
Abstract
In inflammatory bowel diseases (IBD), high mobility group box 1 (HMGB1), as an endogenous inflammatory molecule, can promote inflammatory cytokines secretion by acting on TLR2/4 resulting in tissue damage. The underlying mechanisms remain unclear. Here we report a novel role of HMGB1 in controlling the maintenance and function of intestine-resident group-3 innate lymphoid cells (ILC3s) that are important innate effector cells implicated in mucosal homeostasis and IBD pathogenesis. We showed that mice treated with anti-HMGB1 Ab, or genetically deficient for TLR2–/– or TLR4–/– mice, displayed reduced intestinal inflammation. In these mice, the numbers of colonic ILC3s were significantly reduced, and the levels of IL-17 and IL-22 that can be secreted by ILC3s were also decreased in the colon tissues. Furthermore, HMGB1 promoted DCs via TLR2/4 signaling to produce IL-23, activating ILC3s to produce IL-17 and IL-22. Our data thus indicated that the HMGB1-TLR2/4-DCs-IL-23 cascade pathway enhances the functions of ILC3s to produce IL-17 and IL-22, and this signal way might play a vital role in the development of IBD.
Keywords
Introduction
Inflammatory bowel disease (IBD) is a lifelong disease and has a significant impact on the quality of life. IBD, including Crohn’s disease (CD) and ulcerative colitis (UC), is characterized by chronically relapsing intestinal disorders of complex pathogenesis. 1 In IBD, a great number of inflammatory cells infiltrate intestinal tissues and produce pro-inflammatory cytokines, leading to mucosal damage and persistent intestinal inflammation. 2 At present, millions of patients suffer from IBD in developed countries, and the incidence of this disease is increasing in other parts of the world. 3 The cellular and molecular mechanisms that trigger and exacerbate IBD, however, remain unclear.
High-mobility group box 1 (HMGB1) is an important nuclear non-histone protein that binds to DNA and regulates transcription. 4 In addition to its functions in the nucleus, HMGB1 could be released to the extracellular space by necrotic cells or actively secreted from a variety of immune cells including macrophages, endothelial cells, and dendritic cells (DCs). 5 More recently, HMGB1 has been identified as damage-associated molecular pattern (DAMP) or a pro-inflammatory cytokine in the pathogenesis of several inflammatory disorders, such as systemic lupus erythematosus, 6 rheumatoid arthritis, 7 and septic shock. 8 It also acts as an endogenous adjuvant mediator that activates inflammatory cells through TLR2, TLR4 and receptor for advanced glycan end-products (RAGE).4,9 As evolutionarily conserved innate receptors, TLRs are able to activate the NF-κB pathway, which leads to the synthesis of pro-inflammatory cytokines and chemokines upon the recognition of exogenous (microbial components) or endogenous (ligands generated at sites of injury) ligands. 10 TLRs play important roles in the maintenance of intestinal homeostasis and in the pathogenesis of IBD. Previous genetic studies showed that polymorphisms in TLR2 and TLR4 were associated with risk of IBD.11,12 It was also reported that mice with induced experimental colitis by dextran sulfate sodium (DSS) developed a series of inflammatory responses, and the expression of PRRs such as TLR2 and TLR4 in the intestinal mucosa was significantly increased. 13 In the gastrointestinal tract, as DAMP and endogenous ligand of TLR2/4, abundant HMGB1 was found in the stools of IBD patients 14 and secreted by human inflamed intestinal tissues. 15 This evidence suggests that HMGB1 and its receptors TLR2/4 have important roles in the development of IBD.
Innate lymphoid cells (ILCs) are a group of innate immune cells residing at mucosal barrier sites, such as the intestine, lungs and skin. They are critically involved in lymphoid organogenesis, host defense, and mucosal homeostasis. 16 ILCs have been classified into three groups: group 1 ILCs (ILC1s), group 2 ILCs (ILC2s) and group 3 ILCs (ILC3s). 17 ILC1s express the transcription factor T-box 21, which encodes the T-bet and require IL-15 to develop. ILC1s mainly produce the Th1 cell-associated cytokine IFN-γ. 18 ILC2s depend on GATA-3 for their development and maintenance. ILC2s are capable of producing Th2 cell-associated cytokines, including IL-5 and IL-13. 19 ILC3s depend on the transcription factor retinoic-related orphan receptor (RORγt) for their development and function. ILC3s produce the Th17 cell-associated cytokines IL-17 and IL-22. 20 Several studies suggested that ILC3s were important effector cells for mucosal homeostasis and inflammation in the gastrointestinal tract.21–24 ILC3s accumulate in inflamed colon tissues in patients with CD compared with control individuals, suggesting ILC3s may be involved in immune-pathological processes of IBD. 25 IL-23, a known activator of ILC3s, also plays an important role in intestinal inflammation. 26 Recent evidence suggests that IL-23 promote ILCs responses in murine models of colitis and in human IBD.25–27
Here we show that anti-HMGB1 Ab or TLR2/4 gene knockout attenuates intestinal inflammation of 2,4,6-trinitrobenzenesulfonic acid (TNBS) colitis. Accompanied by inhibition of HMGB1 effects, colonic ILC3s and DCs responses decreased in mice suffering from colitis. Furthermore, HMGB1 stimulates IL-23 production by DCs in a TLR4- or TLR2-dependent manner. These results indicate that increased HMGB1 expression can enhance ILC3s responses in the mouse intestine by promoting DCs to produce IL-23.
Materials and methods
Animals
Male C57/BL6 and C57BL/10 mice were purchased from the Centers for Disease Control (CDC), Wuhan (Hubei, China). C57/B6.129-Tlr2tmIkir/JNju (TLR2–/–) and C57BL/10ScNJNju (TLR4–/–) mice were purchased from Biomedical Research Institute (Nanjing, China). Mice were used at 5–6 wk of age. Mice were kept in specific pathogen-free (SPF) facilities and all the experiments in this study were performed according to the guidelines for animals use in research by Tongji Medical College, Huazhong University of Science and Technology (China).
