
Abstract
Proteinase-activated receptor 2 (PAR2), a 7-transmembrane G protein-coupled receptor, contributes to inflammation either positively or negatively in different experimental systems. Previously, we reported that concurrent activation of PAR2 and TLRs in human lung and colonic epithelial cells resulted in a synergistic increase in NF-κB-mediated gene expression, but a down-regulation of IRF-3-mediated gene expression. In this study, the effect of PAR2 activation on LPS-induced TLR4 signaling was examined in primary murine macrophages. The PAR2 activation of wild-type macrophages enhanced LPS-induced expression of the anti-inflammatory cytokine, IL-10, while suppressing gene expression of pro-inflammatory cytokines, TNF-α, IL-6, and IL-12. Similar PAR2-mediated effects on LPS-stimulated IL-10 and IL-12 mRNA were also observed in vivo. In contrast,
Keywords
Introduction
Pattern-recognition receptors (PRRs) of the innate immune system detect infection by recognizing pathogen-associated molecular patterns (PAMPs), evolutionarily conserved structural motifs that are shared among microbes, e.g. LPS, lipopeptides, flagellin, and microbial nucleic acids.1,2 Pattern-recognition receptors also sense tissue damage by responding to endogenous, host-derived danger-associated molecular patterns (DAMPs). 3 Classical PRRs, e.g. TLRs, provide surveillance by recognizing various PAMPs and DAMPs as ligands. In contrast, non-classical PRRs, 4 such as the protease activated receptors (PARs), respond to infection and tissue damage by sensing pathogen- or host-derived proteolytic enzymes, as well as certain allergens with intrinsic protease activity.5,6 In macrophages, PRR activation primarily drives development of antimicrobial and pro-inflammatory responses that are typically associated with the ‘classically activated’ (‘M1’) differentiation phenotype that favors development of T helper cell type 1 (Th1)-skewed cytokine production, e.g. IL-12 and IFN-γ.2,7 However, a temporally delayed, anti-inflammatory transcriptional program leading to expression of genes such as IL-10, IL-4, and IL-13 is also activated in macrophages by certain pathogens and promotes tissue homeostasis by counteracting induction of pro-inflammatory cytokines and by leading to the development of macrophages with an AA-Mϕ (also referred to as ‘M2’) differentiation phenotype that dampens the pro-inflammatory response. 8 – 11 Such macrophages produce a ‘Th2-like’ cytokine milieu and are often associated with tissue repair, wound healing, and responses to allergens and parasitic infections.
Toll-like receptors represent a large family of germline-encoded, single-transmembrane, classical PRRs. They are distributed ubiquitously in the body and are expressed on many innate immune cell types, including epithelial cells and macrophages.2,12 Direct or indirect binding of TLR-activating ligands to the TLR N-terminal ectodomain induces TLR dimerization that brings the intracytoplasmic ‘Toll/interleukin-1 receptor resistance’ (TIR) domains into close proximity. This interaction facilitates the recruitment of TIR-domain-containing adapters (e.g. MyD88, TRIF), kinases, and other signaling molecules to the ‘signaling platform’ generated by the initiating TLR TIR dimer. Activation of the Gram-negative bacterial LPS transducing receptor, TLR4, results in nuclear translocation of NF-κB through the ‘MyD88-dependent’ signaling pathway and of interferon regulatory factor-3 (IRF-3) via the ‘MyD88-independent’ signaling pathway. These two key transcription factors are required for expression of many inflammatory and immunomodulatory chemokines and cytokines.2,12
Proteinase-activated receptor 2 (PAR2) belongs to a family of four seven-transmembrane G protein-coupled receptors (7-TM GPCRs).5,13 It is expressed highly in the respiratory and gastrointestinal (GI) tracts,14,15 and, like the TLRs, is also expressed on epithelial cells and macrophages. PAR2 mediates the cellular effects of trypsin and trypsin-like serine proteases, including mast cell tryptase, coagulation factors VIIa and Xa, and several pathogen-derived proteinases. The PAR-activating enzymes cleave each PAR irreversibly at a specific site in the extracellular N-terminus to expose a tethered neo-ligand that binds intramolecularly to the second extracellular loop (ECL2) of the GPCR to trigger receptor activation. Synthetic PAR agonist peptides (AP) that bear the hexapeptide sequences of the tethered neo-ligands of PAR1, PAR2, and PAR4 mediate signaling non-enzymatically by binding directly to the ECL2 of their respective native, uncleaved PAR. We have reported that the human PAR2 AP, SLIGKV-NH2, induces both NF-κB and IRF-3 reporter activities in PAR2-expressing HEK293T cells, but not in pcDNA3.1 empty vector-transfected cells, and that PAR2 co-immunoprecipitated with TLR4 in the presence of AP, suggesting that these two receptors may physically associate in response to PAR2 activation.
