
Abstract
LPS can activate the inflammatory cascades by inducing various inflammatory mediators, such as prostaglandin E2 (PGE2) resulting from cyclooxygenase-2 (COX-2), and NO produced by inducible NO synthase (iNOS). Lysophosphatidic acid (LPA) has been demonstrated to participate in inflammation. This study aimed to clarify the impact and the involving mechanisms of LPA on LPS-incurred inflammation in macrophages. First, LPA appeared to attenuate LPS-induced protein and mRNA expression of COX-2 and iNOS genes, as well as production of PGE2 and NO. By using selective inhibitors targeting various signaling players, the inhibitory G protein alpha subunit (Gαi) seemed to be involved in the effect of LPA; p38, ERK and NF-κB were involved in the LPS-mediated COX-2/PGE2 pathway; and p38, JNK, phosphoinositide-3-kinase and NF-κB were involved in the LPS-mediated iNOS/NO pathway. LPA was able to diminish LPS-induced phosphorylation of p38 and Akt, as well as NF-κB p65 nuclear translocation. By utilization of inhibitors of COX-2 and iNOS, there appeared to be no modulation between the COX-2/PGE2 and the iNOS/NO signaling pathways. Our findings demonstrate a clear anti-inflammatory role of LPA acting via Gαi in LPS-mediated inflammatory response in macrophages, owing, at least in part, to its suppressive effect on LPS-induced activation of p38, Akt and NF-κB.
Introduction
Inflammation is an important part of the host defense system in response to internal or external environmental stimuli to counteract the insults exerted by these stimuli to the host.1,2 The common component of the Gram-negative bacterial cell envelope, LPS can incite the host immune system, such as macrophages, to initiate the intracellular signaling pathways to produce multiple inflammatory mediators, so as to eliminate intruding pathogens. 3
Cyclooxygenase (COX) controls the rate-limiting step that catalyzes the biosynthesis of various prostaglandins (PGs) from the arachidonates released from the plasma membrane. 4 In addition to the constitutively expressed isoform cyclooxygenase-1 (COX-1), cyclooxygenase-2 (COX-2) is an inducible isoform usually not detected in most tissues, and its expression is dramatically induced by a variety of stimuli, including cytokines, growth factors and LPS.5–7 Up-regulated COX-2 and the subsequent production of PGs have been widely recognized to be involved in the inflammatory consequences through the regulation of various inflammatory-related signaling cascades. 8
NO is a molecule produced from various cell types and, in addition to its primary role as a vascular relaxing agent, NO is also produced during the inflammatory response and it has been implicated as a pro-inflammatory player. 9 Currently, three types of NO synthase (NOS) isoforms with two constitutively expressed forms and one inducible form known as inducible NOS (iNOS) being expressed when cells are activated. 10 Previous studies have demonstrated a relation of LPS-induced iNOS and NO production to systemic vasodilatation, which may possibly result in septic shock, suggesting a significant role of the iNOS/NO system in pathogen-mediated inflammatory cascades.11,12
Lysophosphatidic acid (LPA), a mixture of phospholipids containing various fatty acid side chains, was first recognized as an intermediate in lipid metabolism; however, it was later noted to regulate multiple physiological events, such as smooth muscle contraction, platelet aggregation, and immune function.13,14 In fact, the cognate LPA receptors have been detected in various immune cells. 15 Some studies reported that LPA can induce COX-2 expression and the production of PGs in ovarian cancer cells and fibroblast-like synovial cells, suggesting a pro-inflammatory role of LPA.16,17 However, other studies indicate that LPA has the ability to attenuate LPS-mediated mice mortality and TNF-α secretion, suggesting an anti-inflammatory role of LPA. 3 This study aimed to decipher how LPA would affect LPS-mediated inflammatory cascades in macrophages by monitoring the COX-2/PGE2 and the iNOS/NO pathways, two critical components in inflammation, and to clarify the involved signaling mechanisms and the interaction profile between COX-2/PGE2 and iNOS/NO.
Materials and methods
Reagents
FBS was obtained from HyClone (Logan, UT, USA). LPS, LPA, pertussis toxin (PTX) and Griess reagent were from Sigma (St. Louis, MO, USA). Reverse transcriptase and Taq polymerase were from Promega (Madison, WI, USA). The selective Gαi/o inhibitor NF023 was from TOCRIS Bioscience (Minneapolis, MN, USA). The pan-LPA receptor antagonist BrP-LPA (1-bromo-3-(S)-hydroxy-4-[palmitoyloxy]butyl phosphonate) was purchased from Echelon Biosciences Inc. (Salt Lake, UT, USA). Rabbit anti-COX-2 polyclonal Ab and rabbit anti-iNOS polyclonal Ab were from Santa Cruz Biotechnology (Dallas, TX, USA). PhosphoPlus Ab kits for MAPKs p38 (Thr180/Try182), JNK (Thr183/Try185) and ERK p44/p42 (Thr202/Try204), as well as for Akt (Ser473), were from Cell Signaling (Danvers, MA, USA). Rabbit anti-NF-κB/p65 (RelA) polyclonal Ab was from Thermo Fisher Scientific (Fremont, CA, USA). Mouse anti-α-tubulin monoclonal Ab and sheep anti-mouse IgG secondary Ab conjugated with HRP were from Sigma. Mouse anti-histone H1 monoclonal Ab was purchased from Santa Cruz Biotechnology. Donkey anti-rabbit IgG secondary Ab conjugated with HRP was from Amersham Life Science (Arlington Heights, IL, USA). PGE2 enzyme immunoassay kit was from Assay Designs (Ann Arbor, MI, USA). Unless otherwise specified, all other chemicals and reagents used in this study were from Sigma.
Cell culture
A mouse macrophage cell line, J774, was obtained from Bioresource Collection and Research Center (BCRC) of Taiwan (BCRC 60140, Lot-01538) and was maintained at 37℃ with DMEM medium (HyClone) containing 10% FBS. The cells were seeded in six-well plates at a density of 8 × 105 cells per well overnight (16 h) prior to various treatments the following day.
Western blotting
Overnight-plated J774 cells were treated with LPS (1 μg/ml) alone or in combination with LPA for the indicated times. The protein concentrations of the harvested total cell lysates, nuclear and cytosolic proteins were determined using Bio-Rad protein assay reagent (Bio-Rad, Hercules, CA, USA), and the total protein concentration was adjusted with SDS-PAGE loading buffer and heated to 100℃ for 10 min and then subjected to regular Western blotting assay to determine the expression profile of various proteins. Samples of equal amounts of proteins (50 μg) were separated on 10% SDS-PAGE, transferred onto a nitrocellulose membrane, blocked with 5% milk for 1 h, and incubated overnight with various specific Abs, including rabbit anti-COX-2 polyclonal Ab (1:1000), rabbit anti-iNOS polyclonal Ab (1:1000), rabbit anti-phospho-p38 and rabbit anti-total-p38 polyclonal Ab (1:1000), rabbit anti-phospho-JNK and rabbit anti-total-JNK Abs (1:1000), rabbit anti-phospho-ERK and rabbit anti-total-ERK polyclonal Abs (1:2000), rabbit anti-phospho-Akt and rabbit anti-total-Akt monoclonal Abs (1:1000), rabbit anti-NF-κB/p65 (Rel A) polyclonal Ab (1:500), mouse anti-histone H1 monoclonal Ab (1:500) or mouse anti-α-tubulin monoclonal Ab (1:5000), followed by incubation for 2 h with the corresponding secondary Abs, such as sheep anti-mouse IgG HRP-coupled Ab (1:5000) or donkey anti-rabbit IgG HRP-coupled Ab (1:2500). The membrane was exposed to film, and the bands of interest on the film were quantified with ImageQuant 5.2 software (Molecular Dynamics, Sunnyvale, CA, USA).
Measurement of PGE2 and NO production
Overnight-plated J774 cells were treated with LPS in the presence or absence of LPA for 24 h. Cultured medium was collected and analyzed for the PGE2 concentrations using an EIA kit (Assay Designs) and for the NO levels by nitrite assay using Griess reagent (Sigma).