Induction of TNBS-colitis and administration of anti-HMGB1 Ab
Mouse colitis was induced by TNBS. Mice were anesthetized with methoxyflurane, and 2.5 mg TNBS (Sigma-Aldrich, St. Louis, MO, USA) was administered in 100 µl of 50% ethanol (EtOH) intrarectally via a polyurethane catheter (Becton Dickinson, San Jose, CA, USA). Control mice were treated with 50% EtOH only. According to a previous study, each mouse was given 100 µg/20 g HMGB1-neutralizing mAb (Anti-HMGB1 Ab; gift from Institute of Biophysics, Chinese Academy of Science, Beijing, China) by intraperitoneal injection every other day, 28 starting 1 d before administration of TNBS. Control group mice were given the same dose of isotype control Ab IgG (Sigma-Aldrich, St. Louis, MO, USA). Mice were monitored daily for body mass. Mice were sacrificed on the 4th d after the last treatment and colons were collected for the following studies.
Histopathological analysis
For histological examinations, small segments of proximal, medial and distal colon were removed and then fixed overnight in 4% (w/v) paraformaldehyde in PBS and embedded in paraffin. Sections (3 µm) were cut and stained with hematoxylin and eosin (H&E). The histological score of H&E-stained sections of the colon was determined by a pathologist in a blinded fashion, according to a previously described method:
29
(1) Extent (0, none; 1, focal; 2, limited to one segment; 3, involving more than one segment); (2) Inflammation (0, none; 1, mild; 2, moderate; 3, severe); (3) Damage (0, none; 1, mild/superficial; 2, moderate/involving muscularis mucosae; 3, severe/transmural); (4) Regeneration (3, none; 2, focal migration and mitotic figures; 1, broad multifocal re-epithelialization; 0, complete re-epithelialization). The histological scores of the individual ranged from 0 to 12. For statistical analysis, we averaged the histological scores of the sections for each colon.
Anti-HMGB1 Ab alleviated intestinal inflammation of TNBS-induced colitis. C57/BL6 mice were given 2.5 mg/mouse TNBS (dissolved in 50% ethanol) intrarectally on d 0 and treated with anti-HMGB1 Ab (HMGB1 Ab) or IgG (100 µg/20 g) once every 2 d, for 4 d. All the mice were sacrificed on d 4. (a) Illustration of experimental protocol. (b) Body mass changes were monitored between d 0 and d 4 in different groups of mice. (c) Colon lengths were measured after mice had been sacrificed on d 4. (d) Histologic analysis (hematoxylin and eosin staining) of mice colons was performed microscopically (original magnification, 200×). (e) Histological scores of mice colons were recorded. EtOH: mice treated with 50% ethanol; TNBS + HMGB1 Ab: TNBS-induced colitis mice treated with anti-HMGB1 Ab; TNBS + IgG: TNBS-induced colitis mice treated with an isotype IgG control; *P < 0.05, vs. EtOH control; #P < 0.05 vs. TNBS + IgG treatment group. Data are presented as means + SD of three to six individual mice per group and represent >3 independent experimental repeats.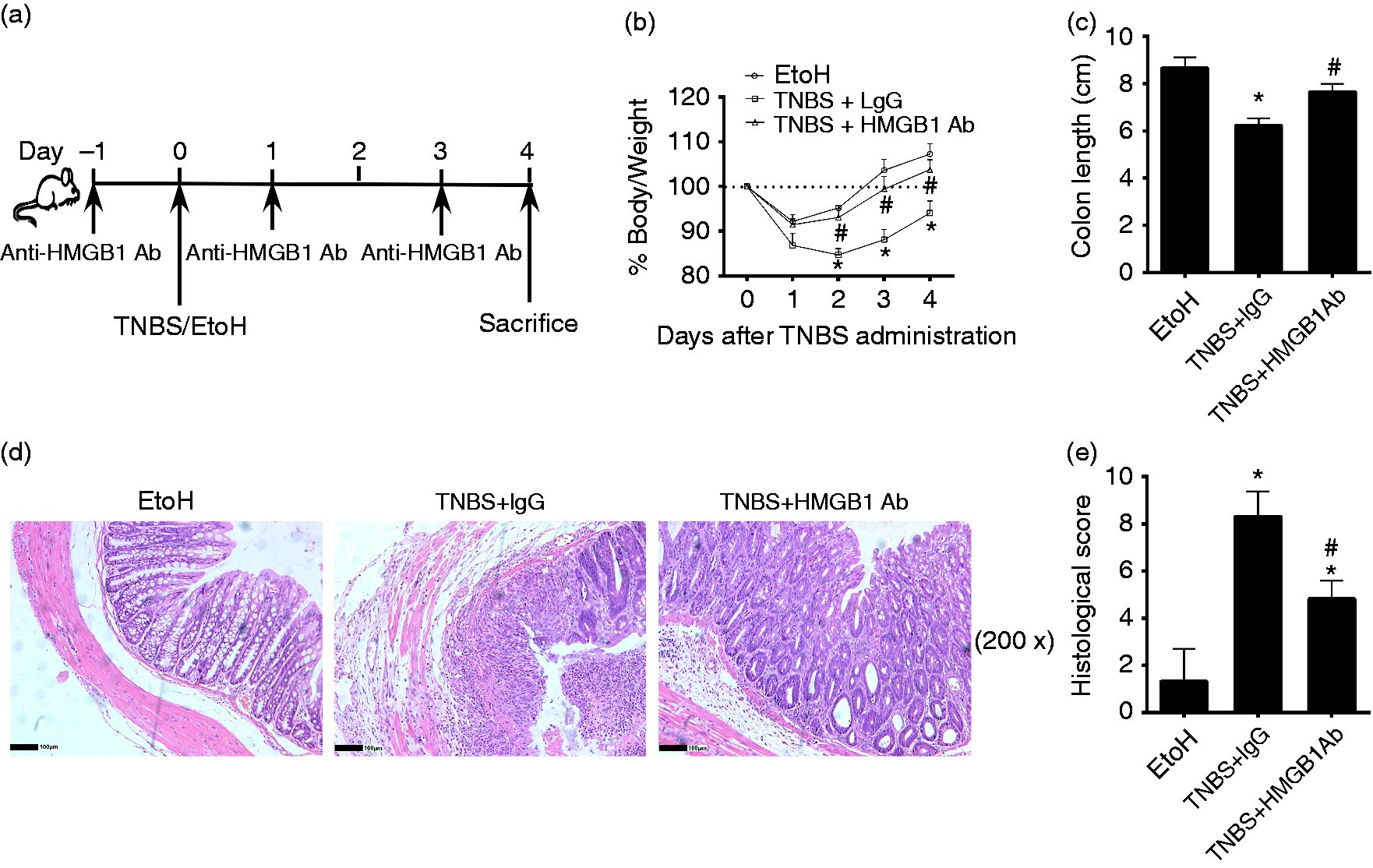
Colon tissue processing
Full-thickness strips of colonic segments weighed 0.08–0.12 g per strip were homogenized on ice in PBS of 0.05% of Triton X-100, protease inhibitor PMSF and aprotinin. Homogenates were then centrifuged for 5 min at 4℃ and a speed of 13,800 g. After centrifugation, the supernatant was collected and stored at –80℃. The supernatant samples were used for cytokine assay in subsequent experiments.
