
Abstract
Purpose
Demonstration of the role of autocrine motility factor receptor (AMFR) in atrial fibrillation (AF) mice.
Methods
The AF model was established by administering Ang II to mice and transfected with AMFR knockdown or AMFR overexpression plasmids by tail vein injection of the AAV vector. Atrial fibrosis was examined by Masson staining. The mRNA expression of inflammatory factors TNF-α, IL-1β, and IL-6 in atrial tissue was detected by PCR. Reactive oxygen species (ROS) production in atrial tissue was examined by dihydroethidium staining. Apoptotic cells of atrial tissue were examined by TUNEL staining. The expression levels of fibrosis-related genes (COL1A1 and α-SMA), apoptosis-related genes (cleaved-caspase3 and cleaved-PARP), and SOD1 were detected by western blot. The ubiquitination level of superoxide dismutase 1 (SOD1) was detected by ubiquitination assay.
Results
Ang II resulted in increased AMFR expression in mouse atrial tissue, and knockdown of AMFR inhibited atrial fibrosis, inflammatory factors, and ROS production, as well as apoptosis in mice. In addition, the knockdown of AMFR inhibited the ubiquitination level of SOD1 and increased the protein expression level of SOD1, whereas overexpression of AMFR exerted the opposite effect and aggravated Ang II-induced AF.
Conclusion
Knockdown of AMFR promotes SOD1 expression by inhibiting SOD1 ubiquitination levels and attenuates atrial fibrosis in AF mice by regulating SOD1.
Introduction
Atrial fibrosis has been widely recognized as an important predictor of the occurrence and development of atrial fibrillation (AF). It is significantly associated with multiple cardiovascular complications of AF and is a key pathological feature of atrial cardiomyopathy. Experimental and clinical studies have confirmed that there is a bidirectional interaction between AF and atrial fibrosis: on the one hand, AF can promote the process of fibrosis; on the other hand, atrial structural remodeling caused by fibrosis will further increase the risk of AF and accelerate its progression.1,2
More and more research points to oxidative stress as a major pathophysiological reason for AF. In the context of AF, oxidative stress 3 and mitochondrial dysfunction have been identified in the atrial tissue of AF patients. 4 Tachycardia-induced rise of reactive oxygen species (ROS) in ventricles results in apoptosis. Gene expression profiling in the atrial tissue of AF patients showed an increase in ROS-producing genes and a decrease in the expression of anti-oxidative genes. 5
E3 ubiquitin ligases have been shown to impact the course of myocardial infarction and AF oxidative stress. Tripartite motif-containing 21 (TRIM21) may mechanistically stimulate Nox2 expression by activating the NF-κB pathway, which causes inflammation, atrial remodeling, and myocardial oxidative damage. 6 Autocrine motility factor receptor (AMFR), which is often referred to as Gp78, is a ubiquitin E3 ligase that is involved in the endoplasmic reticulum protein degradation. 7 Asthmatic lung macrophages overexpress AMFR, and a reduction in AMFR dramatically lessens eosinophilic and Th2-mediated allergic inflammation, mechanistically, Lys48-linked polyubiquitination of cytokine-induced SH2 protein (CIS) is triggered by AMFR through direct binding to CIS. 8
In this study, by querying the GSE79768 expression profile, it was found that AMFR expression in AF atrial tissue was significantly higher than that in atrial tissue in sinus rhythm. Based on the previous literature reports that AMFR affects lung fibrosis, 9 it is speculated that AMFR may affect atrial fibrosis in AF. Therefore, this study confirmed for the first time the role and related mechanisms of AMFR in AF.
Methods
Bioinformatics Analysis
The differential gene analysis was performed on the GSE79768 expression profile, with logFC = 0.5 and adj P < .05 as the cutoff value, and the volcano map was drawn using the “ggplot2” package in R language. The AMFR expression values in the normal group and AF group in the expression profile were statistically analyzed and box plots were drawn.
Animals
For three weeks, male C57BL/6 mice that were eight weeks old were injected with Ang II (2000 ng/kg/min) using osmotic minipumps (n = 6). Mice requiring gene transfection were injected with AAV-AMFR (n = 6), AAV-sh-AMFR (n = 6), AAV-SOD1 (n = 6), and AAV-sh-AMFR + sh-SOD1 (n = 6) via tail vein one week prior to Ang II injection. The control group (n = 6) was a normal control group without any treatment. The Dalian Medical University Animal Care and Use Committee approved this study (Approval No. 20230439). Every experiment for the Care and Use of Laboratory Animals was carried out according to the guidance.