16
In addition, the PAR2 AP, SLIGKV-NH2, but not a scrambled control peptide, induced footpad edema in wild-type C57BL/6J mice, but not in
Previous studies indicate that PAR2 is involved in pro-inflammatory and allergic responses at anatomical sites that interact with protease-rich environments, e.g. inflamed tissues, the gut lumen, and the respiratory tract. 21 – 23 In addition, PAR2 signaling intersects with TLRs to augment pro-inflammatory responses in epithelial cells, endothelial cells, and monocytes.4,16,24– 26 However, cytoprotective and anti-inflammatory functions have also been ascribed to PAR2 signaling. For example, PAR2 activation exerts powerful bronchoprotection in the airway and also protects the gastric mucosa against the injurious effects of non-steroidal anti-inflammatory drugs and acid or ethanol solutions.27,28 In vivo, PAR2 AP treatment inhibits Th1 cytokine production and protects mice from 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis and lethality. 29 Intranasal PAR2 AP administration inhibits LPS-induced pulmonary neutrophil influx. 30 In this study, we hypothesized that the cytoprotective, anti-inflammatory outcomes observed in vivo for PAR2 may depend on PAR2-mediated activation of macrophages, as members of several other 7-TM GPCR families, e.g. β-adrenergic and adenosine A2a receptors, have been shown to dampen inflammation induced by LPS via TLR4 in myeloid cells.7,31– 33 In fact, activation of these GPCRs in macrophages has been shown to augment TLR4-induced expression of the anti-inflammatory cytokine, IL-10, while down-regulating the expression of various pro-inflammatory mediators, e.g. IL-1β, IL-6, TNF-α, and IL-12.7,31– 33 This study sought to define the capacity of PAR2 to modulate TLR4 signaling in murine macrophages.
Materials and methods
Reagents, mice, and tissue culture
Human PAR2 AP, SLIGKV-NH2, and an inactive, control reverse peptide (RP), VKGILS-NH2, were synthesized (>96% purity) by Phoenix Pharmaceuticals (Belmont, CA, USA). PAR2 fAP (2-furoyl-LIGRLO-NH2; >98% purity), a stabilized PAR2 agonist, was purchased from Calbiochem (San Diego, CA, USA). Protein-free, phenol/water-extracted LPS from Escherichia coli K235 was purified as described previously. 34 Murine recombinant IL-4 (rIL-4) was purchased from R&D Systems (Minneapolis, MN, USA).
Wild-type C57BL/6J mice and
Quantitative real-time PCR (qPCR)
Total RNA from tissue cultures or organs was extracted, and oligo(dT)-primed cDNA was synthesized as previously described. 35 The qPCR primers were designed and synthesized as previously described.9,35 qPCR was carried out on ABI Prism® 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA) as previously described. 35
Flow cytometry analysis
C57BL/6J and
Western analysis
At the indicated times, whole-cell lysates were prepared and resolved by gel electrophoresis, transferred to polyvinylidene difluoride membranes, probed with antibodies, and target protein bands detected by enhanced chemoluminescence as described. 35 Primary and horseradish peroxidase-conjugated secondary antibodies used in this study were purchased from Cell Signaling Technology (Danvers, MA, USA) and used at 1 : 1000 and 1 : 2000 dilutions, respectively. Densitometric analysis of Western blots was carried out using NIH software, Image J (<http://rsbweb.nih.gov/nih-image/>).
Analysis of secreted proteins by ELISA
The concentrations of secreted proteins were determined by enzyme-linked immunosorbent assay (ELISA) using kits from R&D Systems or through the Cytokine Core Laboratory (UMB).
Statistical analysis
Using GraphPad PRISM v4.0 (GraphPad Software, San Diego, CA, USA), one-way analysis of variance (ANOVA) with Tukey’s post-test or two-way ANOVA with Bonferroni post-test was performed to assess statistical significance (P values < 0.05).