Semi-quantitative RT-PCR
Primers used and the sizes of PCR products.

Statistical analysis
Experimental data are expressed as mean ± SEM and were analyzed by ANOVA followed by the least-significant difference test in order to compare the differences between each of the treatment groups and the control group. Difference with a P-value < 0.05 was considered to be statistically significant. To test the dose-dependent effect of the compounds used in more than two concentrations in this study, a linear regression analysis was performed to determine if these compounds generated a dose-dependent effect.
Results
LPA attenuation of LPS-induced protein and mRNA expression of COX-2 and iNOS, as well as the production of PGE2 and NO
We first examined whether LPA was able to regulate LPS-mediated COX-2 and iNOS protein expression, as well as PGE2 and NO production, in J774 mouse macrophage cells. Exposure of J774 cells to LPS (1 μg/ml) in the absence or presence of LPA (20, 10, 5 μM) for 24 h demonstrated that COX-2 and iNOS protein expression, as well as PGE2 and NO production, were low in control and LPA alone treatment groups; indeed, LPS dramatically induced COX-2 and iNOS expression, as well as PGE2 and NO secretion (Figure 1A,B). Notably, LPA (20, 10 μM) appeared to reduce LPS-mediated COX-2 protein expression and PGE2 secretion in a dose-dependent manner (R
2
= 0.99 and 0.91, respectively, after linear regression analysis; Figure 1A). A dramatic reduction of LPS-induced iNOS protein expression and NO production by LPA at all doses was also noted (Figure 1B); the impact of LPA on LPS-induced iNOS expression was also dose-dependent (R
2
= 0.98).
Attenuation of LPS-induced COX-2 and iNOS protein and mRNA expression, as well as PGE2 and NO production by LPA. Overnight-plated mouse J774 macrophage cells were treated with LPS (1 μg/ml) in the absence or presence of LPA (20, 10, 5 μM) for 24 h (A, B) or 12 h (C, D). The COX-2 (A) and iNOS (B) protein levels were determined by Western blotting using α-tubulin as an internal control. The PGE2 (A) and NO (B) production in the cultured medium was determined by ELISA and nitrite assay, respectively. The mRNA profiles of COX-2 (C) and iNOS (D) were examined by semi-quantitative RT-PCR using GAPDH as an internal control. The results are expressed as mean ± SEM from four separate experiments. *P < 0.05 compared with 24 h control treatment; #P < 0.05 compared with LPS treatment.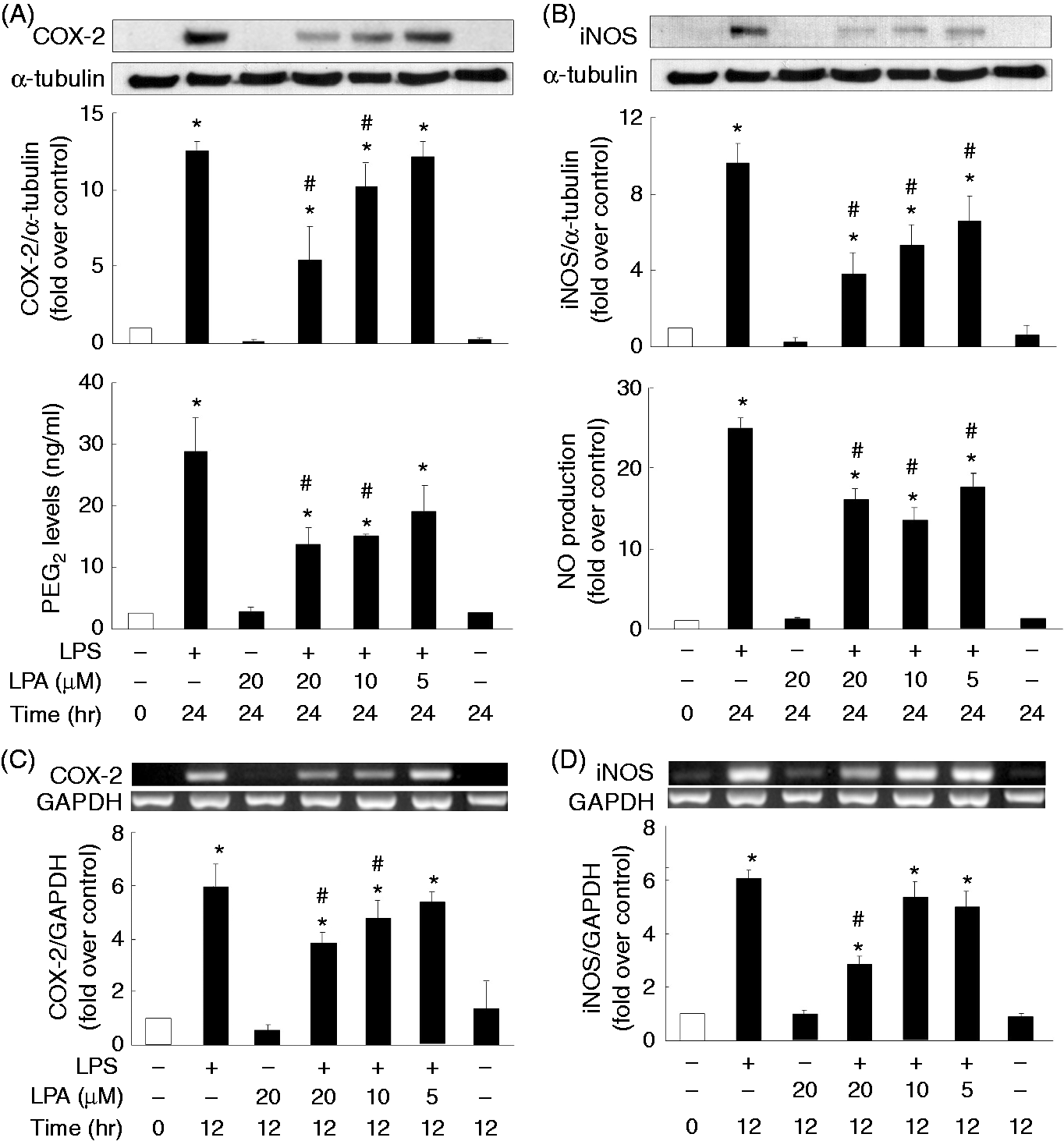
To clarify whether regulation of COX-2 and iNOS by LPS and LPA occurs at the transcriptional stage, we determined the mRNA expression levels after LPS (1 μg/ml) incubation with or without LPA (20, 10, 5 μM) for 12 h. Expression of COX-2 and iNOS was undetected in the control or LPA alone treatment groups, and the LPS treatment resulted in an elevation of COX-2 and iNOS mRNA levels (Figure 1C,D); LPA appeared to dose dependently reduce the LPS-induced COX-2 (R 2 = 0.95) and iNOS mRNA (R 2 = 0.81) expression (Figure 1C,D).
Involvement of Gαi protein and LPA receptors in LPA regulation of LPS-induced COX-2 and iNOS expression
It was reported previously that LPS-mediated secretion of cytokines and chemokines was increased in Gαi protein-deficient mice compared with wild type animals, and the cognate LPA receptors were well-recognized as G protein-coupled receptors (GPCRs).15,18 These prompted us to examine whether the Gαi was involved in LPA modulation of LPS-induced inflammation. J774 cells were pre-treated with a Gαi selective inhibitor, PTX, before the inclusion of LPS and LPA.