Isolation of colonic lamina propria lymphocytes
Lamina propria mononuclear cells (LPMCs) were isolated and collected from colons according to methods published previously. 22 Briefly, mucus and epithelial cells were removed from colons, then sliced, and digested in Roswell Park Memorial Institute (RPMI)-1640 medium containing 2.5% FBS, 0.2 mg/ml Collagenase IV (Sigma-Aldrich, St. Louis, MO, USA), and 0.1 mg/ml DNase I (Sigma-Aldrich) by shaking (230 rpm) at 37℃ for 60 min. Resultant cells were re-suspended in 40% Percoll and layered over a 75% Percoll prior to centrifugation at 738 g for 30 min. The cells at the interface between 40% and 75% Percoll were collected, washed with PBS three times and used for flow cytometry analysis.
In vitro generation of DCs and rHMGB1 stimulation
Mouse bone marrow-derived DCs (BMDCs) were obtained by bone marrow cells culture. Briefly, bone marrow cells were isolated from femurs and tibias of TLR2–/– and TLR4–/– mice. A total of 2 × 105 cells were cultured in complete RPMI1640 medium containing 10% FCS, 10 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) and 5 ng/ml IL-4. After 8 d of culture, DCs were treated with medium in the absence or presence of various concentrations of recombinant HMGB1 (rHMGB1) (0, 100 or 500 ng/ml) (Sigma-Aldrich) for 2 d. IL-23 in the cell culture supernatants was measured using an ELISA kit.
ELISA
HMGB1 was measured in duplicates with a commercially available ELISA kit (Uscn Life Science, Wuhan, China). Quantification of IL-22, IL-17 and IL-23 in colon tissue homogenate supernatant and cell culture supernatant were measured by ELISA kit (BioLegend, San Diego, CA). ELISA analyses were performed according to the manufacturer’s instructions.
Flow cytometry
Cells were prepared from colonic lamina propria. For ILC3s, cells were first stained with Abs to following the common lineage markers: CD3ε, CD5, CD8α, CD19, B220, CD11b, CD11c, Ter119, F4/80, Gr-1, TCRβ, TCRγδ, CD49b, and FcɛR1α and other Ags (CD90.2, and CD127), and then fixed and permeabilized for further staining for intracellular Ag (RORγt) using a transcription factor buffer set (BD Biosystems, San Jose, CA, USA). For cytokine detection, LPMCs were stimulated in the presence of 25 ng/ml phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich, St Louis, MO, USA), 1 µg/ml Ionomycin (Sigma), and 50 ng/ml IL-23 (Peprotech, Rocky Hill, NJ, USA), in the presence of GolgiStop (BD). After 6 h of stimulation, cells were harvested and stained for surface markers, fixed, permeabilized with Cytofix/Cytoperm and Perm/Wash, and then stained for intracellular Ags (IL-17 and IL-22). Flow cytometry data were collected by flow cytometer (FACS Calibur; BD Biosystems, San Jose, CA, USA) and analyzed using FlowJo 7.6 software (Acresso). All Abs were from BioLegend or eBioscience unless otherwise indicated.
Statistical analysis
Results are shown as mean ± SD. Statistical significance of differences were analyzed by a one-way ANOVA or Student t test. The software package GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA) was used for data analysis. Values of P < 0.05 were considered significant.
Results
Anti-HMGB1 Ab treatment alleviates TNBS colitis
Previous studies demonstrated that HMGB1 was an important mediator of colonic inflammation in mice and human colitis.14,30 Therefore, we first assessed whether anti-HMGB1 Ab protected against development of colitis induced by TNBS. Anti-HMGB1 Ab or control IgG was administered intraperitoneally into mice (Figure 1a). As expected, mice treated with control IgG experienced severe mass loss. In contrast, mice receiving anti-HMGB1 Ab experienced less mass loss (Figure 1b). The shortening of colons was alleviated in mice treated with anti-HMGB1 Ab (Figure 1c). Mice treated with anti-HMGB1 Ab also exhibited ameliorated transmural inflammation of the bowel wall and injury with ulceration, fewer infiltrated immune cells, and less-severe histological features of colitis (Figure 1d and e). Taken together, these results demonstrated that HMGB1 is a critical regulator in murine colitis, and that anti-HMGB1 Ab treatment can ameliorate intestinal inflammation in experimental colitis.
Anti-HMGB1 Ab treatment down-regulates the functions of colonic ILC3s in mouse colitis
ILC3s reside predominantly in mucosal tissues, particularly in the intestinal tract. ILC3s have an important role in mediating the balance between the symbiotic microbiota and the intestinal immune system.
31
ILC3s accumulate in the human intestine under the inflammatory conditions that occur in CD.
25
Next, we investigated the regulatory role of HMGB1 in ILC3s function. We first neutralized HMGB1 in vivo by administration of anti-HMGB1 Ab, and analyzed ILC3s in the colon tissues by flow cytometry after the mice had developed colitis (Figure 2a). Anti-HMGB1 mAb administration significantly reduced ILC3s expansion in TNBS-treated mice (Figure 2b). Mice treated with anti-HMGB1 Ab also showed decreased frequencies of IL-17+ILC3s or IL-22+ILC3s in the inflammatory colons compared with IgG-treated mice (Figure 2c). Together, these data indicated that HMGB1 promotes the maintenance and functions of intestinal ILC3s.