Results
Activation of PAR2 mediates an anti-inflammatory response in LPS-stimulated murine macrophages
PAR2 and TLR4 are expressed on many cell types in the body, including epithelial cells and macrophages.2,5,12,13 Like TLR4, PAR2 has been strongly implicated in the induction of pro-inflammatory responses in a variety of cell types in various species.5,13 We reported recently that PAR2 activation in HEK293T-PAR2 transfectants by its activating enzyme, trypsin, or its AP, SLIGKV-NH2, elicited NF-κB-dependent and IRF-3-dependent luciferase reporter activities. 16 We also reported that PAR2 activation in HEK293T-PAR2 transfectants and in human A549 lung and SW620 colonic epithelial cell lines induced expression of the NF-κB-dependent neutrophil chemokine, IL-8.4,16 However, when PAR2 is activated in the presence of a TLR agonist in these epithelial cell lines, NF-κB-dependent gene expression and secretion was further increased, while IRF-3-dependent gene expression induced by LPS (a TLR4 agonist) or poly I:C (a TLR3 agonist) was repressed. 4
In contrast to the positive co-operation that was observed between PAR2 AP and LPS for the induction of pro-inflammatory responses in mucosal epithelial cell lines, simultaneous treatment of primary murine peritoneal macrophages with PAR2 AP (SLIGKV-NH2) and LPS resulted in a dose-dependent down-regulation of several key LPS-inducible pro-inflammatory cytokines, e.g. TNF-α, IL-6, and IL-12 p40 (Figure 1). In contrast, LPS-induced production of the potent anti-inflammatory cytokine, IL-10, was increased in the presence of PAR2 AP (Figure 1). We confirmed and extended these findings in murine macrophages using a chemically modified PAR2 AP, 2-furoyl-LIGRLO-NH2 (fAP).
37
PAR2 fAP is metabolically stable, resistant to aminopeptidases, and exhibits high PAR2 selectivity and potency.
37
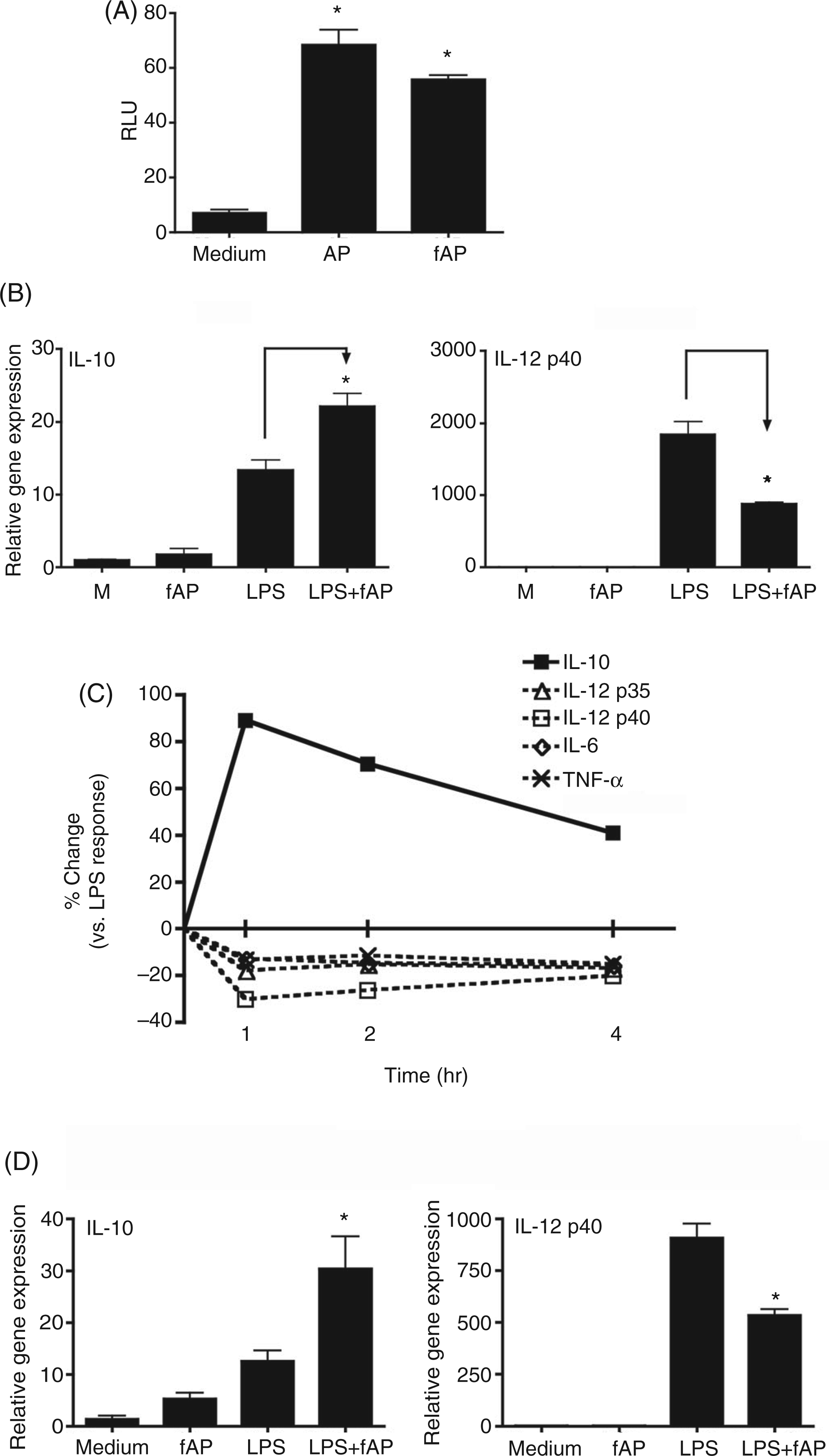
Like native PAR2 AP, fAP induced comparable NF-κB-luciferase reporter activities in HEK293T-PAR2 transfectants (Figure 2A), but not in pcDNA3.1-transfected HEK293T cells (data not shown). In agreement with the results obtained using the native PAR2 AP, SLIGKV-NH2, PAR2 fAP also enhanced LPS-induced IL-10 mRNA expression synergistically, while attenuating LPS-induced IL-12 p40 (Figure 2B), IL-12 p35, IL-6, and TNF-α mRNA levels (Figure 2C). The control reverse peptide (RP), 2-furoyl-OLRGIL-NH2, had no effect on the LPS response (data not shown). This same pattern of gene expression, i.e. increased IL-10 mRNA and decreased IL-12 p40 mRNA, was also observed in bone marrow-derived macrophages (Figure 2D).