Masson Staining
Decapitation killed mice, and atrial tissues were removed and fixed with paraformaldehyde for 4 h. The fixed atrial tissues were embedded in paraffin and cut into thin slices of 4 μm thickness by sectioning, deparaffinized in xylene and rehydrated in alcohol, and then stained with Masson staining, and finally observed the atrial fibrosis under the microscope.
ROS Detection
The DHE kit was used to identify the formation of ROS in atrial myocytes. Mouse atrial tissues encased in paraffin were frozen at −80 °C and sliced into 5 μm sections. After being stained with the DHE reaction mixture, the slices were seen under a laser confocal microscope.
TUNEL Staining
The atrial tissue samples were fixed and dehydrated. They were fixed with 4% paraformaldehyde at room temperature and then rinsed with PBS. They were then dehydrated with gradient ethanol. The tissues were pretreated with proteinase K to expose DNA fragments. The samples were then treated with 0.1% Triton X-100 and 0.1% sodium citrate solution to increase the permeability of the cell membrane. Finally, they were incubated with TUNEL reaction solution at 37 °C for 60 min. DAPI staining solution was added to the samples and incubated at room temperature in the dark. Finally, they were observed under a fluorescence microscope.
RT-PCR
Utilizing TRizol, total RNA was isolated from the atrial tissues of mice and transcribed backward into complementary DNA (cDNA). In an ABI Prism 7700 real-time PCR system, the mRNA was amplified by RT-qPCR using the SYBR Green reagent. The following were the primer sequences: TNF-α forward primer: 5'-CCCTCACACTCAGATCATCTTCT-3', reverse primer: 5'-GCTACGACGTGGGGCTACAG-3'. IL-1β forward primer: 5'-CTTCCAGGATGAGGACATGAG-3', reverse primer: 5'-TCACACACCAGCAGGTTATCG-3'. IL-6 forward primer: 5'-CTTCCATCCAGTTGCCTTCT-3', reverse primer: 5'-CCTTCTGTGACTCCAGCTTATC-3'.
Western-Blot
Using RIPA buffer, total protein isolation was carried out in cells or tissues, and the BCA kit was used to determine the concentration. Protein was electroblotted onto PVDF membranes that had been blocked with BSA for 1 h at room temperature after being uploaded onto SDS-PAGE. Membranes were probed overnight at 4 °C using primary antibodies, and then they were incubated for 1.5 h at room temperature with HRP-tagged goat anti-rabbit IgG (1:20000, ab205718, Abcam). Membranes were then treated with a developing solution, and image software was used to measure the band intensities. 10 Antibody information: AMFR (1:1000, 16675-1-AP, Proteintech). α-SMA (1:6000, 14395-1-AP, Proteintech). COL1A1 (1:1000, ab270993, Abcam). Cleaved-caspase3 (1:5000, ab214430, Abcam). Cleaved-PARP (1:1000, ab32064, Abcam). SOD1 (1:1000, ab308181, Abcam). GAPDH (1:2500, ab9485, Abcam).
Co-IP
Take 50 mg tissue, add 1 mL RIPA lysis buffer, and homogenize on ice. Centrifuge at 4 °C, 12,000g for 15 min, and take the supernatant to get the total protein. Determine the protein concentration by the BCA method. Take 500 μg total protein + 20 μL Protein A/G magnetic beads, and incubate at 4 °C with rotation for 1 h to reduce nonspecific binding). Incubate with SOD1 antibody or IgG at 4 °C with rotation for 4 h. Add 20 μL Protein A/G magnetic beads, and incubate at 4 °C with rotation for 2 h. Wash the magnetic beads 3 to 4 times with cold PBS to remove unbound proteins. Add 2 × SDS loading buffer, boil at 95 °C for 5 min, and centrifuge to take the supernatant (for WB, detection of AMFR bands).
SOD1 Ubiquitination
SDS lysis buffer was used to lyse the atrial tissue. Following ten minutes of heat denaturation at 95 °C, the lysate was diluted ten times using IP buffer. The lysate was then centrifuged for 10 min at 4 °C at 15,000 r/min, immunoprecipitated using anti-SOD1 antibody, and immunoblotted using an anti-Ub antibody to check for SOD1 ubiquitination.