Anti-HMGB1 Ab treatment decreased the numbers and cytokines production of ILC3s in mice colons. LPMCs were isolated from the colons of mice sacrificed on d 4 after intrarectal administration of ethanol or TNBS. ILC3s in the colonic lamina propria (cLP) were determined by flow cytometry. Mice colon tissues were collected and homogenized to evaluate the cytokine levels using ELISA. (a) Gating strategies for ILC3s (Lin–CD127+CD90.1+RORγt+). (b) The numbers of ILC3s separated from mice cLP were determined. (c) The frequencies of IL-17- or IL-22-positive ILC3s in mice cLP were evaluated. (d) The levels of cytokines IL-23, IL-17 and IL-22 in mice colons were quantified. EtoH: mice treated with 50% ethanol; TNBS + HMGB1 Ab: TNBS-induced colitis mice treated with anti-HMGB1 Ab; TNBS + IgG: TNBS-induced colitis mice treated with an isotype IgG control; *P < 0.05, vs. EtOH control; #P < 0.05 vs. TNBS + IgG treatment group. Data were presented as means + SD of three to six individual mice per group and represent > 3 independent experimental repeats.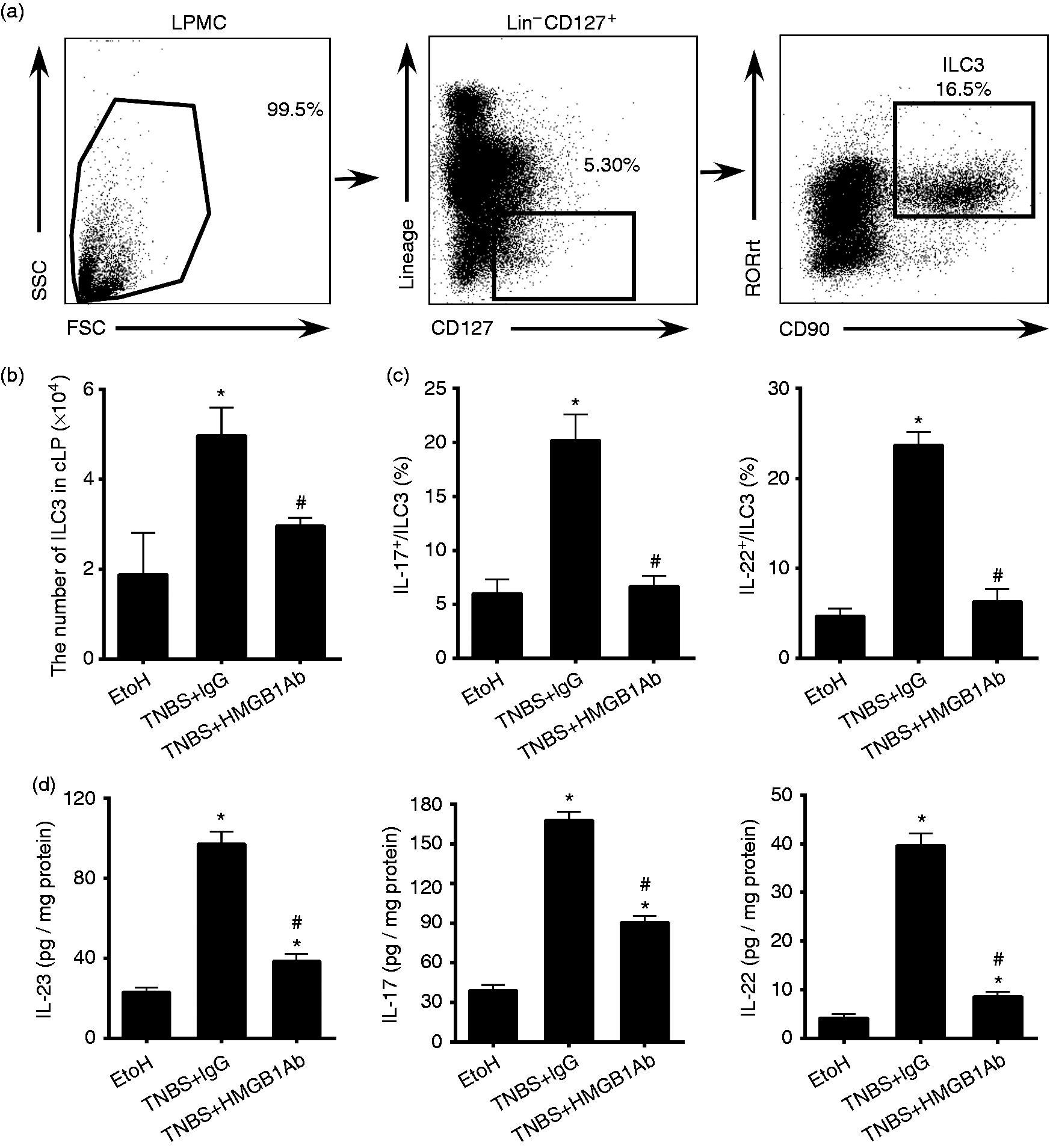
ILC3s secrete IL-17 A and IL-22 in response to IL-23. 28 IL-23, IL-17 and IL-22 have essential roles in IBD. 28 In our study, the cytokines of IL-23, IL-17 and IL-22 in colon tissues from each group were detected by ELISA. It was noted that the levels of IL-17 and IL-22 in colon homogenate increased while the mice suffered colitis. And both cytokine levels in the anti-HMGB1 Ab-treated group were lower than those in the IgG group (Figure 2d). Moreover, the levels of IL-23 in the colon homogenate decreased after anti-HMGB1 Ab administration (Figure 2d). Thus, HMGB1 might promote ILC3s responses by enhancing IL-23 levels.
Contribution of TLR2 and TLR4 to development of TNBS-induced colitis
When acting as an inflammatory cytokine, HMGB1 transduces its signals by ligating with TLR2 and TLR4. TLR2 and TLR4 ligation induces the activation of ERK1/2 and NF-κB signaling, which triggers cytokine production. As receptors of HMGB1, TLR2 and TLR4 gene expression may be important in the biological pathogenesis of IBD. 32 We thus asked whether the extent of mice colitis might depend on TLR2 and/or TLR4.