PAR2 AP differentially modulates LPS-induced cytokine production in primary murine macrophages to promote an anti-inflammatory response. Thioglycollate-elicited peritoneal macrophages from C57BL/6J mice were stimulated for 24 h with medium only (Med), PAR2 AP (SLIGKV-NH2; 200 µ Both PAR2 AP and fAP differentially modulate LPS-induced gene expression and cytokine production in primary murine macrophages to promote an anti-inflammatory response. (A) Induction of NF-κB activity by PAR2 AP and fAP in PAR2-expressing HEK293T transfectants. HEK293T cells were transiently transfected with PAR2, together with NF-κB-luciferase and β-galactosidase reporter constructs. Cells were treated with PAR2 AP (SLIGKV-NH2; 200 µ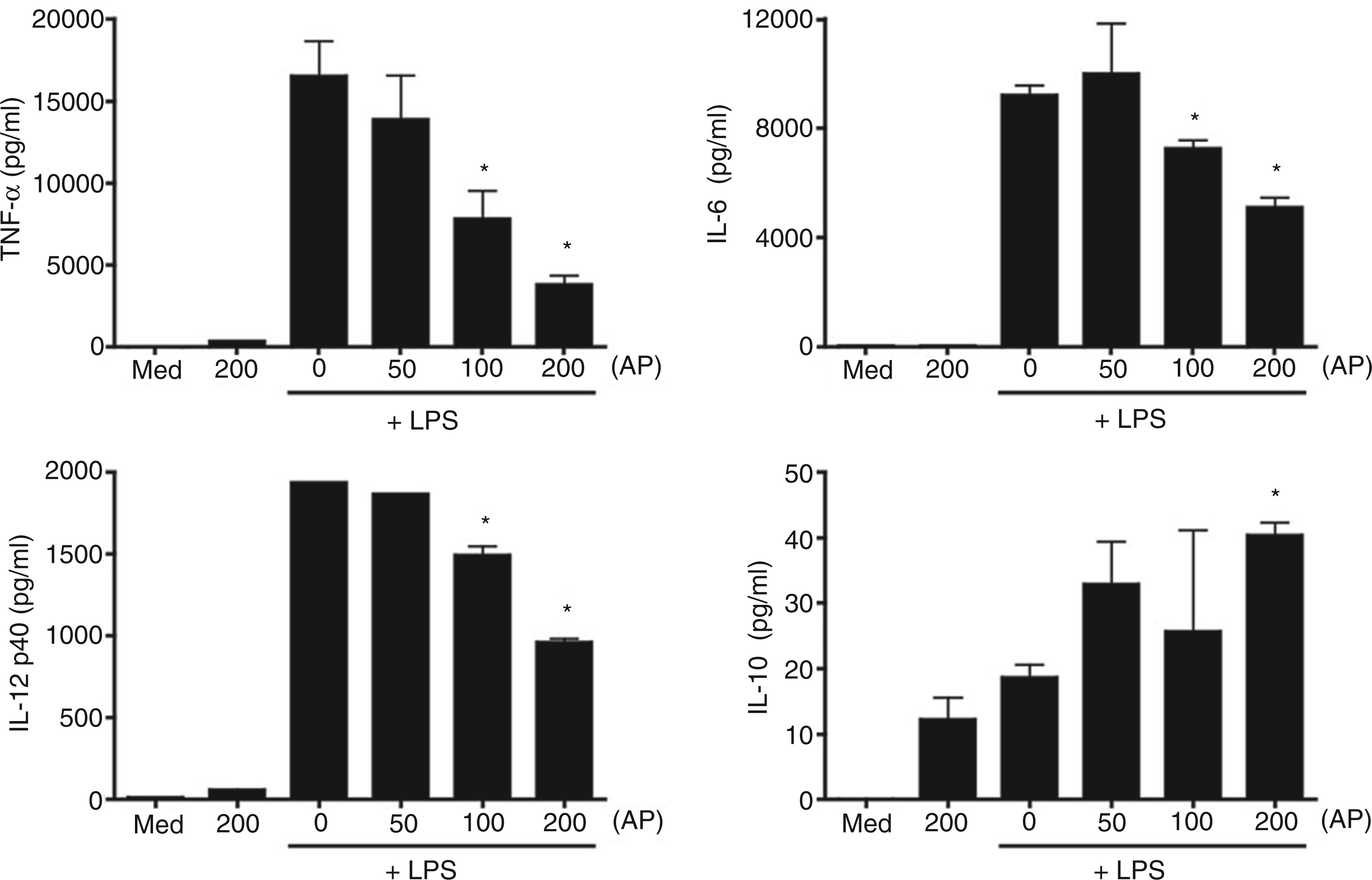
Next, the extent of PAR2 and TLR4 signaling crosstalk was examined in vivo. Analysis of livers of mice injected i.p. with LPS in the absence or