Statistical Analysis
The Shapiro-Wilk test was used to determine whether the data obeyed normality, and SPSS 22.0 was utilized for statistical analysis. The data were presented as mean ± standard deviation (SD). Student's t-test was used for equal variance (two groups) and Welch's t-test was used for unequal variance (two groups). After determining the significance of differences between several groups using a one-way ANOVA, the Bonferroni post hoc test was conducted. *P< .05, ***P < .001 were considered statistically significant.
Results
AMFR is Highly Expressed in AF
The expression of AMFR in the GSE79768 expression profile in sinus rhythm atrial (n = 12) and AF patients (n = 14) was examined, and the differential gene results were projected into a volcano plot (Figure 1A). Plotting the AMFR expression values into a box plot revealed that the atrial tissue of AF patients had high levels of AMFR expression (Figure 1B).

AMFR is Highly Expressed in AF. (A) Volcano Plot of Differentially Expressed Genes in GSE79768 Expression Profile. (B)AMFR Expression in Atrial Tissue of Patients with Sinus Rhythm and AF in GSE79768 Expression Profile.
Knockdown of AMFR Ameliorates Atrial Fibrosis in AF
The expression of AMFR in the atrial tissues of mice in each group was detected by western-blot assay, and the protein expression of AMFR in the atrial tissues of Ang II-induced AF mice was significantly increased, and the protein expression was further increased after transfection with AMFR. However, after transfection of sh-AMFR, AMFR expression in the atrial tissues of AF mice was significantly reduced, indicating a better transfection effect (Figure 2A). Masson staining results showed that the fibrotic lesions of atrial hemorrhage in mice in the AF group were aggravated after AMFR