19
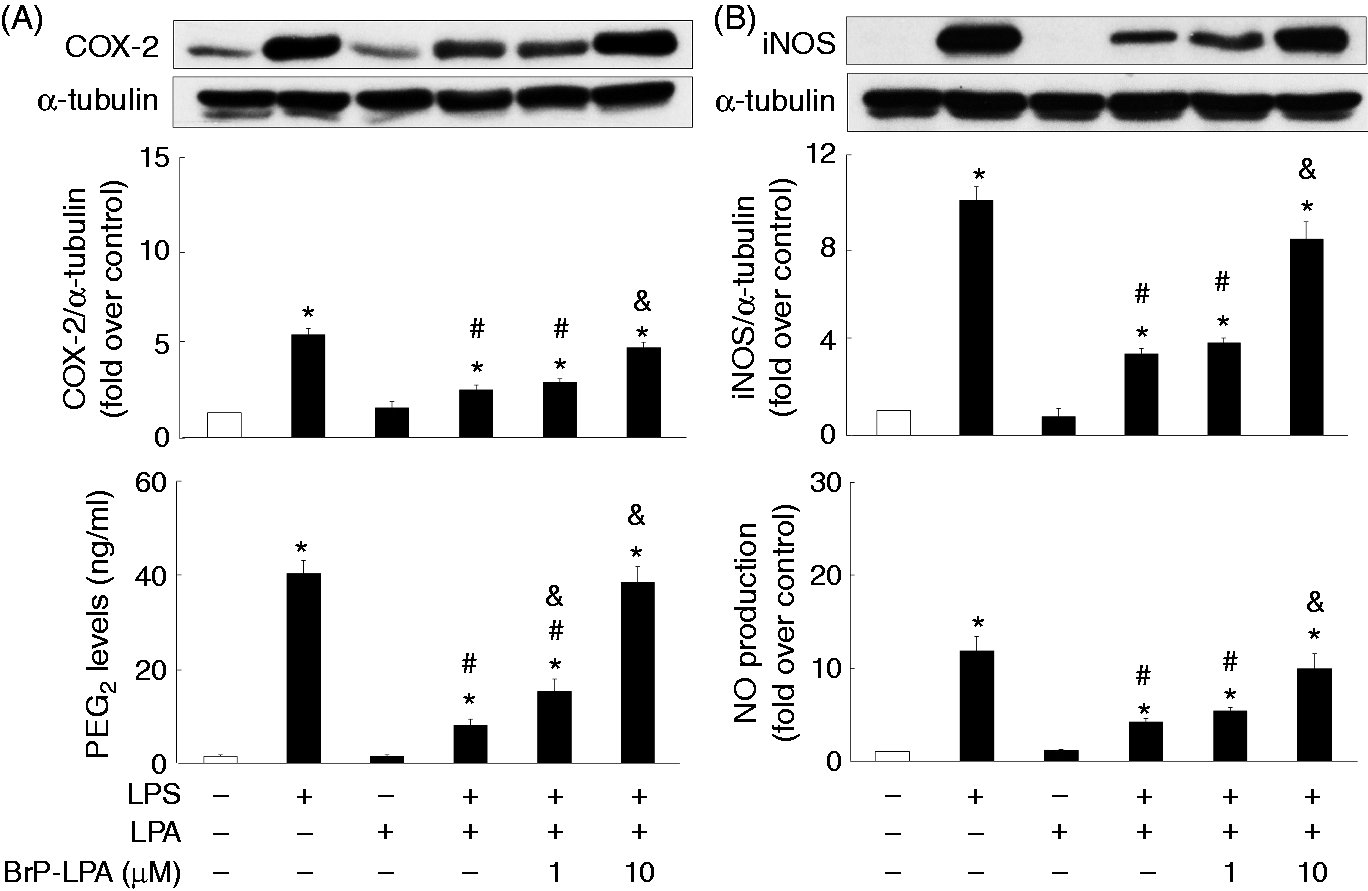
PTX (200 ng/ml) appeared to reverse the inhibitory effect of LPA on LPS-induced COX-2 and iNOS expression, as well as the accompanied production of PGE2 and NO (Figure 2A,B). Besides, another selective Gαi/o inhibitor NF023 was also used to further confirm the involvement of Gαi in the action of LPA action; NF023 (10 μM) did, indeed, significantly neutralize the LPA attenuation of LPS-induced expression of COX-2 and iNOS, and the production of PGE2 and NO (Figure 2C,D).20,21 In addition, to examine the involvement of LPA receptors in LPA-mediated suppression on LPS-induced expression of COX-2 and iNOS, as well as the production of PGE2 and NO, a pan-LPA receptor antagonist, BrP-LPA, appeared to significantly block the LPA effect on LPS-induced COX-2 and iNOS expression, as well as the production of PGE2 and NO (Figure 3), suggesting that LPA may act through LPA receptors and Gαi to perform its anti-inflammatory function.22,23
Involvement of the Gαi in LPA-regulated LPS-mediated COX-2 and iNOS expression. Overnight-plated J774 cells were pre-treated with PTX (200 ng/ml) or NF023 (1, 10 μM) for 4 h, followed by the treatment with LPS (1 μg/ml) and LPA (20 μM) for an additional 24 h to determine COX-2 (A, C) and iNOS (B, D) protein expression, and PGE2 (A, C) and NO (B, D) production in the cultured medium. Data are expressed as mean ± SEM of four individual experiments. *P < 0.05 compared with 24 h control treatment; #P < 0.05 compared with LPS treatment; &P < 0.05 compared with LPS and LPA combination treatment. Involvement of the LPA receptors in LPA attenuation of LPS-mediated COX-2/PGE2 and iNOS/NO production. Overnight-plated