presence of PAR2 AP (SLIGKV-NH2) revealed enhanced expression of IL-10 mRNA that was followed by a significant decrease in IL-12 p40 mRNA levels in mice that received both PAR2 AP and LPS (Figure 3). Co-administration of PAR2 AP and LPS also led to a modest reduction in the expression of TNF-α, IL-6, and IL-12 p35 compared to that induced by LPS alone (data not shown). Taken together, our findings in murine macrophages in vitro and in livers, where the Kupffer cell resident macrophages represent the predominant LPS-responsive cells leading to the expression of IL-10 and IL-12 p40,
38
show that PAR2 and TLR4 signaling pathways intersect such that PAR2 promotes development of an anti-inflammatory IL-10 response while dampening the Th1-like pro-inflammatory response induced by LPS.
PAR2 AP differentially modulates LPS-induced cytokine gene expression to promote an anti-inflammatory response in vivo. C57BL/6J mice were injected i.p. with 0.5 ml of saline, PAR2 AP (SLIGKV-NH2; 500 µ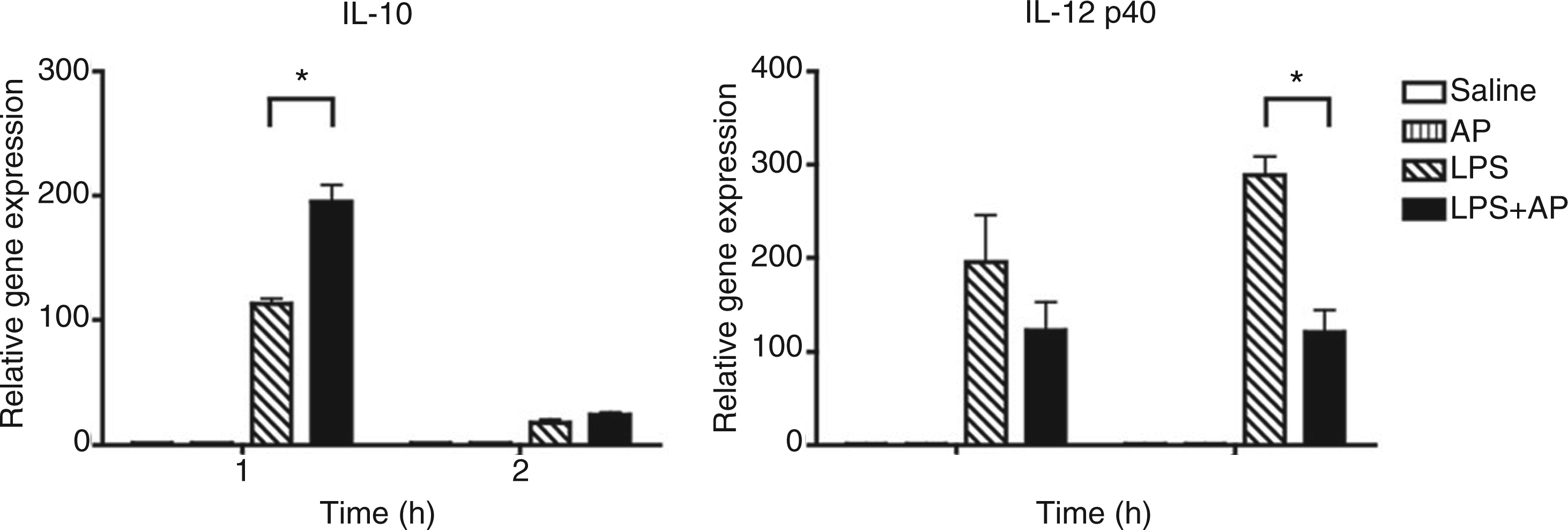
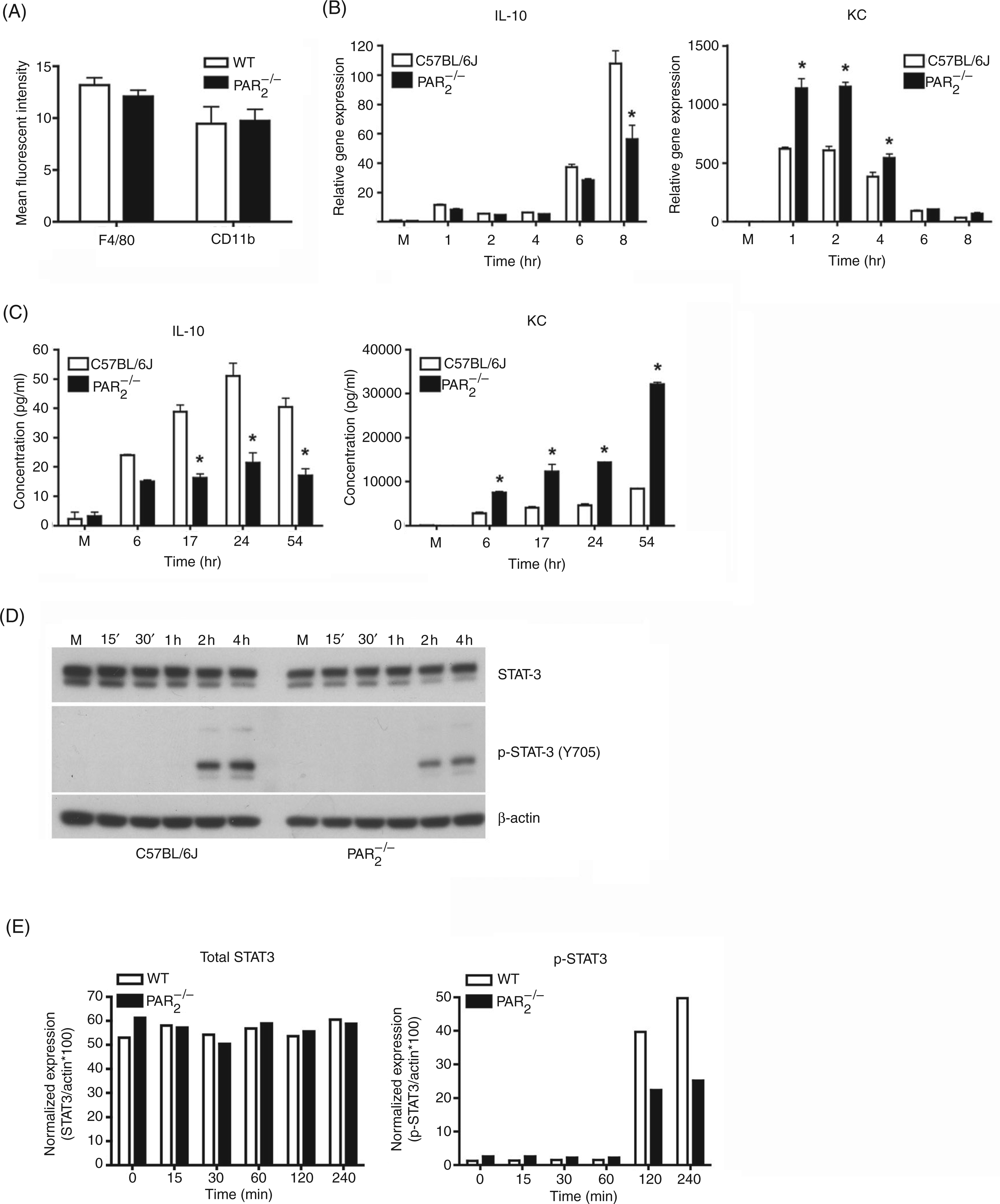
macrophages exhibit altered LPS responses
In the next series of experiments,
Activation of PAR2 synergistically augments expression of alternatively-activated/M2 macrophage markers
Recently, Porta et al.
40
reported that LPS-treated macrophages exhibited elevated expression of AA-Mϕ markers, e.g. IL-10, CCL2, CCL17, CCL22, and arginase-1 that followed the induction of pro-inflammatory cytokine gene expression. Since IL-4 and IL-13 drive the induction of AA-Mϕ,
8
–
11
we first measured the effect of PAR2 AP and/or rIL-4 or LPS on the production of IL-4 and IL-13. PAR2 fAP was a poor inducer of IL-4 and IL-13 by macrophages, but significantly enhanced production of both IL-4 and IL-13 induced by rIL-4 or LPS at 48 h (Figure 5A).
