
Abstract
Intestinal ischemia is a serious condition that may lead to both local and systemic inflammatory responses. Restoration of blood supply (reperfusion) to ischemic tissues often increases the extent of the tissue injury. Cysteine-rich angiogenic inducer 61 (Cyr61)/CCN1 is an extracellular matrix-associated signaling protein that has diverse functions. CCN1 is highly expressed at sites of inflammation and wound repair, and may modify cell responses. This study aimed to investigate regulation and cellular distribution of CCN1 in intestinal ischemia/reperfusion (I/R) injury in pigs. After intestinal I/R, increased expression of CCN1 was detected by quantitative RT-PCR, Western blot analysis and immunohistochemistry compared with non-ischemic intestine. Immunoflorescence staining revealed that CCN1 was mainly up-regulated in intestinal mucosa after intestinal I/R. Microvillus epithelial cells and vascular endothelial cells were strongly positive for CCN1 in intestinal I/R, while natural killer cells and/or subsets of neutrophils were only modestly positive for CCN1. Furthermore, blood samples taken from the portal and caval veins during ischemia and after reperfusion showed no change of the CCN1 levels, indicating that CCN1 was locally regulated. In conclusion, these observations show, for the first time, that the CCN1 molecule is up-regulated in response to intestinal I/R in a local manner.
Introduction
The intestines are very sensitive to ischemia/reperfusion (I/R) injury and play a pivotal role in the induction of systemic inflammatory response syndrome and multiple organ dysfunction syndrome following intestinal necrosis.1,2 Mesenteric ischemia may occur owing to a restriction of intra-abdominal blood supply as a result of physiological conditions (e.g. exercise, stress) or in pathophysiological events such as strangulation, adhesions, thrombosis, embolies, trauma, shock, complications to surgery or organ transplantation. 3 Untreated ischemia leads to cellular dysfunction and necrosis. However, restoration of blood flow after ischemia (reperfusion) may result in an increase in tissue damage. The sudden reintroduction of oxygen stimulates the production of free radicals that promote or accelerate necrosis. Present protective treatment of I/R damage includes the application of various extracellular signaling molecules and the manipulation of a variety of intracellular signaling pathways. 4 Therefore, increasing knowledge of the cellular and molecular events that take place during I/R-induced intestinal injury may be important for better I/R treatment. 5
Cells are exposed to a number of extracellular proteins in the extracellular matrix (ECM). In turn, these proteins provide structural and biochemical support to the surrounding cells and trigger signals in cells expressing the corresponding receptors. A subset of ECM, known as matricellular proteins, embraces structurally unrelated extracellular macromolecules that do not play a primary role in tissue architecture, but modulate cell–cell and cell–matrix interactions. 6 Expression of matricellular proteins is generally low in normal adult tissues but is up-regulated during development and in response to injury. 7 The cysteine-rich angiogenic inducer 61 (Cyr61)/CCN1 protein belongs to the matricellular CCN family, which comprises six members in mammals.8,9 In close similarity to the other members, it has four conserved domains, with similarity to insulin-like growth factor-binding protein (IGFBP), the von Willebrand factor type C repeat (vWC), the thrombospondin type I repeat (TSR) and the carboxyl-terminal (CT) cysteine knot motif, respectively. vWC, CT and TSR domains apparently provide several integrin-binding sites that may mediate the functions of CCN1. 10 Hence, CCN1 can exhibit diverse and different functions based on its modular structure and its ability to bind different integrins, thereby involving it in distinct and complex processes such as cell adhesion, migration, proliferation, apoptosis, and senescence. 9 For instance, CCN1 induces apoptosis in fibroblasts through α6β1-heparan sulfate proteoglycan-mediated interaction but promotes survival of endothelial cells by interacting with αvβ3. 10 CCN1 may act as an autocrine/paracrine mediator to activate the expression of key inflammatory cytokines.11–13 Studies of Ccn1-deficent mice have revealed that CCN1 plays an essential role in embryonic development. 14 CCN1 has been shown to be transcriptionally activated by various factors such as growth hormone, vitamin D3 and environmental stress such as hypoxia, ultraviolet light and mechanical stretch in different experimental settings. CCN1 is also induced by bacterial and viral infections and by pro-inflammatory cytokines such as IL-1 and TNF-α. 10 Under homeostatic conditions, CCN1 is expressed at low levels in most adult tissues by different cell types, including endothelial cells, epithelial cells, fibroblasts and vascular smooth muscle cells. 9 In addition, flow cytometry studies have revealed constitutive expression of CCN1 in circulating human leukocytes. 15 More recent reports have also described its role in immune cell migration, inflammation and ischemic injury.9,16–20 However, the expression and role of CCN1 in the intestinal tract has been poorly investigated. 21 Our current studies investigated the regulation of CCN1 in intestinal tissue subjected to I/R in pigs. CCN1 protein was up-regulated in the intestinal tissue that was subjected to ischemia but unchanged in the portal or the systemic circulation, suggesting that CCN1 was locally regulated. The up-regulation of CCN1 was more concentrated in the mucosal area of intestinal I/R where epithelial—and vascular endothelial cells were identified to be among the cell types that were most strongly positive for CCN1.