J774 cells were pre-treated with BrP-LPA (1, 10 μM) for 4 h, followed by the treatment with LPS (1 μg/ml) and LPA (20 μM) for an additional 24 h to determine COX-2 (A) and iNOS (B) protein expression, and PGE2 (A) and NO (B) production in the cultured medium. Data are expressed as the mean ± SEM of four individual experiments. *P < 0.05 compared with control treatment; #P < 0.05 compared with LPS treatment; &P < 0.05 compared with LPS and LPA combination treatment.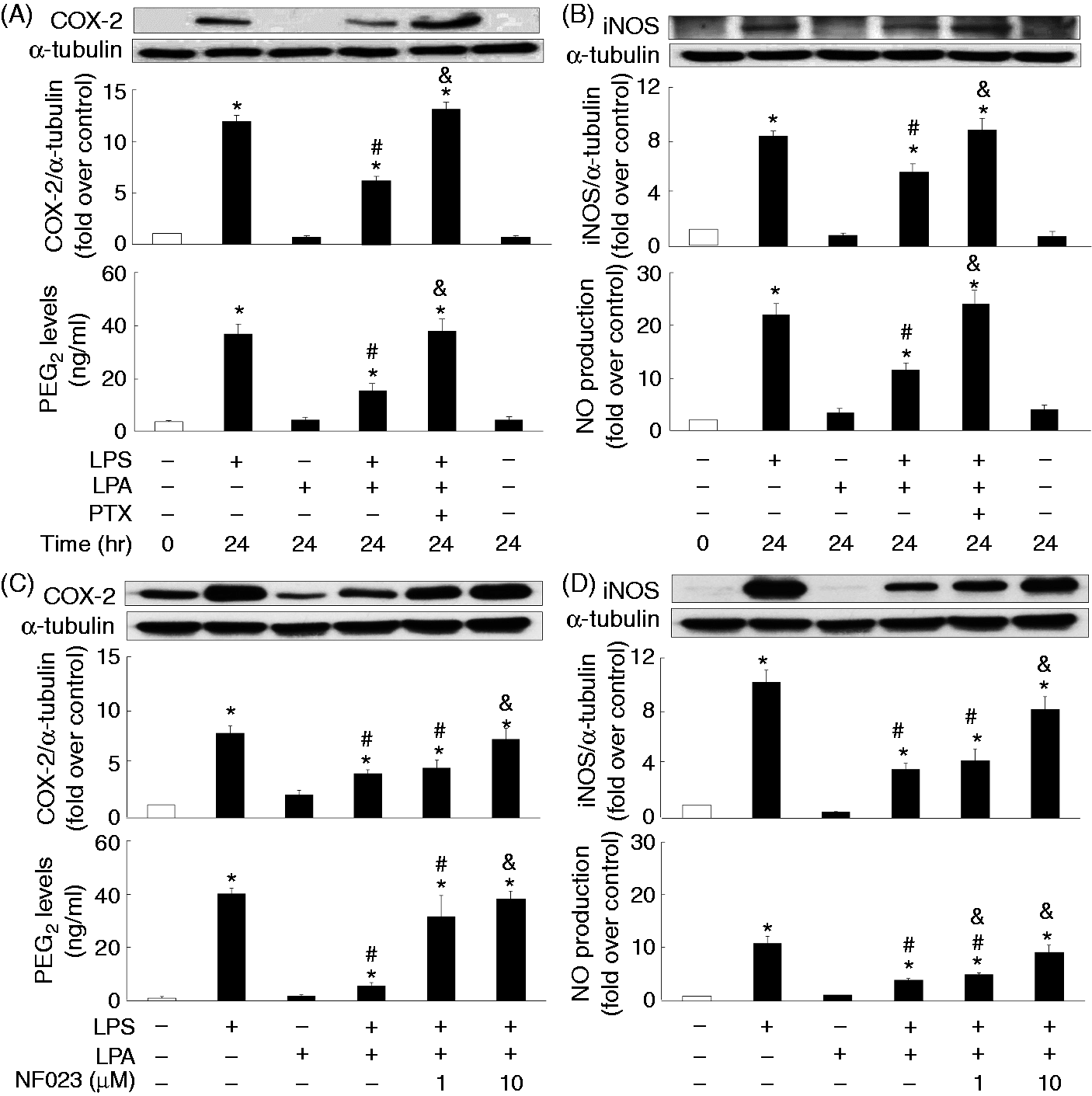
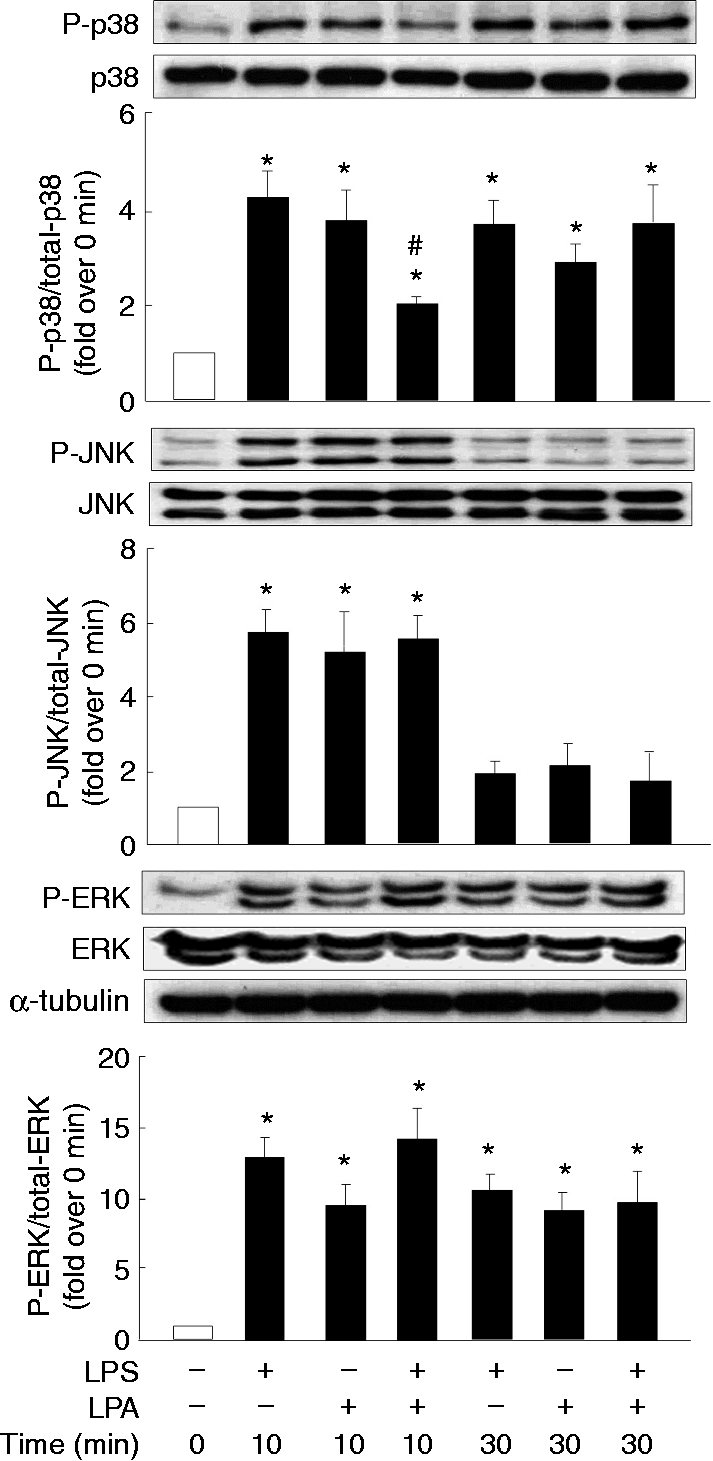
LPA attenuation of LPS-induced p38 phosphorylation in relation to LPS-mediated COX-2/PGE2 and iNOS/NO regulation
A previous study has reported that MAPKs may act upstream to mediate the expression of various inflammatory genes.
24
Thus, inhibitors of MAPKs (p38, JNK, ERK) were used to first clarify the signaling pathways post-LPS activation on the induction of COX-2 and iNOS in macrophages. LPS-mediated COX-2 expression and PGE2 production were suppressed by the inhibitors of p38 (SB2030580) and ERK (PD98059) (Figure 4A); however, LPS-mediated iNOS expression and NO production were attenuated by inhibitors of p38 and JNK (SP600125) (Figure 4B). Consequently, we further examined whether LPA would affect LPS-induced activation (phosphorylation) of such important MAPKs, and we noted that LPS significantly induced p38 and ERK phosphorylation at 10 and 30 min, and JNK phosphorylation at 10 min; LPA also appeared to attenuate LPS-induced p38 phosphorylation at 10 min (Figure 5). Interestingly, LPA treatment alone also induced JNK phosphorylation at 10 min, as well as p38 and ERK phosphorylation at both 10 and 30 min (Figure 5).