PAR2 fAP synergistically augments rIL-4- or LPS-induction of IL-4, IL-13, and alternative activation of murine macrophages. (A) PAR2 fAP synergizes with rIL-4 and LPS to induce IL-4 and IL-13. Thioglycollate-elicited peritoneal macrophage from C57BL/6J mice were stimulated for 48 h with medium (M), PAR2 fAP (2-furoyl-LIGRLO-NH2; 200 µ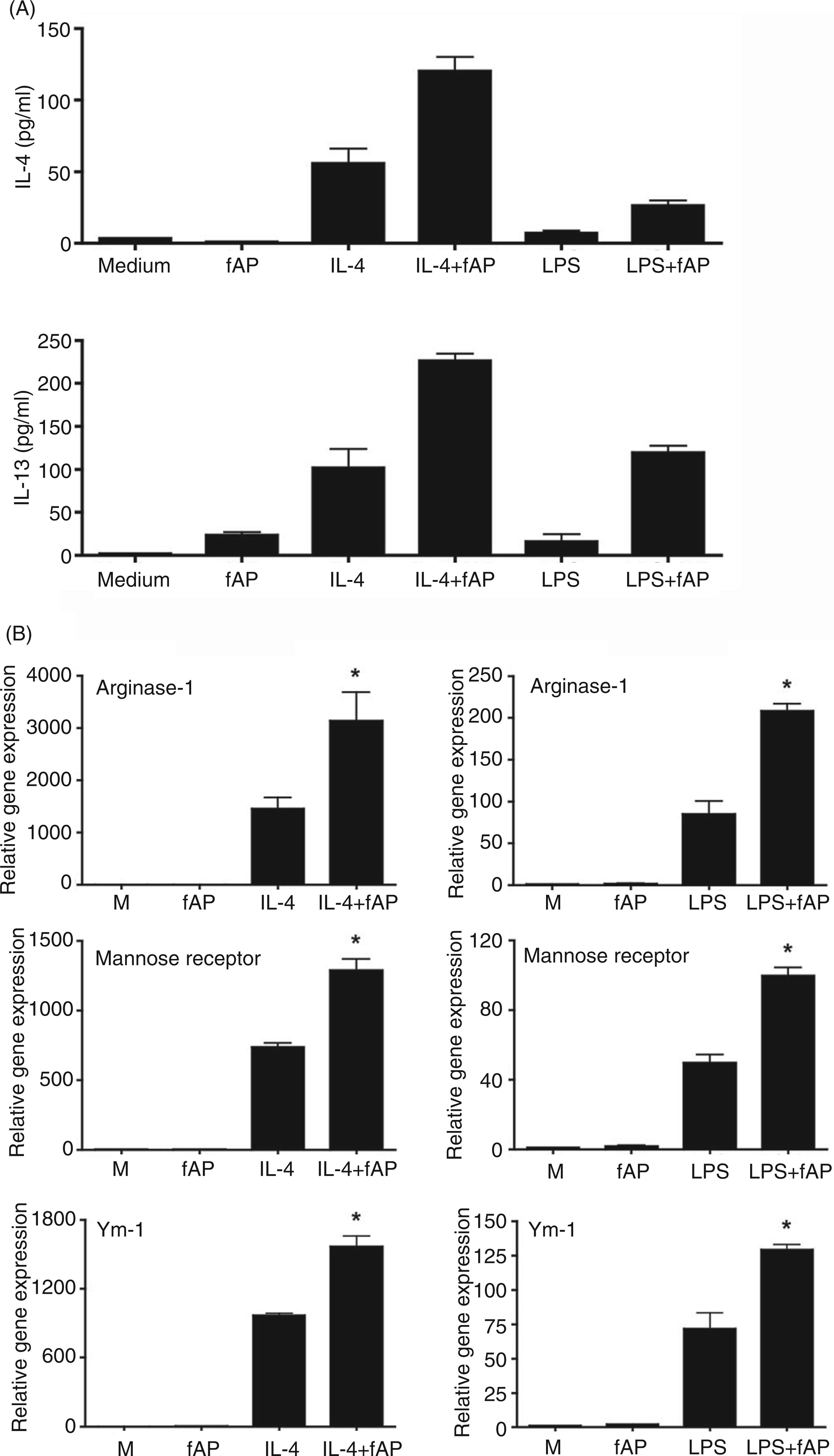
Figure 5B illustrates that macrophages stimulated with rIL-4 (5 ng/ml) or LPS (10 ng/ml) for 48 h exhibited increased mRNA expression of the prototypical AA-Mϕ markers, i.e. arginase-1, Ym-1, and mannose receptor. 8 The LPS-induced mRNA expression of the AA-Mϕ markers was significantly lower than that induced by treatment with rIL-4. Nevertheless, these data confirm the findings of Porta et al. 40 and indicate that, like rIL-4, LPS has the capacity to induce AA-Mϕ differentiation in primary murine macrophages.
