer cells. 47 Consistent with our initial findings in MB231 cells, bufalin also has an impact on the TPA-mediated COX-2 and IL-8 production in MCF-7 cells; however, a lower dose at 0.01 µM seems to act most potently in TPA-induced COX-2 and IL-8 expression (Supplementary Figure S1A). Interestingly, the impact of bufalin appears different in Hs578T cells (Supplementary Figure S2). The Hs578T cell line belongs to the primary carcinoma of breast cancer; however, MCF-7 and MB231 lines are from the pleural effusion of metastasized adenocarcinoma. Whether the impact of bufalin to breast cancer depends on the stage of breast cancer will need further investigation. However, there was no obvious effect of exogenous human recombinant IL-8 on MB-231 cell proliferation and migration (data not shown), notwithstanding the fact that IL-8 has been demonstrated to be involved in the regulation of angiogenesis and in immune cell recruitment during breast tumor development.48,49
Previous studies have demonstrated that bufalin is able to regulate PKC activity and induce leukemia cell apoptosis. 50 However, in our study, bufalin did not appear to affect PKC activity (Supplementary Figure S3) and neither was it able to inhibit the cell proliferation of MB-231 cells (data not shown). Using biochemical analysis, our findings show that bufalin is not able to affect TPA-induced PKC activation (Supplementary Figure S3), but it is able to regulate the MAPK and NF-κB signaling cascades via increased PKC-mediated phosphorylation of all three MAPKs (Figure 4), as well as via PKC-induced NF-κB nuclear accumulation (Figure 5). In fact, there are NF-κB responsive elements within the promoter region of both the human COX-2 gene and the human IL-8 gene.51,52 In addition, similar to our findings, it has been shown previously that bufalin is able to induce the phosphorylation of JNK in human hepatoma cells, of p38 and JNK in human pancreatic and oral cancer cells, and of all three MAPKs, p38, JNK and ERK, in MB-231 cells, as well as bring about NF-κB activation in human promyelocytic leukemia HL-60 cells and in the rat brain.53–57 However, some other studies have demonstrated that bufalin in osteosarcoma cells is able to suppress the activation of the JNK and ERK pathways and that bufalin is also able to suppress the NF-κB pathway in rheumatoid arthritis fibroblast-like synoviocytes.58,59 This inconsistency across different cell types/tissues suggests that the action of bufalin in relation to the MAPKs and NF-κB signaling pathways is highly dependent on cell type. Regardless of this variation, our findings clearly confirm that, in breast cancer cells in particular, bufalin seems to be able to act post-PKC activation and upregulate at various downstream points the PKC-mediated p38, JNK and ERK signaling cascades, as well as the NF-κB signaling cascade.
Notably in MB-231 cells, bufalin treatment alone at 1 μM slightly increased COX-2 protein expression, although the effect was not as potent as TPA treatment (Figure 1). In addition, bufalin at 0.1 or 1 μM dramatically promoted TPA-induced COX-2 and IL-8 expression in MB-231 cells (Figure 1a, b), but bufalin was only able to promote TPA-mediated COX-2 and IL-8 expression in MCF-7 cells at 0.01 μM (Supplementary Figure S1A). The reason for this difference in response of MB-231 and MCF-7 cells to bufalin possibly involves the differences between the two cell lines, but is currently a puzzle to us. In addition, in a normal non-tumorigenic human mammary epithelial cell line H184, the protein levels of COX-2 were found to be very low and when these cells were treated with either TPA or bufalin there did not seem to be any effect on COX-2 expression (Supplementary Figure S1B). Similarly, although IL-8 expression could be detected, this was also not affected by TPA or bufalin (Supplementary Figure S1B). 60 When all our findings are taken together, they clearly suggest that bufalin has a pro-inflammatory effect in human breast cancer cells but not in normal mammary epithelial cells. The anti-tumor activity of bufalin has been reported previously, specifically an inhibition of the growth of endometrial and ovarian cancers. 61 In addition, recent studies have shown that the dose range of bufalin used to treat a variety of cancers varies between 0.01 and 1 μM. 27 However, another study has indicated that bufalin at 250 nM has no apoptotic effect on MB-231 breast cancer cells. 