Involvement of the MAPK pathways in LPS-induced COX-2/PGE2 and iNOS/NO production. Overnight-seeded J774 cells were pre-treated with MAPKs inhibitors, including SB2030580 (SB, 20 µM) for p38, SP600125 (SP, 10 µM) for JNK and PD98059 (PD, 20 µM) for ERK for 1 h, followed by LPS (1 μg/ml) treatment for an additional 24 h to analyze protein expression of COX-2 (A) and iNOS (B), and PGE2 (A) and NO (B) production in the cultured medium. Data are expressed as mean ± SEM of four individual experiments. *P < 0.05 compared with control treatment; #P < 0.05 compared with LPS treatment. The impact of LPA on LPS-induced MAPK activation. Overnight-seeded J774 cells were treated with LPS (1 μg/ml) in the presence or absence of LPA (20 μM) for 10 or 30 min to monitor phosphorylation of MAPKs (p38, JNK and ERK). Data are expressed as mean ± SEM of four individual experiments. *P < 0.05 compared with control treatment; #P < 0.05 compared with LPS treatment at the same time point.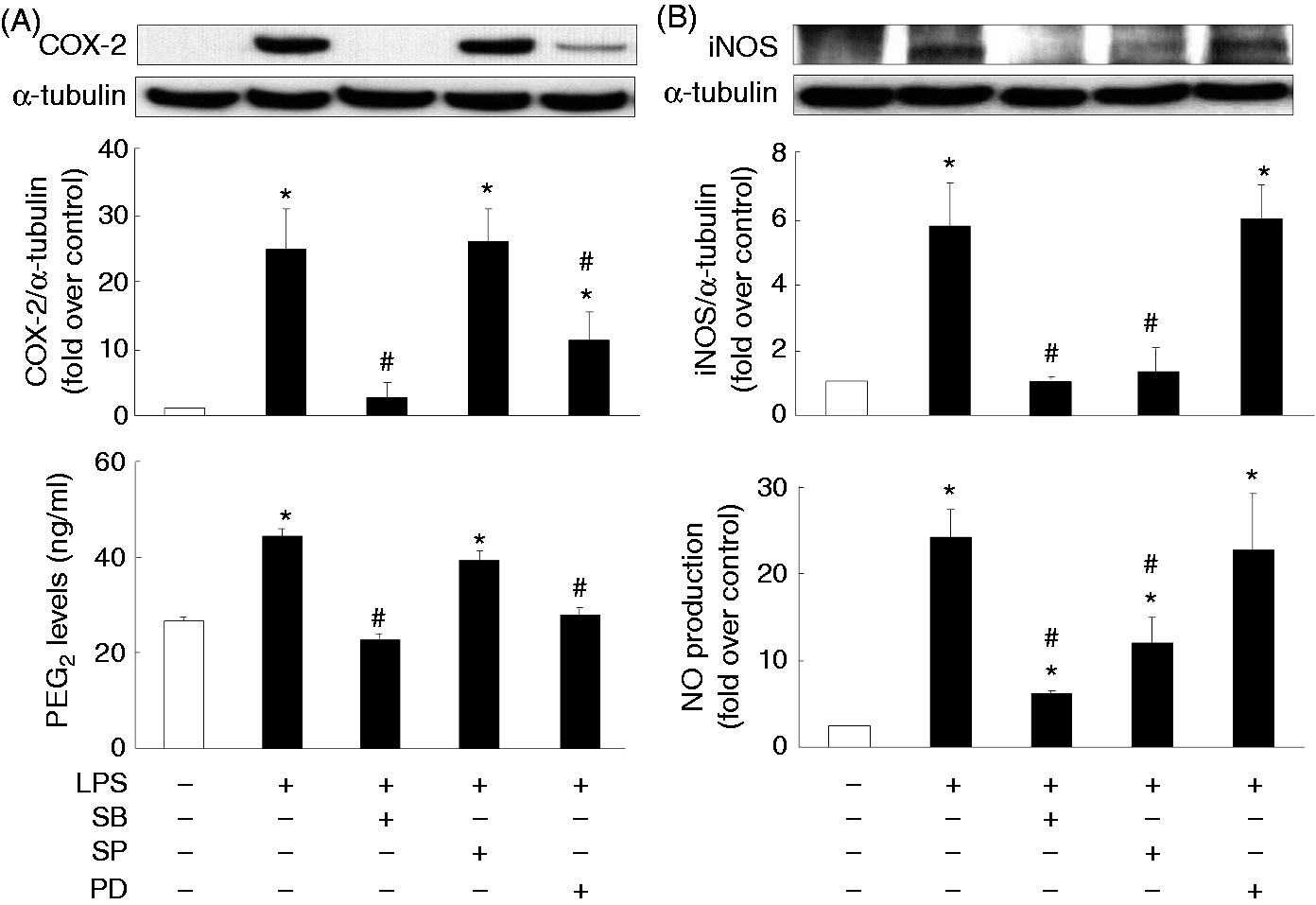
LPA attenuation of LPS-induced Akt phosphorylation in relation to LPS-mediated COX-2/PGE2 and iNOS/NO regulation
A previous report indicated that the PI3K signaling pathway could be activated by LPS.
25
Thus, a PI3K inhibitor LY294002 (20, 10, 5 μM) was used to evaluate whether the PI3K pathway could be the target of LPA to perform its inhibitory effect on LPS-induced inflammation. LY294002 did not have any effect on LPS-induced COX-2 protein expression or PGE2 production (Figure 6A) but it dose-dependently inhibited LPS-induced iNOS protein expression (R
2
= 0.99) and NO production (R
2
= 0.98) (Figure 6B). Therefore, we went on to examine the impact of LPA on LPS-mediated activation (phosphorylation) of a PI3K downstream signaling player, Akt. LPS treatment resulted in Akt phosphorylation at both 10 and 30 min (Figure 6C), and LPA significantly attenuated the LPS-mediated Akt phosphorylation at 30 min (Figure 6C).