After treatment with TNBS, less mass loss was detected in TLR2−/− and TLR4−/− mice compared to the wild type (wt) (Figure 3(a)). In addition, although there were no significant differences in colon lengths between wt and TLR2−/−, and TLR4−/− mice treated with TNBS, histological changes showed that colon tissue damage in TLR2−/− and TLR4−/− mice were relieved in comparison with wt mice (Figure 3b). As evidenced on H&E staining, inflammatory cell infiltration and crypt damage was less apparent in TLR2−/− and TLR4−/− mice (Figure 3c and d). These results suggested that TLR2 and TLR4 promote the inflammatory responses in TNBS-challenged mice.
TLR2–/– and TLR4–/– colitis mice displayed decreased intestinal inflammation in comparison with wt mice. Wt, TLR2–/– and TLR4–/– mice were colitis-induced by administrating with 2.5 mg/mouse TNBS (dissolved in 50% ethanol) intrarectally on d 0. The EtOH control mice were treated with 50% ethanol. All the mice were sacrificed on d 4. (a) Body mass changes were monitored between d 0 and d 4 in different groups of mice. (b) Colon lengths were measured after mice had been sacrificed on d 4. (c) Histologic analysis (hematoxylin and eosin staining) of mice colons was performed microscopically (original magnification, 200×). (d) Histological scores of mice colons were recorded. TNBS: mice treated with TNBS; EtOH: mice treated with 50% ethanol instead of TNBS; (*P ≤ 0.05 vs. wt + TNBS.) Data are presented as means + SD of three to six individual mice per group and represent > 3 independent experimental repeats.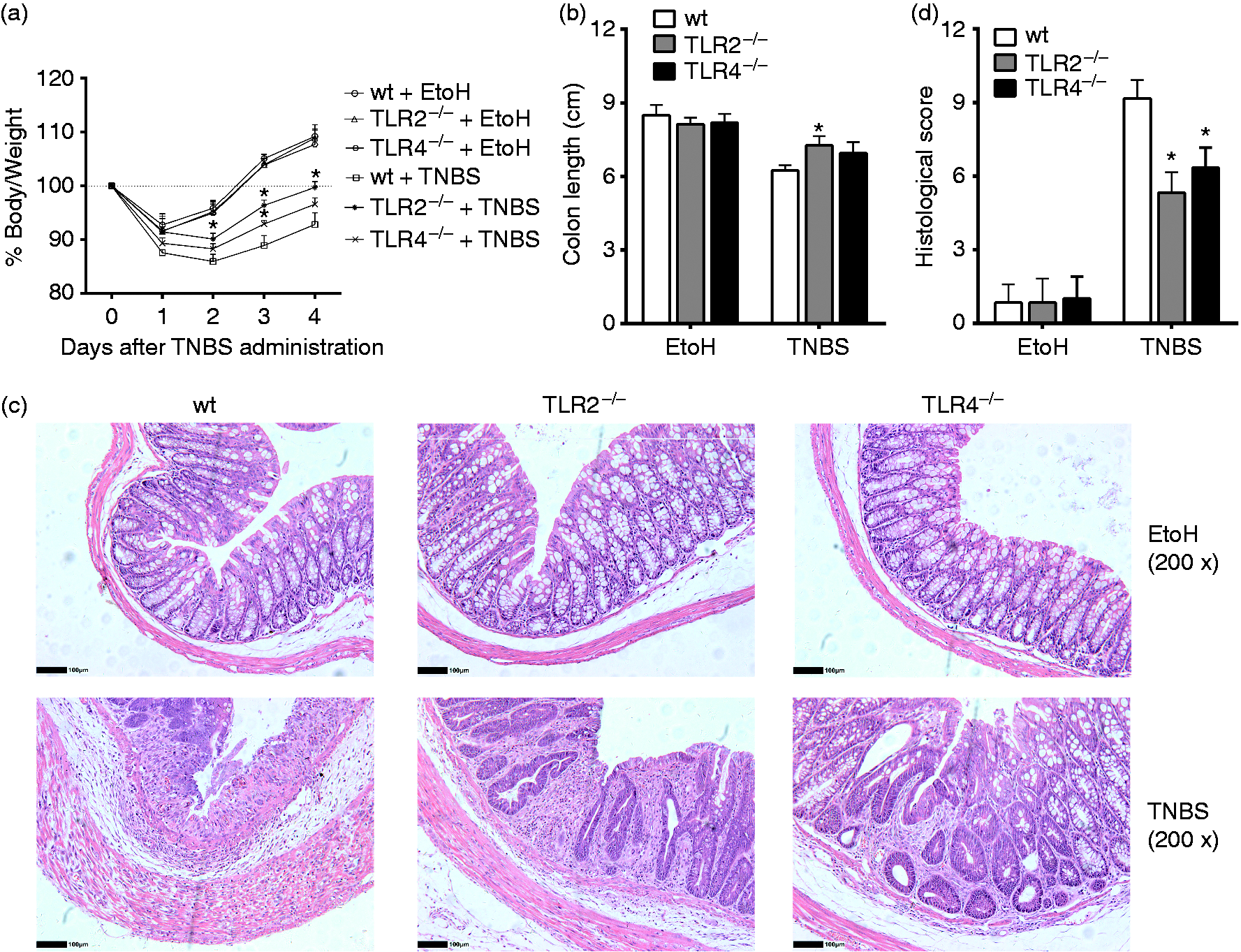
TLR2 and TLR4 deficiency results in down-regulation of colonic ILC3s in mice colitis
We showed that HMGB1 could regulate the numbers and functions of colonic ILC3s in colitis mice. We thus asked whether HMGB1 might control ILC3s responses in part through TLR2 and/or TLR4. The numbers of ILC3s decreased in the colons of TLR2−/− and TLR4−/− mice compared with wt mice following treatment with TNBS (Figure 4a). Interestingly, in the colonic ILC3s, the proportions of IL-17 - or IL-22-positive cells were significantly lower in the TLR2−/− and TLR4−/− mice than in the wt mice (Figure 4b). We also found that the levels of HMGB1 expression were significantly elevated in the colons of wt mice with TNBS treatment; however, the elevation was substantially attenuated in TLR2−/− and TLR4−/− mice (Figure 4c). Strikingly, the levels of IL-23, IL-17 and IL-22 expression were much lower in TNBS-treated TLR2−/− and TLR4−/− mice compared with wt mice. The results showed that knockout of TLR2 or TLR4 gene inhibited the functions of ILC3s compared with wt mice (Figure 4d). These data suggest that TLR2- or TLR4-dependent signaling pathway was involved in regulation of HMGB1 on colonic ILC3s.