transfection, while the fibrosis was alleviated after sh-AMFR transfection (Figure 2B). In addition, the expression of fibrosis-related proteins α-SMA and COL1A1 was detected. Consistent with the results of Masson staining, the expression of α-SMA and COL1A1 in the atrial tissue of mice in the AF group increased, AMFR further promoted the expression of the protein, and sh-AMFR reduced the expression of the protein (Figure 2C).
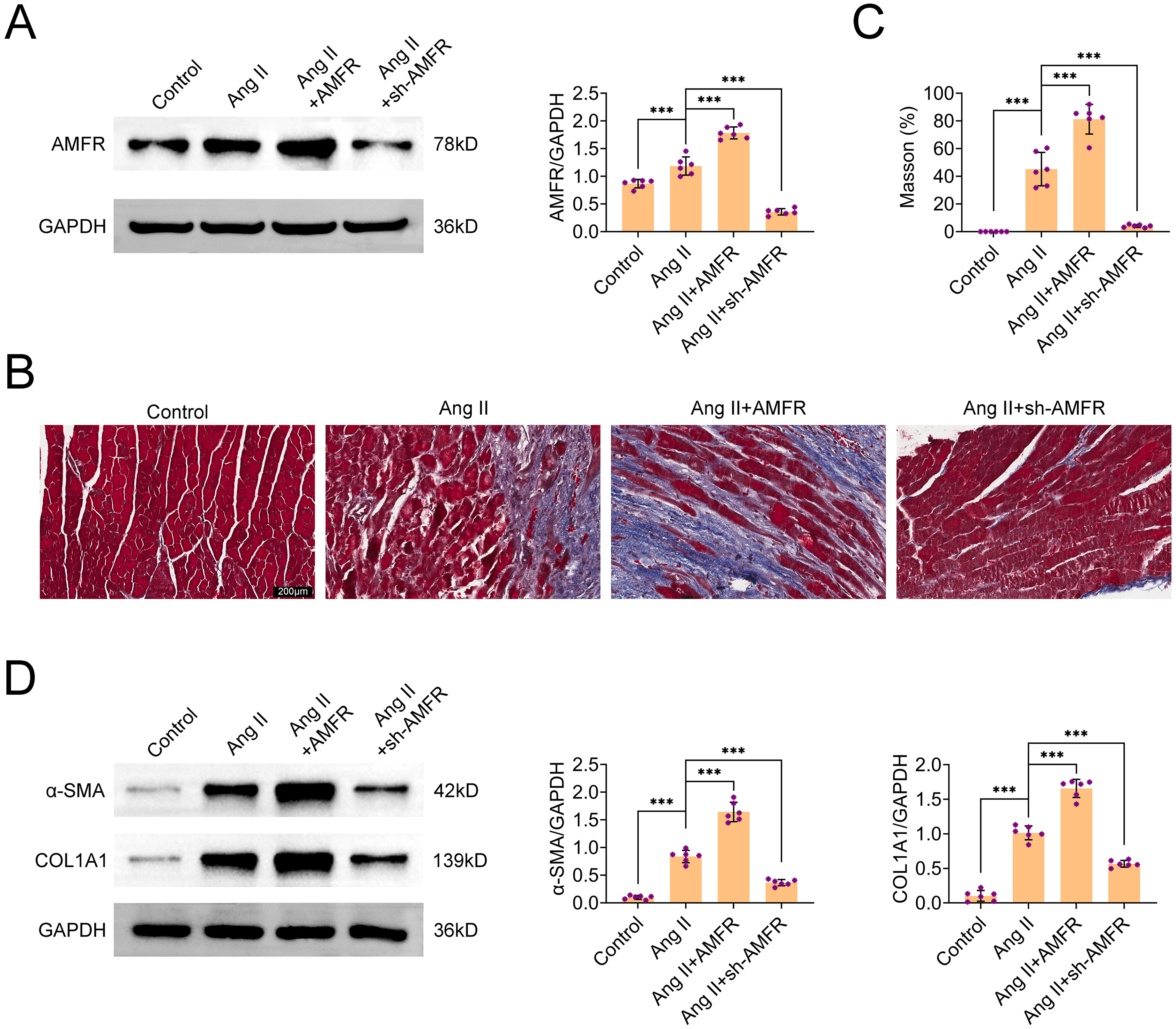
Knockdown of AMFR Ameliorates Atrial Fibrosis in AF. (A) Western-Blot Detection of AMFR Protein Expression in Atrial Tissue of Each Group of Mice After Transfection. (B) Masson Staining was Used to Examine the Fibrosis of Atrial Tissue in Each Group of Mice After Transfection. (C) Western-Blot Detection of COL1A1 and α-SMA Protein Expressions in Atrial Tissues of Mice in Each Group After Transfection.
Knockdown of AMFR Improves Inflammation and ROS Production in AF Tissue
The findings demonstrated a considerable increase in the mRNA expression of several inflammatory factors in the atrial tissue of mice in the AF group. Knocking down AMFR prevented atrial tissue inflammation in AF mice, as evidenced by the increased expression of inflammatory factors in mice transfected with AMFR compared to the AF group and the lower expression of inflammatory factors in mice transfected with sh-AMFR (Figure 3A). To demonstrate the effect of AMFR on atrial ROS in AF mice, ROS content in the atrial tissues of mice in each group was examined by the DHE method, and the results showed that the atrial tissues of mice in the AF group showed an increase in ROS, and transfection of AMFR exacerbated ROS production in the atrial tissues of AF mice, while knockdown of AMFR decreased ROS production in the atrial tissues of AF mice (Figure 3B).