Involvement of the PI3K signaling pathway in LPS-induced COX-2 and iNOS expression, as well as PGE2 and NO production, and attenuation of LPS-induced Akt phosphorylation by LPA. Overnight-plated J774 cells were pre-treated with LY294002 (LY) as PI3K inhibitor (20, 10, 5 μM) for 1 h, followed by LPS (1 μg/ml) treatment for an additional 24 h to determine COX-2 (A) and iNOS (B) protein expression, as well as PGE2 (A) and NO (B) production in the cultured medium, or were treated with LPS (1 μg/ml) in the presence or absence of LPA (20 μM) for 10 or 30 min to determine phosphorylation of Akt protein (C). Data are expressed as the mean ± SEM of four individual experiments. *P < 0.05 compared with 24 h control treatment; #P < 0.05 compared with LPS treatment.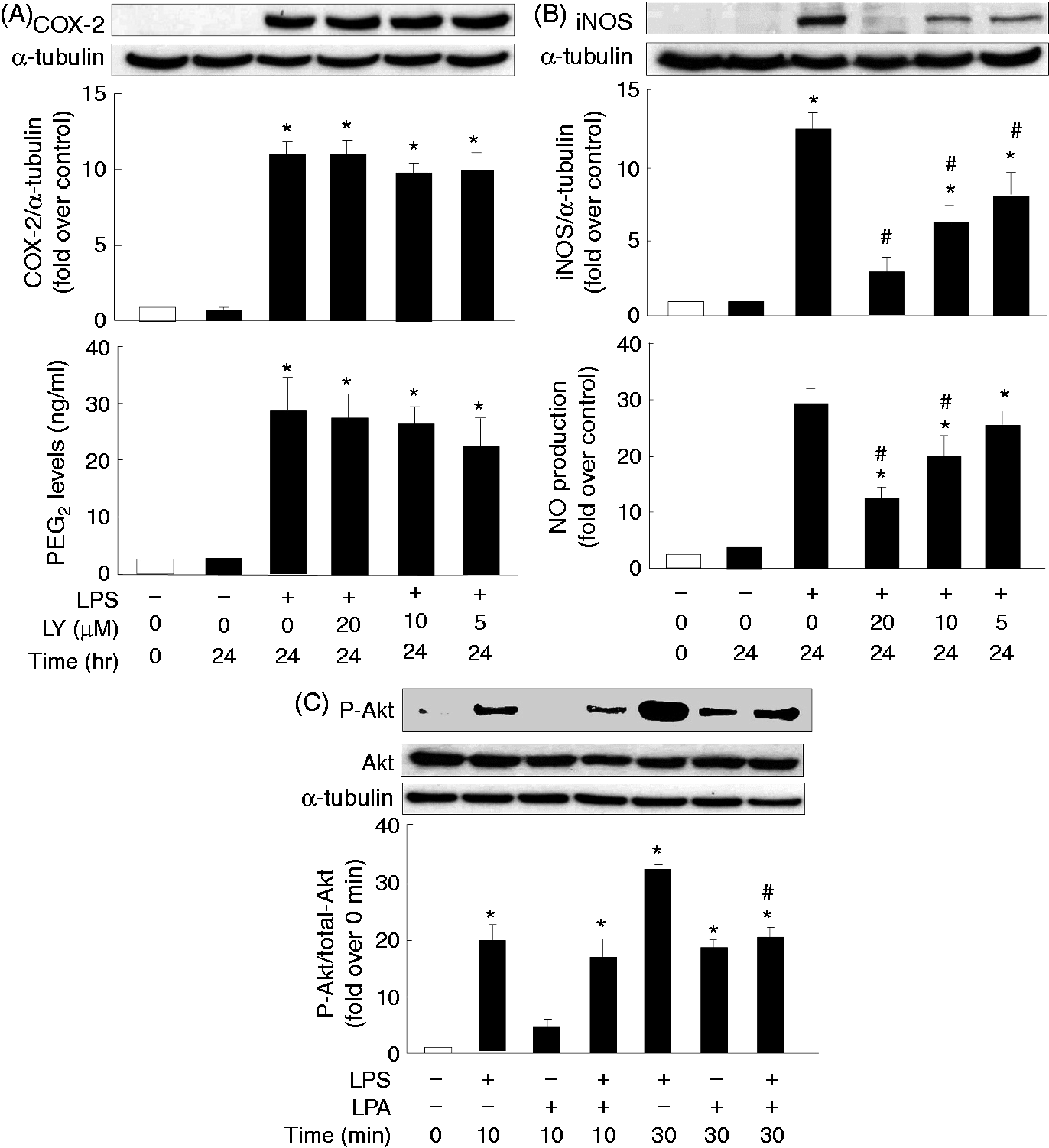
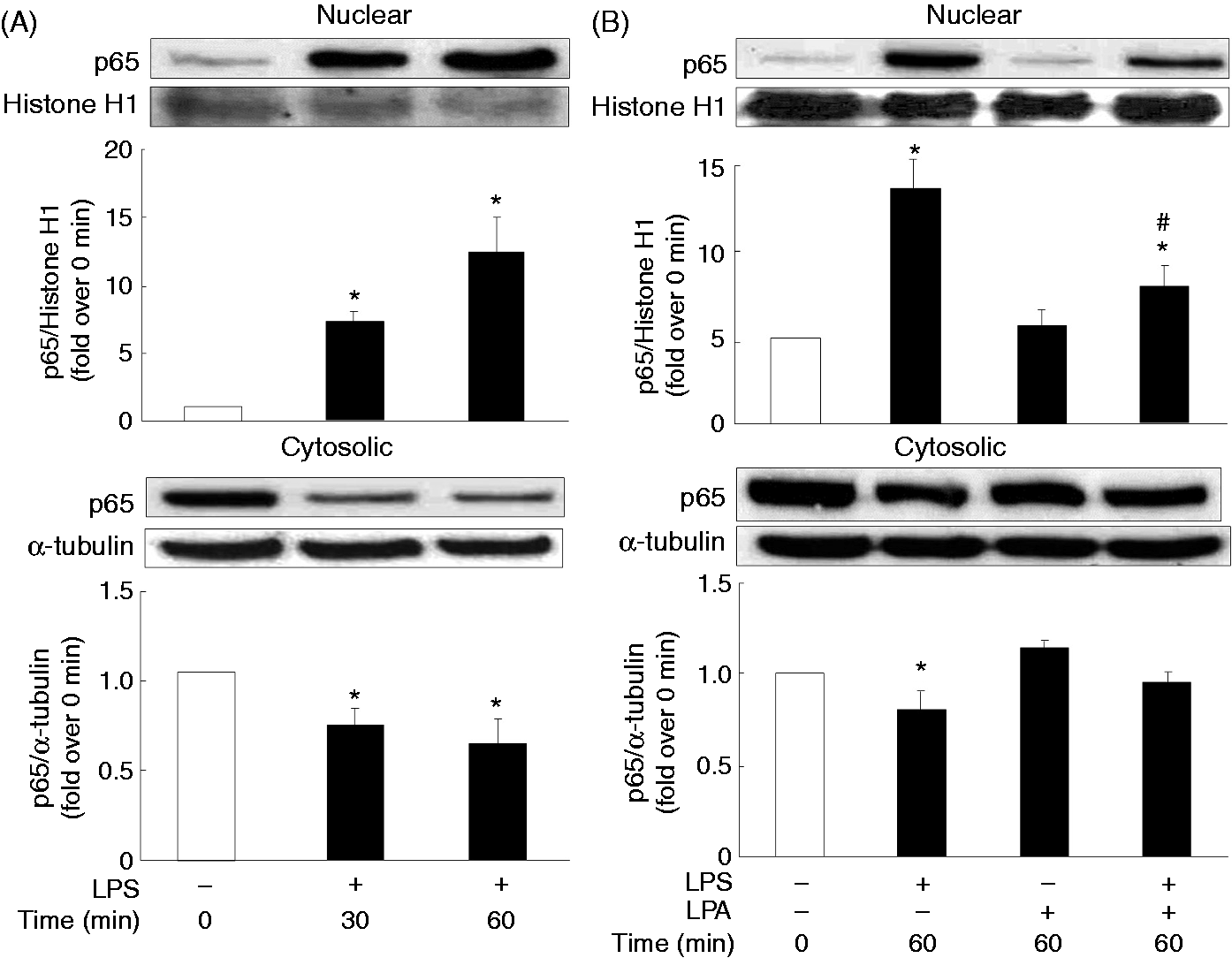
LPA attenuation of LPS-induced NF-κB p65 nuclear translocation in relation to LPS-mediated COX-2/PGE2 and iNOS/NO regulation
It has been reported that COX-2 and iNOS gene expression could be regulated by the involvement of the transcription factor NF-κB.
26
Thus, we went on to clarify the role of NF-κB in LPS-mediated COX-2 and iNOS expression. Pre-treatment with the NF-κB inhibitor parthenolide significantly reduced LPS-induced COX-2 (R
2
= 0.94) and iNOS (R
2
= 0.91) expression, as well as PGE2 (R
2
= 0.97) and NO (R
2
= 0.96) production (Figure 7), in a dose-dependent manner; LPS dramatically induced translocation of the NF-κB p65 subunit from the cytosol to the nucleus at 30 and 60 min (Figure 8A); LPA alone did not affect p65 translocation but significantly reduced LPS-mediated p65 nuclear translocation (Figure 8B).
Involvement of the NF-κB in LPS-induced COX-2 and iNOS protein, as well as PGE2 and NO production. Overnight-plated J774 cells were pre-treated with a NF-κB inhibitor parthenolide (PAR) (1, 0.3, 0.1 μM) for 1 h, followed by LPS (1 μg/ml) treatment for an additional 24 h to analyze COX-2 (A) and iNOS (B) protein expression, and PGE2 (A) and NO (B) levels in the cultured medium. Data are expressed as mean ± SEM of four individual experiments. *P < 0.05 compared with 24 h control treatment; #P < 0.05 compared with LPS treatment. Attenuation of LPS-induced NF-κB translocation by LPA. Overnight-plated J774 cells were incubated with (A) LPS (1 μg/ml) for 30 or 60 min or (B) LPS (1 μg/ml) in the presence or absence of LPA (20 μM) for 60 min to monitor LPS-induced nuclear and cytosolic protein levels of NF-κB p65 subunit. Data are expressed as mean ± SEM of four individual experiments. *P < 0.05 compared with control treatment; #P < 0.05 compared with LPS treatment.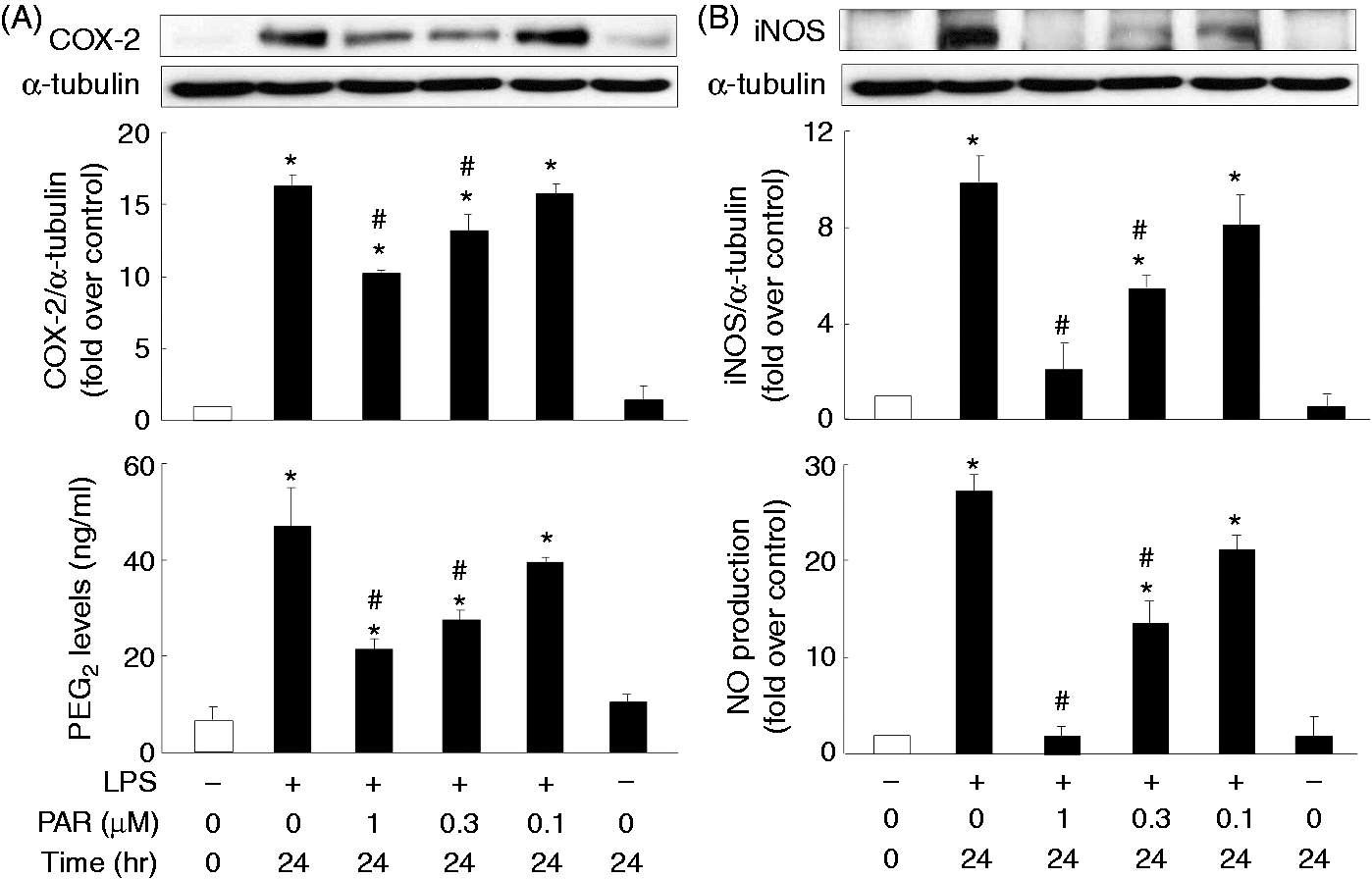