The numbers of ILC3s and levels of IL-23, IL-17 or IL-22 decreased in the colons of TLR2–/– or TLR4–/– mice with colitis. LPMCs were isolated from the colons of wt, TLR2–/– and TLR4–/– mice sacrificed on d 4 after intrarectal administration of EtOH or TNBS. Colonic ILC3s were determined by flow cytometry. Mice colon tissues were collected and homogenized to evaluate the levels of cytokines using ELISA. (a) The numbers of ILC3s separated from mice cLP were determined. (b) The frequencies of IL-17- or IL-22-positive ILC3s in mice cLP were evaluated by flow cytometry. (c) The levels of HMGB1 in mice colons were quantified by ELISA. (d) The levels of cytokines IL-23, IL-17 or IL-22 in mice colon were quantified by ELISA. (*P ≤ 0.05 vs. wt + TNBS). Data are presented as means + SD of three to six individual mice per group and represent > 3 independent experimental repeats. EtOH: mice treated with 50% ethanol.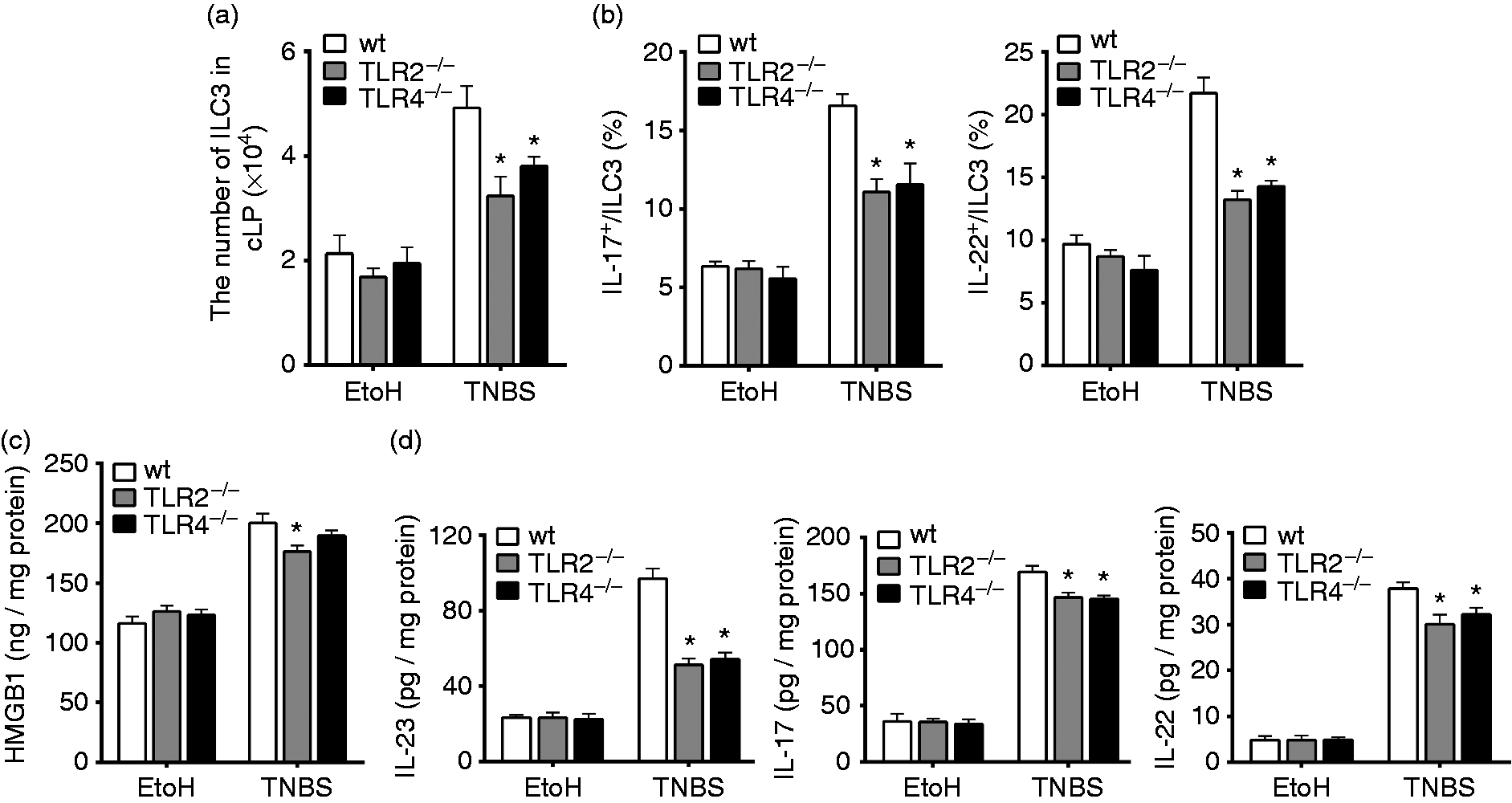
DCs play a vital role in regulation of HMGB1 on ILC3s
Our results demonstrated that the axis of HMGB1-TLR2/4 played a significant role in regulation of colonic ILC3s. However, it had been demonstrated that the ILC3s mice did not express TLR2 and TLR4.
33
It is well known that ILC3s produce cytokines IL-17 and IL-22 in response to IL-23. And a previous study has demonstrated that DCs have the ability to produce IL-23 in response to TLRs signaling stimulation.
34
To examine whether colonic DCs had important roles in the cascade signaling way of HMGB1-TLR2/4-IL-23-ILC3s in mouse colitis, we analyzed the number changes of colonic DCs while HMGB1 was inhibited or after TLR2/4 genes had been deleted (Figure 5a). As expected, inhibition of HMGB1-TLR2/4 signaling with anti-HMGB1 Ab or TLR2/4 genes knockout resulted in a decrease of the number of colonic DCs in colitis mice (Figure 5b and c).