62 In our study, we tested a dose range of bufalin from 1 nM to 1 μM and our findings also indicate that, within this range, bufalin does not appear to have a cytotoxic effect on MB-231 cells after 24 h incubation (data not shown). Interestingly, a previous study has investigated the cytotoxic effect of the cardiotonic steroids (CTS), including bufalin in normal human breast cells and breast cancer cells, and it was revealed that, in general, the CTS appears to inhibit the membrane Na,K-ATPase activity in all cell lines of normal or cancer breast cells. However, the cellular responses to these tested drugs in terms of the impact on cell proliferation are different in non-tumor and tumor cells, as CTS seemed to mediate a greater inhibition of proliferation, as well as a more extensive apoptosis in non-tumor breast cells than in tumor cells. Specifically, the bufalin treatment at 50 or 100 nM for 2 h was able to reduce the proliferation of the non-tumor cells but not the tumor cells, and even for 24 h neither dose was able to affect the proliferation of the tumor cell lines, including the MB231 line. Based on the relative resistance of breast cancer cell lines to CTS, it was therefore concluded that CTS might not be a good choice with which to treat cancers. 63 Similar to our finding in this study, bufalin at certain doses may further enhance inflammatory response and breast tumor growth in vivo. In our current study, whether the enhancement effect of bufalin on PKC-mediated inflammation is dependent on its inhibition on the membrane Na,K-ATPase activity requires further investigation. Regarding the dosage of bufalin in an animal model, according to the information provided on the Material Safety Data Sheet, the median i.v. lethal dose (LD50) for mice is 0.74 mg/kg. A previous study administered i.p. bufalin to nude mice bearing BEL-7402 human hepatocellular carcinoma cells at 0.5, 1.0 or 1.5 mg/kg daily for 10 d (18–20 g mouse body mass) and demonstrated an anti-tumor effect of bufalin. These doses are equivalent to 10–30 µg/mouse, whereas in our study, the intra-tumor injections used were 10 μl of 1 μM bufalin three times a week, which is equivalent to 3.87 ng/mouse. 64 Thus, there appears to be a more than 1000-fold difference between the earlier report and our current study. Whether the contrasting findings regarding the effect of bufalin on tumor growth in the earlier study and our findings regarding tumor growth promotion are due to either the different doses used or the different cancer cell lines used is currently not clear and will need further investigation. However, when these earlier findings are considered together with our findings, they suggest that a more in-depth evaluation is needed when treating cancers with bufalin.
In conclusion, our study reveals a novel role for bufalin that it is likely able to act as a pro-inflammation molecule in vitro and in vivo during breast cancer progression. Our study provides new findings in relation to breast cancer development and how bufalin affects the expression of COX-2 and IL-8. In addition, we have also shown that bufalin modifies the production of PGE2 and IL-8 in response to PKC activation; these effects of bufalin occur, at least in part, via targeting of the MAPK and NF-κB signaling cascades.
Supplemental Material
Supplemental material for Novel augmentation by bufalin of protein kinase C-induced cyclooxygenase-2 and IL-8 production in human breast cancer cells
Supplemental Material for Novel augmentation by bufalin of protein kinase C-induced cyclooxygenase-2 and IL-8 production in human breast cancer cells by Hsiao-Ting Chen, David Sun, Yen-Chun Peng, Pu-Hong Kao and Yuh-Lin Wu in Innate Immunity
Footnotes
Acknowledgements
We thank Dr Ralph Kirby, Department of Life Sciences, National Yang-Ming University, for his help with language editing.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Ministry of Science and Technology (MOST 100-2320-B-010-018-MY3; MOST 102-2320-B-010-010-MY3), the Department of Chinese Medicine and Pharmacy, Ministry of Health and Welfare (CCMP100-RD-012), the Cheng Hsin General Hospital (104F003C04, 105F003C04), and the Taiwan Ministry of Education, Aim for the Top University Plan.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.