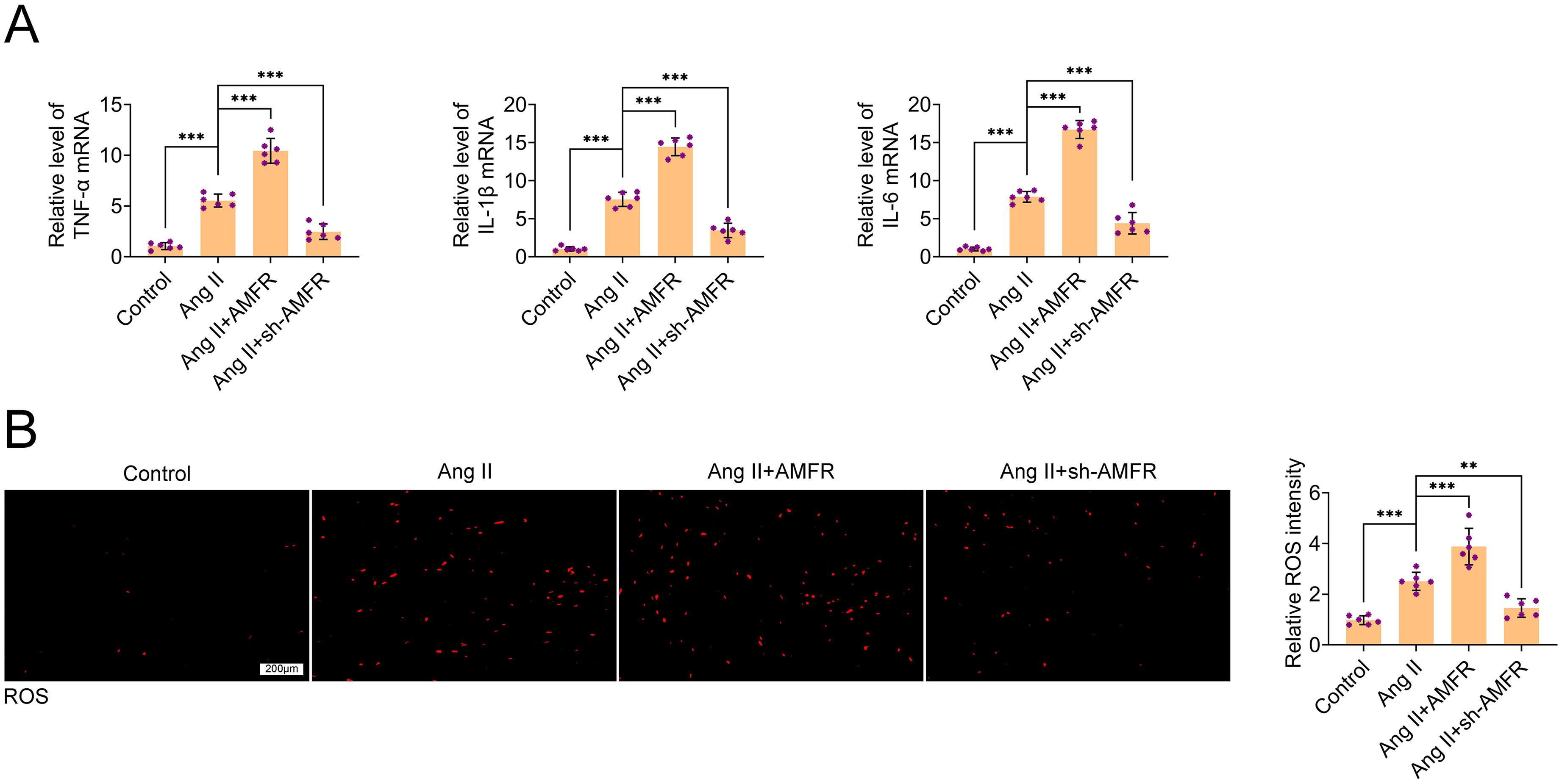
Knockdown of AMFR Improves Inflammation and ROS Production in AF Tissue. (A) PCR was Used to Detect the mRNA Expression of TNF-α, IL-1β, and IL-6 in the Atrial Tissue of Each Group of Mice After Transfection. (B) DHE Staining to Examine the Production of ROS in the Atrial Tissue of Each Group of Mice after Transfection.
Knockdown of AMFR Attenuates Apoptosis in AF Tissues
The effect of AMFR on apoptosis in atrial tissue cells was detected by TUNEL staining, and the number of apoptotic cells in atrial tissue cells was increased in AF mice. The number of apoptotic cells in the atrial tissue of mice in the AMFR group was greater than that of mice in the AF group. In contrast, the number of apoptotic cells in the atrial tissue of mice in the sh-AMFR group was decreased (Figure 4A). In addition, protein expression of apoptosis-related proteins cleaved-caspase3 and cleaved-PARP in atrial tissues was also detected by western-blot, and in agreement with the results of TUNEL staining, protein expression of cleaved-caspase3 and cleaved-PARP in atrial tissues of mice in the AF group was increased. AMFR aggravated this phenomenon, whereas sh-AMFR attenuated it (Figure 4B), suggesting that the knockdown of AMFR inhibited apoptosis in the atrial cells of AF mice.

Knockdown of AMFR Attenuates Apoptosis in AF Tissues. (A) TUNEL Staining was Used to Examine Apoptotic Cells in the Atria of Each Group of Mice After Transfection. (B) Western-Blot Detection of Protein Expression of Cleaved-Caspase3 and Cleaved-PARP in Atrial Tissue of Each Group of Mice After Transfection.