The numbers of colonic DCs decreased while HMGB1-TLR2/4 signaling was inhibited by anti-HMGB1 Ab administration or TLR2/4 genes deletion. LPMCs were isolated from the colons of mice sacrificed on d 4 after intrarectal administration of EtOH or TNBS. Colonic DCs were determined by flow cytometry. (a) Gating strategies for DCs (CD11c+MHCII+). (b) The numbers of colonic DCs were determined after treatment with anti-HMGB1 Ab or IgG. (*K ≤ 0.05 vs. EtOH, #P ≤ 0.05 vs. TNBS + IgG.) (c) The numbers of DCs separated from wt, TLR2–/– and TLR4–/– mice cLP were determined. (*P ≤ 0.05 vs. wt + TNBS). Data are presented as means + SD of three to six individual mice per group and represent > 3 independent experimental repeats. EtOH: mice treated with 50% ethanol.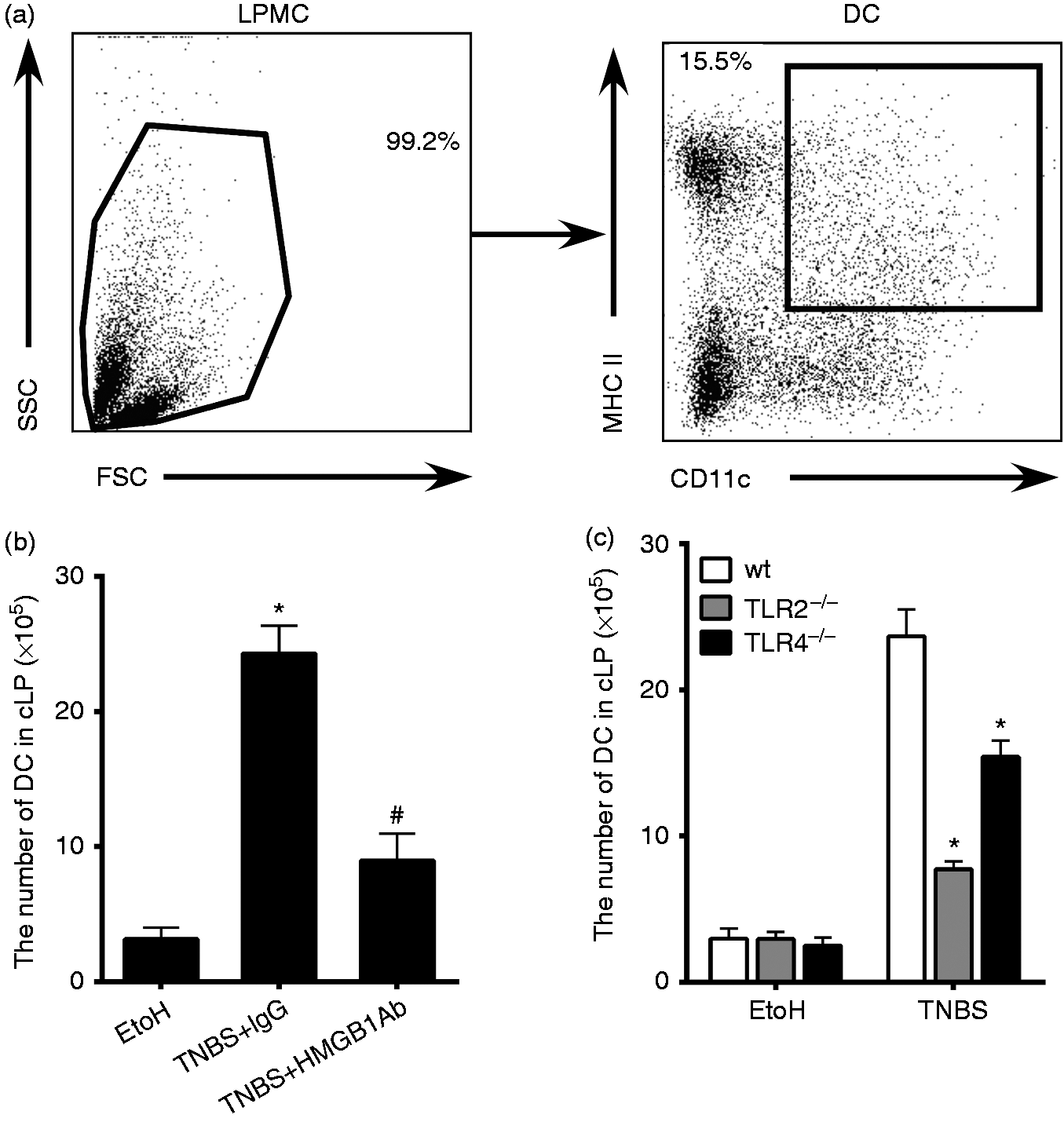
To further directly define that HMGB1 can promote DCs to produce IL-23 via TLR2 or TLR4, we cultured BMDCs from wt, TLR2−/− and TLR4−/− mice and treated these with recombined HMGB1 protein (rHMGB1). We initially exposed BMDCs to various concentrations of rHMGB1 and measured their capacity to produce IL-23 in culture supernatants. The results showed that rHMGB1 significantly increased the level of IL-23 in wt BMDC culture supernatants. However, the levels of IL-23 in TLR2−/− and TLR4−/− BMDC culture supernatants were lower than in wt BMDC culture supernatants. These results indicated that rHMGB1-stimulated DCs (via TLR2/4) had the potential to secrete ILC3s activation-related factor IL-23 (Figure 6).