No regulation between the COX-2/PGE2 and the iNOS/NO signaling pathways
To examine whether regulation between the COX-2/PGE2- and iNOS/NO-mediated signaling pathways exists in LPS-treated macrophages, inhibitors of iNOS (L-NAME) and COX-2 (NS-398) were employed. NS-398 dramatically inhibited LPS-induced PGE2 production but did not affect LPS-mediated expression of COX-2 and iNOS, or NO production (Figure 9A). L-NAME potently suppressed LPS-induced NO production but did not affect LPS-mediated expression of iNOS and COX-2, or PGE2 production (Figure 9B), suggesting that no apparent regulation between the COX-2/PGE2 and the iNOS/NO signaling pathways appears in LPS-activated macrophages.
Examination of the regulation between COX-2- and iNOS-mediated signaling pathways. Overnight-plated J774 cells were treated with LPS (1 μg/ml), in combination with a COX-2 inhibitor, NS-398 (5 μM), or an iNOS inhibitor, L-NAME (0.4, 1 μM), for 24 h to analyze COX-2 (A) and iNOS (B) protein expression, and PGE2 (A) and NO (B) production in the cultured medium. Data are expressed as mean ± SEM of four individual experiments. *P < 0.05 compared with 24 h control treatment; #P < 0.05 compared with LPS treatment.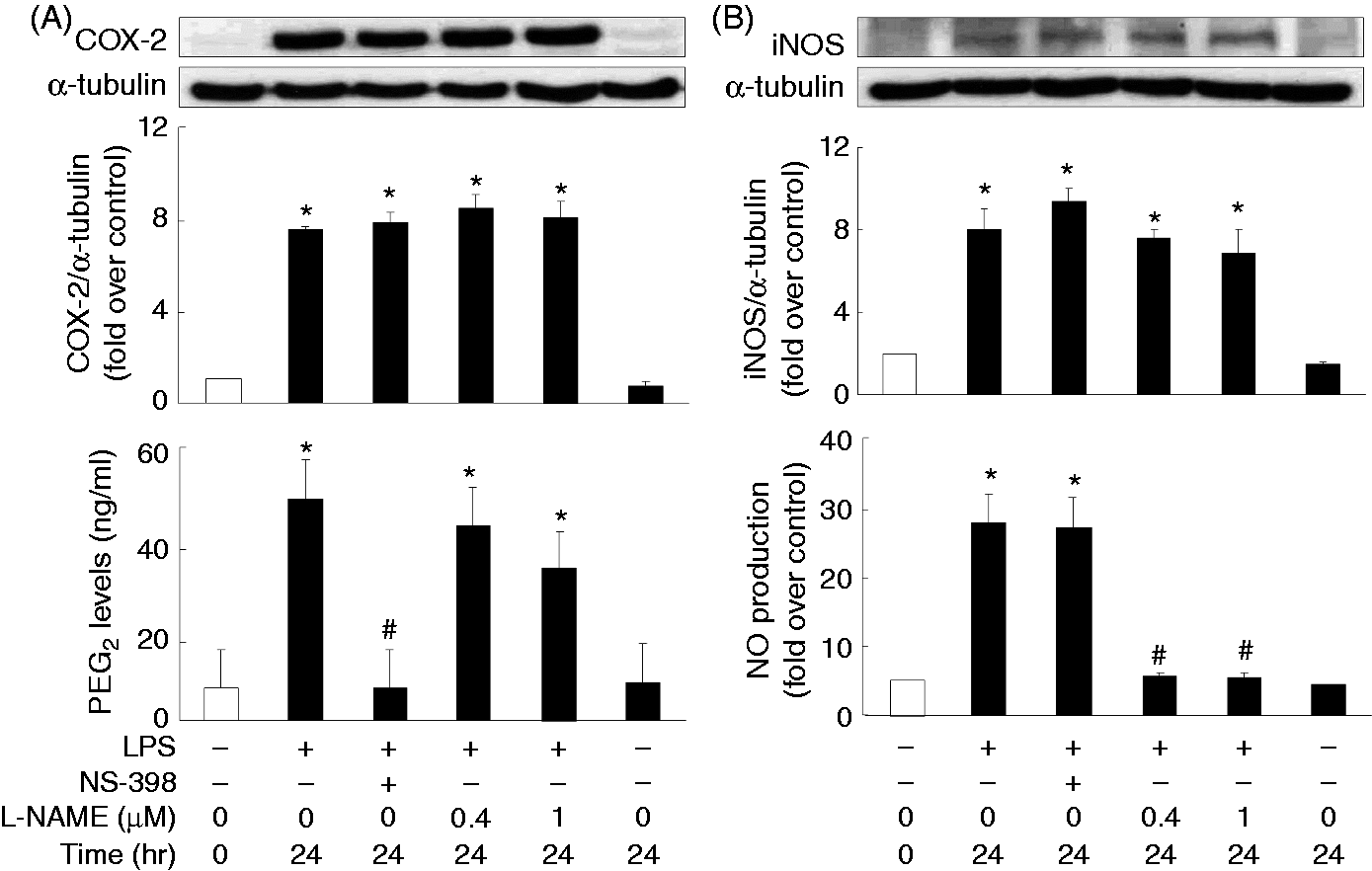
Discussion
This study mimics a bacteria-mediated inflammation in macrophages in order to investigate the potential role of LPA on LPS-mediated COX-2/PGE2 and iNOS/NO production and the underlying mechanisms involved in such a LPA action. Our results demonstrate that LPA was able to reverse the induction of protein and mRNA expression of COX-2 and iNOS, as well as the corresponding production of PGE2 and NO by LPS. LPS-mediated expression/production of COX-2/PGE2 and iNOS/NO involve, at least in part, the MAPKs p38 and ERK, as well as the NF-κB pathways in COX-2/PGE2, and p38, JNK and PI3K/Akt, as well as the NF-κB pathways in iNOS/NO. LPA may target p38, PI3K/Akt and NF-κB to downregulate LPS-mediated activation of these players to subsequently attenuate LPS-mediated COX-2/PGE2 and iNOS/NO responses.