AMFR Ubiquitin-Degrades SOD1 Expression
Western-blot analysis of the protein expression of SOD1 in atrial tissues revealed that mice in the AF group had lower levels of SOD1 expression in atrial tissues. The AMFR group of mice had lower atrial tissue SOD1 expression than the AF group, while the sh-AMFR group of mice had considerably higher SOD1 protein expression (Figure 5A). Immunoprecipitation experiments confirmed the interaction between AMFR and SOD1 (Figure 5B). Compared with the control group, the ubiquitination level of SOD1 increased in the Ang II group. Transfection of sh-AMFR on the basis of Ang II treatment reduced the ubiquitination level of SOD1. Transfection of AMFR aggravated the ubiquitination level of SOD1. After treatment with Ang II + AMFR + MG132, the ubiquitination level of SOD1 increased further. This is because MG132 is a proteasome inhibitor that blocks the degradation of proteins in the ubiquitin-proteasome pathway. The ubiquitin-tagged protein (SOD1) cannot be degraded, resulting in the accumulation of ubiquitinated proteins and an increase in total SOD1 (Figure 5C). This result also clarifies the mechanism by which AMFR regulates SOD1 through the ubiquitin-proteasome pathway.

AMFR Ubiquitin-Degrades SOD1 Expression. (A) Western-Blot Detection of SOD1 Protein Expression in Atrial Tissue of Each Group of Mice After Transfection. (B) Immunoprecipitation Experiments Confirmed the Interaction Between AMFR and SOD1. (C) Ubiquitination Assay to Detect the Ubiquitination Level of SOD1 in the Atrial Tissue of Each Group of Mice After transfection.
Knockdown of AMFR Improves AF Injury by Stabilizing the Expression of SOD1
To demonstrate that AMFR acts through SOD1 in AF, sh-AMFR and sh-SOD1 were cotransfected in mice, and a significant reduction in both AMFR and SOD1 levels was seen (Figure 6A). The expression of fibrosis-related genes (α-SMA and COL1A1) in the atrial tissues of mice in each group was examined by western blot. The results showed that both knockdowns of AMFR and overexpression of SOD1 significantly reduced fibrosis-related genes compared with the expression of fibrotic proteins was increased in atrial tissues of mice co-transfected with sh-AMFR + sh-SOD1 compared with the two groups mentioned above (Figure 6B), suggesting that knockdown of AMFR was attenuating fibrosis through SOD1 in AF mice. In addition, inflammatory factors and ROS production were also detected by PCR and DHE, respectively, and both knockdown of AMFR and overexpression of SOD1 significantly decreased inflammatory factors (Figure 6C) and ROS production (Figure 6D), inflammation and ROS production were increased in atrial tissues of mice co-transfected with sh-AMFR + sh-SOD1, suggesting that knockdown of AMFR inhibits inflammation and oxidative stress through SOD1 in AF mice. Finally, the expression of apoptosis-related proteins (cleaved-caspase3 and cleaved-PARP) was detected. The results showed that both the knockdown of AMFR and the overexpression of SOD1 significantly reduced the expression of apoptosis-related proteins, while the expression of apoptosis proteins in the atrial tissue of mice co-transfected with sh-AMFR + sh-SOD1 increased (Figure 6E), suggesting that the knockdown of AMFR alleviated cell apoptosis in AF mice through SOD1.

Knockdown of AMFR Improves AF Injury by Stabilizing the Expression of SOD1. (A) Western Blot was Used to Detect the Protein Expression of AMFR and SOD1 in the Atrial Tissue of Each Group of Mice After Transfection. (B) Western-Blot Detection of COL1A1 and α-SMA Protein Expressions in Atrial Tissues of Mice in Each Group After Transfection. (C) PCR was Used to Detect the mRNA Expression of TNF-α, IL-1β, and IL-6 in the Atrial Tissue of Each Group of Mice After Transfection. (D) DHE Staining to Examine the Production of ROS in the Atrial Tissue of Each Group of Mice After Transfection. (E) Western-Blot Detection of Protein Expression of Cleaved-Caspase3 and Cleaved-PARP in Atrial Tissue of Each Group of Mice After Transfection.