HMGB1 recombinant proteins promoted BMDCs to produce IL-23 via TLR2 or TLR4 signaling. BMDCs were derived from femurs and tibias of 6 - to 8-wk-old wt, TLR2–/– and TLR4–/– mice. After stimulation of BMDCs with different doses of rHMGB1 protein (0, 100, 500 ng/ml) for 48 h, the levels of cytokines IL-23 in the cell culture supernatants were determined by ELISA. *P ≤ 0.05 vs. wt. Results were representative of the three experiments performed. Data are presented as mean ± SD.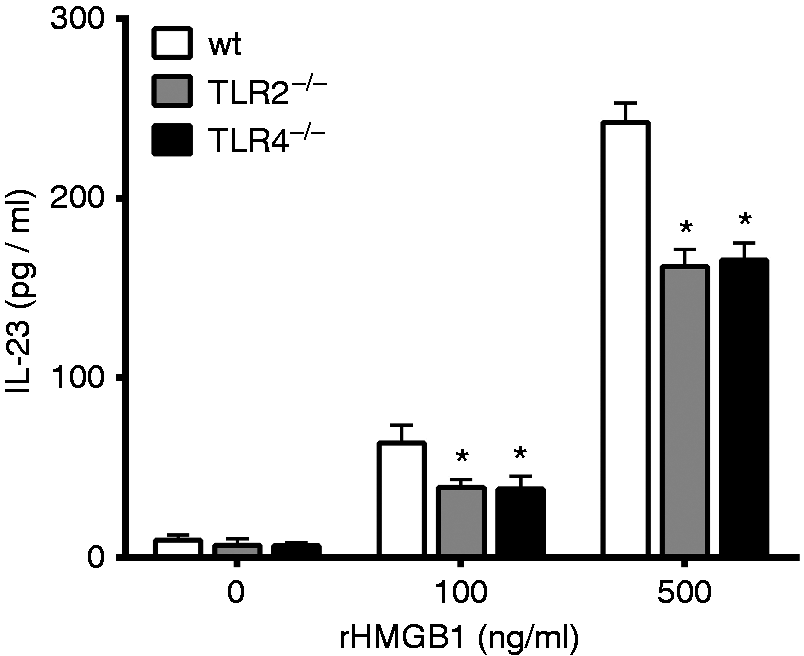
Discussion
This study showed that, correlated with inhibition of HMGB1 secretion, anti-HMGB1 Ab administration ameliorated intestinal inflammation in murine colitis models. Furthermore, HMGB1 is a ligand for the PRR TLR2 and TLR4, since deletion of these PRR genes attenuated intestinal inflammation of TNBS colitis. Therefore, the HMGB1-TLR2/4 axis played an important role in the development of enteric inflammation. Surprisingly, inhibition of the HMGB1-TLR2/4 axis by using anti-HMGB1 Ab or deletion of genes TLR2/4 decreased the numbers and functions of colonic ILC3s and DCs. The levels of cytokines IL-17, IL-22 and IL-23, which can be produced by ILC3s and DCs, decreased along with the changes of colonic ILC3s and DCs. In addition, HMGB1 promoted DCs to produce IL-23 via TLR2/4 in an in vitro experiment. Therefore, the cascade reaction of HMGB1-TLR2/4-DC-IL-23-ILC3s-IL-17, IL-22 had a vital role in the development of colitis.
HMGB1 is an important inflammatory mediator and is related to the onset of a wide variety of autoimmune and inflammatory diseases and for its cascade amplification effect by autocrine and modulation of other cytokines secretion. 4 Previous studies demonstrated that increased HMGB1 in colon tissue was associated with the disruption of the intestinal barrier and colonic inflammation.30,35,36 Studies also suggested that HMGB1 may serve as an important molecular signal in the regulation of immunological mechanisms of intestinal mucosa damage. 37 Our experiments showed that application of anti-HMGB1 Ab, which inhibited HMGB1 function in TNBS-induced colitis, could alleviate intestinal inflammation. It has been demonstrated that HMGB1 had stimulatory effects on innate immune cells including DCs, NK cells, and macrophages, and could assist stimulating the activation of adaptive immune cells, such as T and B cells. 38 However, effects of HMGB1 on ILC3s had not been reported previously. In our experiments, we found that administration of anti-HMGB1 Ab decreased the numbers of colonic ILC3s and the levels of IL-17 and IL-22 that can be released by ILC3s in experimental colitis. The results also demonstrated that a gene knockout of the receptors of HMGB1, TLR2 or TLR4 resulted in a decrease of the colonic ILC3s numbers and IL-17 and IL-22 levels. Therefore, HMGB1 influenced colonic ILC3s via TLR2/4 in the development of mouse colitis.
ILC3s are characterized by secreting a series of cytokines and chemokines to resist infection and promote tissue repair at mucosal barriers. Based on previous data, it was illustrated that cytokines IL-22 and IL-17 secreted by ILC3s played important roles in the pathogenesis of colitis. IL-22 and IL-17 are important components of ILC3s responses and act as a double-edged sword in the intestinal inflammatory response. 39 IL-22 and IL-17 secreted by ILC3s are involved in either pro-inflammatory or anti-inflammatory reactions depending on the cellular and cytokine environment. IL-22 and IL-17 maintain the integrity of intestinal mucus under normal circumstances, but high levels of IL-22 and IL-17 also cause colitis or aggravate inflammation during bacterial infection or intestinal injury. 40 In our experiment, the frequencies of IL-22- or IL-17-positive ILC3s increased in the injured colon tissues. However, inhibition of the HMGB1-TLR2/4 axis by using anti-HMGB1 Ab or TLR2/4 gene deletion resulted in decreased frequencies of IL-22+ or IL-17+ ILC3s. These results demonstrated that the HMGB1-TLR2/4 axis was involved in promoting colonic ILC3s to produce cytokines IL-22 and IL-17 in experimental colitis.
Previous studies demonstrated that mouse ILC3s did not express TLR2 and TLR4, but in our experiments the HMGB1-TLR2/4 axis promoted the function of colonic ILC3s. 33 Therefore, the HMGB1-TLR2/4 axis indirectly enhances ILC3s response in a different way. In our experiments, we found that anti-HMGB1 Ab or TLR2/4 deletion could decrease the level of IL-23, which can stimulate ILC3s to produce IL-22 and IL-17. Our results also showed that the numbers of colonic DCs were reduced after administration of anti-HMGB1 Ab or deletion of TLR2/4 gene in mouse colitis. One earlier study had demonstrated that DCs produce IL-23 while TLR2 or TLR4 are activated by their PAMP ligands. 41 Furthermore, our results demonstrated that HMGB1 promoted DCs to produce IL-23 via TLR2/4 in vitro.
In conclusion, we demonstrated in this study that HMGB1 mediated the pathogenesis of colitis when binding to either TLR2 or TLR4, and promoted DCs to produce IL-23, which enhanced the response of ILC3s in murine colitis. When the signaling pathway of HMGB1-TLR2/4 was inhibited by treatment of anti-HMGB1 Ab or TLR2/4 gene deletion, intestinal inflammation was alleviated and the response of colonic ILC3s was inhibited. Taken together, HMGB1-TLR2/4-DC-IL-23-ILC3s-IL-17, IL-22 cascade response played a vital role in the development of colitis.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (grant number 91542110 to M. Fang), the Ministry of Science and Technology of China (grant number 2013CB530505) and the National Natural Science Foundation of China (grant number 81373167 to M. Fang).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.