Macrophages are crucial cellular components of the innate immune system, and its contribution to inflammation is one of the well-recognized functions. The involvement of macrophage activation by LPS, a critical component of Gram-negative bacteria, has been demonstrated to be important in endotoxemia. 27 In endotoxemia, macrophages are activated to produce a large amount of pro-inflammatory mediators, including iNOS and COX-2. These findings suggest the critical roles of iNOS/NO and COX-2/PGE2 signaling pathways in the pathogenesis of endotoxemia.28–30 Previous studies have suggested an ability of LPA to reduce the organ injury in endotoxemia and to inhibit bacterial endotoxin-induced TNF-α expression.3,31 In fact, LPA receptors have been detected in both human and mouse macrophages, suggesting a potential role of LPA in regulating macrophage functions.32,33 LPA is a normal constituent of serum with a concentration range of 2–20 μM. 34 Hence, we studied the effect of LPA from 5 to 20 μM and noted an anti-inflammatory role of LPA in LPS-mediated inflammation in terms of LPS-mediated COX-2 and iNOS protein and mRNA expression, as well as production of PGE2 and NO (Figure 1). Consistent with our findings, a previous study also stated that LPA was able to reduce the LPS shock-mediated inflammatory response. 31
It has been demonstrated that LPA can exert its effect via its cognate GPCRs and the subsequent activation of various Gα subunits: LPA1 couples with Gαi/o, Gαq/11 and Gα12/13 subtypes; LPA2 couples with Gαi/o, Gαq/11 and Gα12/13 subtypes; LPA3 couples with Gαi/o and Gαq/11; LPA4 couples with Gαq/11.35–37 The involvement of GPCR LPA receptors in LPA anti-inflammatory function was confirmed, as the inhibitory role of LPA on LPS-induced COX-2/PGE2 and iNOS/NO production was blocked by a pan-LPA receptor antagonist BrP-LPA (Figure 3). In fact, the Gαi-deficient mice were noted to exhibit an augmented inflammatory response to LPS stimulation. 18 Consistently, our finding that the inhibitory role of LPA on LPS-induced COX-2/PGE2 and iNOS/NO production was blocked by the Gαi inhibitors PTX and NF023 (Figure 2), indicates a crucial role of Gαi in LPA-attenuated LPS-mediated inflammation in macrophages. Similar to our finding, the involvement of Gαi in LPA action in J774 macrophages have been demonstrated previously to mediate oxidized low-density lipoprotein uptake. 32
With TLR4 being regarded as the major receptor for the LPS molecule, macrophages play an important role, resulting in elevation of inflammatory mediators, such as COX-2/PGE2 and iNOS/NO, leading to the recruitment of more immune cells to fulfill host defense functions against the infection.38–40 A variety of TLR4-triggered signaling pathways, such as MAPK (p38, JNK and ERK), PI3K and NF-κB, have been shown to be involved in the transmission of the primary signal from the extracellular pathogenic agents to target cytokine genes in the nucleus. 41 Therefore, we pinpointed the possible LPA targets within these LPS-induced signaling pathways. Our results revealed that the intracellular signaling pathways mediating LPS-elicited COX-2/PGE2 induction appeared to depend on p38, ERK and NF-κB (Figures 4A and 7A) and iNOS/NO induction appeared to depend on p38, JNK, PI3K and NF-κB (Figures 4B, 6B and 7B). We also demonstrated a LPA-mediated reduction on the LPS-induced phosphorylation of p38 (Figure 5) and Akt (Figure 6C), as well as p65 nuclear accumulation (Figure 8); this impact correlates with the inhibitory effect of LPA on LPS-induced production of the inflammatory mediators analyzed (Figure 1). Similar to our findings, LPA was also reported to perform an anti-inflammatory role by inhibiting cytokine-induced inflammatory signaling in human bronchial epithelial cells. 42 LPA administration alone was found to induce phosphorylation of p38, JNK and ERK (Figure 5), as well as Akt (Figure 6C). In fact, previous publications have also demonstrated that LPA alone can induce ERK phosphorylation in rat aortic smooth muscle cells, p38 and JNK phosphorylation in human bronchial epithelial cells, and Akt phosphorylation in murine hepatocytes.43–45 Nevertheless, in our study, when in combination with LPS, LPA clearly suppressed the LPS-mediated phosphorylation of p38 and Akt. The observation that LPA appears to result in phosphorylation of p38, JNK and ERK (Figure 5), but the regulation of COX-2/PGE2 or iNOS/NO was not affected by LPA treatment alone (Figure 2), is currently still a puzzle. However, a few previous studies have reported the involvement of calcium-dependent PKC and Src-family tyrosine kinase activation but MAPK-independent induction of COX-2 in vascular smooth muscle cells, and a NF-κB-dependent but MAPK-independent regulation of COX-2 translation in intestinal epithelial cells, as well as a NF-κB-dependent but MAPK-independent manner of iNOS induction in intestinal epithelial cells.46,47 Whether such LPA-mediated MAPK activation (phosphorylation) without the induction of COX-2/PGE2 or iNOS/NO in our study plays a significant role in physiology would need further investigation. Regarding the transcriptional regulation of COX-2 and iNOS genes, consistent with our findings, the binding site for NF-κB was identified within the promoter sequences of both COX-2 and iNOS genes.48,49 Whether this crucial NF-κB element is the target of LPA to perform its inhibitory effect on LPS-induced COX-2 and iNOS transcription needs further investigation.
Modulation between the COX-2/PGE2 and iNOS/NO signaling pathways has previously been proposed.50–52 However, such a manner of regulation did not appear to exist in the LPS-regulated COX-2/PGE2 and iNOS/NO signaling pathways in our study (Figure 9). The discrepancy may be ascribed to these two different stimuli resulting in different regulation mechanisms of the COX-2/PGE2 and iNOS/NO pathways, as the Mycobacterium bovis bacillus Calmette-Guerin (BCG) was from Gram-positive bacteria in the study by Marnett et al., 51 while in our study the LPS component was from Gram-negative bacteria. 50 Besides, the study conducted by Marnett et al. 51 on LPS-mediated COX-2 expression and PGE2 production under an iNOS-deficiency circumstance in mouse peritoneal macrophages revealed no significant difference in COX-2 protein expression between normal and iNOS-deficient animals in response to LPS administration; however, a dramatic 80% decrease in PGE2 production was noted in the iNOS-deficiency group. 51 They concluded that NO and/or NO-derived molecules may activate COX activity to result directly in the production of PGE2. 51 The difference between our findings and the study of Marnett et al. 51 could be owing to a different sensitivity to LPS insult, possibly that thioglycolate treatment in vivo may have resulted in higher responsiveness of primary peritoneal macrophages in Marnett et al. 51 than the mouse macrophage cell line J774 used in our study while exposed to LPS. 51 In addition, a previous study reported that peritoneal macrophages purified from EP2 knockout mice only weakly up-regulated iNOS gene expression upon stimulation with LPS compared with wild type peritoneal macrophages, suggesting a potential regulation between COX-2/PGE2 and iNOS/NO. 52 In this study, the LPS dose (100 μg/ml) used was higher than the dose (1 μg/ml) used in the present one. It is likely that such a high dose of LPS may cause more diverse and complicated inflammatory signaling pathways. Nevertheless, the significant role of NF-κB in regulation of COX-2 and iNOS was noted in the previous study and in our findings (Figures 7 and 8). 50
In conclusion, our study demonstrates a novel role for LPA as a potential anti-inflammatory molecule in the alleviation of bacterial -induced inflammation in macrophages. These effects occur, with the involvement of Gαi, at least in part by targeting LPS-induced activation of p38, Akt and NF-κB signaling molecules.
Footnotes
Funding
This work was supported by grants from the Taiwan Ministry of Science and Technology (MOST 100-2320-B-010-018-MY3; MOST 102-2320-B-010-010-MY3), the Cheng Hsin General Hospital (99F167CY17, 102F218C21, 103F003C03) and the Ministry of Education, Aim for the Top University Plan.
Acknowledgements
We thank Dr Ralph Kirby, Department of Life Sciences, National Yang-Ming University, for his help with language editing.
Conflict of interest
The authors do not have any potential conflicts of interest to declare.