Discussion
In this study, AMFR expression was increased in the atrial tissue of patients with AF compared with those in sinus rhythm. Furthermore, injection of Ang II in mice resulted in increased AMFR expression. AMFR overexpression exacerbated Ang II-induced AF in mice, whereas reduced AMFR expression attenuated atrial fibrosis, apoptosis, inflammation, and ROS production in AF mice. In addition, AMFR was found to ubiquitinate SOD1 and reduce SOD1 expression, and the knockdown of SOD1 counteracted the attenuating effect of sh-AMFR on AF.
Atrial fibrosis is a clear manifestation of structural remodeling in patients with AF and is the structural basis for the maintenance of AF. Most of the pathological changes associated with atrial fibrosis are caused by abnormal collagen metabolism and aberrant expression of fibrogenic genes. 11 Knockdown of AMFR inhibited atrial fibrosis in AF mice by suppressing the expression of COL1A1 and α-SMA.
Ang II is a contributing factor to AF and can increase the inducibility of AF by inducing inflammation, 12 oxidative stress, 13 and atrial fibrosis. 14 The pathogenesis of AF is linked to inflammation, and too many inflammatory mediators can change the structural and electrical characteristics of atrial tissue. 15 AMFR transfection resulted in a further increase in Ang II-induced TNF-α, IL-1β, and IL-6 mRNA levels, whereas sh-AMFR transfection decreased the levels of the above inflammatory factors, suggesting that knockdown of AMFR inhibits inflammation in atrial tissues of AF mice.
ROS are not only a causative agent of AF, but also a factor in the maintenance of arrhythmias after the onset of AF. Effective treatment approaches for AF include methods to increase antioxidant enzymes or genetically or pharmacologically inhibit oxidative enzymes, which lowers ROS in atrial myocytes. According to this study, AMFR increased the generation of ROS produced by Ang II, but sh-AMFR decreased this production. This suggests that AMFR knockdown prevents atrial oxidative stress in AF mice.
SOD1 is an important enzyme that is mainly responsible for catalyzing the conversion of superoxide into oxygen and hydrogen peroxide, thus acting as an antioxidant in cells. SOD1 is essential in scavenging cellular ROS and cardiac autonomic remodeling due to AF can be modulated by regulating SOD1. 16 Overexpression of SOD1 attenuates apoptosis and inflammatory responses during myocardial ischemia-reperfusion injury. 17 This study further supported the role of SOD1 in Ang II-induced AF in mice by showing that overexpression of SOD1 decreased the levels of expression of genes linked to apoptosis and fibrosis, as well as suppressed the generation of ROS and inflammatory factors.
AMFR acts as an E3 ubiquitin ligase with ubiquitination. In hepatocellular carcinoma, AMFR mediates ubiquitination and degradation of PDL1. 18 In this study, it was found that AMFR may have a role in ubiquitinating SOD1 by querying ubiquitination websites, so SOD1 ubiquitination assay experiments were carried out, and it was found that AMFR transfection exacerbated the ubiquitination level of SOD1, while sh-AMFR transfection attenuated the ubiquitination level of SOD1, suggesting that AMFR mediates the ubiquitination of SOD1.
In conclusion, this study demonstrates for the first time that knockdown of AMFR promotes SOD1 expression by inhibiting SOD1 ubiquitination levels and attenuates atrial fibrosis in AF mice by regulating SOD1. However, this study has some shortcomings. Due to objective limitations, no clinical samples were collected to test AMFR expression. Cellular experiments were also not performed to conduct an in-depth study of the ubiquitination mechanism, which will be included in future studies.
The conclusion in this study may not fully extend to paroxysmal AF driven by electrophysiological triggers. We also highlight the need for future validation in other models (eg, rapid atrial pacing or spontaneous AF models).
Footnotes
Ethical Considerations
Ethical approval was obtained from the Ethics Committee of the Dalian Medical University (Approval No. 20230439). All animal experiments should comply with the ARRIVE guidelines and conducted in accordance with the UK Animals (Scientific Procedures) Act, 1986 and associated guidelines, EU Directive 2010/63/EU for animal experiments/National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978).
Author Contributions
All authors contributed to the study conception and design. Material preparation and the experiments were performed by Shanshan Geng. Data collection and analysis were performed by Zhongbao Ruan and Lianghong Ying. The first draft of the manuscript was written by Zhou Liu and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.The present study are available from the corresponding author on reasonable request.