Since concurrent PAR2 activation up-regulated LPS-induced IL-4, IL-13, and IL-10 production, while decreasing pro-inflammatory cytokine expression, we sought to determine if PAR2 activation would also augment rIL-4- or LPS-induced differentiation of AA-Mϕ. Consistent with the data shown in Figure 5A, Figure 5B also shows that PAR2 fAP synergistically enhanced both LPS- and rIL-4-induced mRNA expression of the AA-Mϕ markers, arginase-1, Ym-1, and mannose receptor.
Discussion
Previous studies have demonstrated that concurrent stimulation of mucosal epithelial cells through PAR2 and TLR4 enhanced expression of many NF-κB-dependent, pro-inflammatory genes, while inhibiting IRF-3-driven gene expression.4,16,25 However, in contrast to the epithelial cell response, concurrent PAR2 and TLR4 stimulation of murine macrophages resulted in a down-regulation of TLR4-induced pro-inflammatory gene and protein expression, augmented TLR4-driven production of the potent Th2-skewing, anti-inflammatory cytokine, IL-10, and promoted TLR4- or IL-4 receptor-mediated differentiation of AA-Mϕ phenotype, that has been strongly associated with wound healing, tissue repair, allergic and anti-parasitic Th2 immune responses.8,10 These findings were confirmed and extended by showing that in PAR2-null macrophages, the response to LPS was skewed toward a pro-inflammatory cytokine profile. Collectively, these data suggest that PAR2 activation occurs in response to LPS signaling and attenuates the macrophage response to LPS both in vitro and in vivo through the production of counter-regulatory cytokines such as IL-10.
Activation of PAR2 has been reported to exhibit opposite effects in several models of inflammation,21,22,28,41 and perhaps this is attributable to the balance of synergistic or antagonistic effects reported herein between PAR2 and activation of other immune modulating receptors in different cell types. Our results in macrophages showing a preferential induction of an anti-inflammatory phenotype mediated by concurrent PAR2 and TLR4 activation support and extend earlier studies. For example, a study using an ovalbumin-induced asthma model, in which CD4+ Th2 cells are strongly implicated, reported that
The induction of Th2 differentiation is not limited to the intercellular interactions between antigen-presenting cells, e.g. macrophages and dendritic cells, and T cells; epithelial cell-derived thymic stromal lymphopoietin (TSLP) can also trigger dendritic cell-mediated Th2-type inflammation. 44 The Th2-promoting role for PAR2 is further supported by two recent studies showing that PAR2 activation in epithelial cells induces TSLP.45,46 In addition, allergens that possess intrinsic protease activity can activate PAR2 on epithelial cells to induce a Th2-type allergic inflammatory response.6,47 Taken together, the results reported herein and by others21,42,43,46 indicate that PAR2 activation favors an immune deviation from the classical Th1-like pro-inflammatory response to one that is more Th2-like.
Clearly, these observations suggest that PAR2 signaling in different cell types can differentially affect the outcome of an inflammatory response. PAR2 activation has been observed to exert both protective and pathogenic effects in different cell types, i.e. glial cells and neurons, in Alzheimer’s disease. 48 Differential expression of PAR2 on different cell types, coupled with the co-expression of other PAR2-interacting adapter proteins, e.g. G proteins, β-arrestins, 49 – 51 and the TLR adapters, 16 or PAR2-interacting receptors, e.g. TLR4, 16 may also dictate the cell type-specific outcome of an inflammatory response. Future experiments will be required to dissect the relative contributions of these various interacting partners of PAR2 to signaling in different cell types.
Conclusions
The results from this study provide a mechanism that potentially explains the opposite inflammatory roles observed for PAR2 in different experimental models. We propose that the types of cell being activated and the timing of the experimental outcome likely contribute to the dual roles observed for PAR2 signaling in inflammation. The results from our study suggest that manipulation of the extracellular protease/anti-protease balance, as well as cell-type specific inhibition or activation of PAR2 signaling, may represent future novel therapeutic approaches to treating inflammatory PAR2-dependent disorders.
Footnotes
Funding
This work was supported by National Institutes of Health (NIH) Grant AI18797 (SNV) and AI49316 (JS). QMN was supported, in part, by NIH Training Grant (T32 AI-07540) and JS was supported by NIH Training Grant (T32 DK067872). This work was carried out in partial fulfilment of the PhD requirements (QMN, JS).
Acknowledgements
Quan M. Nhu and Kari Ann Shirey contributed equally to this manuscript. The authors thank Dr Swamy Polumuri for helpful discussions and Drs Aiping Zhao, Terez Shea-Donohue, Sarah Netzel-Arnett, and Toni Antalis for generously providing materials used in this study and for their helpful suggestions.