Materials and methods
Anesthesia
Anesthesia was induced with an i.m. injection of ketamine 15 mg/kg (Warner Lambert, Morris Plains, NJ, USA), azaperone 1 mg/kg (Janssen-Cilag Pharma, Vienna, Austria) and atropine 0.02 mg/kg (Nycomed Pharma, Asker, Norway). After tracheotomy, anesthesia was maintained with a continuous inhalation of isoflurane (1–1.5%) (Abbott Scandinavia AB, Kista, Sweden) and a mixture of N2O (30%) and O2 (70%). Fentanyl 160 µg/kg/h (Alpharma, Oslo, Norway) was administered as a continuous i.v. infusion. The animals were ventilated with a Servo Ventilator 900 C (Siemens-Elema, Solna, Sweden). Ventilation was adjusted to a pCO2 of 5–6 kPa (37.5–45.0 mmHg). A continuous infusion of 0.9% saline (20 ml/kg/h) was administered as fluid replacement.
Surgical procedures
Surgery was performed under sterile conditions. Initially, a tracheotomy was performed for mechanical ventilation. Both internal jugular veins and the portal vein were cannulated for blood sampling, measuring of central venous pressure (CVP) and infusions of fluids. Arterial pressure (AP) was measured through a catheter placed in a carotid artery, and rectal temperature was measured with a thermistor probe. A Foley catheter was placed in the urinary bladder via a cystotomy to measure urine output. The animals were placed on a heating pad adjusted to 38℃. Arterial and venous blood gases, in addition to hemodynamic recordings (CVP and AP), were regularly measured throughout the 6.5h observation period.
Animals and experimental design
Four juvenile Norwegian Landrace pigs, two females and two males, weighing 26.5–30.0 kg were subjected to jejunal ischemia/reperfusion (JI/R) as follows. The pigs were fasted 12 h with water ad libitum prior to surgery. The abdominal cavity was accessed through a midline laparotomy and jejunum made accessible. Local ischemia was provoked by clamping a part of the segmental jejunal blood supply resulting in a 30-cm central zone of complete ischemia (IS) and two surrounding edge zones of marginal tissue hypoxia (EZ). Following 6 h of occlusion, the vascular clamp was removed and 30 min of recirculation allowed before intestinal biopsies were taken from the IS, the EZ and a distant non-ischemic jejunal intestine (NIS) segment. Each biopsy was divided in two parts and fixated overnight in 10% formalin, and snap frozen in liquid nitrogen until further analysis. Portal and caval vein blood samples were collected: before induction of ischemia [baseline (BS)], at 3 and 6 h after ischemia, and 5, 10 and 30 min after reperfusion (Figure 1). For CCN1 analysis, whole blood was collected into citrate-treated Vacutainers (Becton Dickinson, Franklin Lakes, NJ, USA). For TNF-α analysis heparin-treated Vacutainers were used for whole blood collection. Samples were immediately centrifuged at 2300 g for 5 min at room temperature (20–22℃). The plasma was harvested and centrifuged at 16,000 g for 5 min at room temperature and frozen in sealed polypropylene tubes at −70℃ until later analysis. At the end of the experiment, the animals were killed with a lethal dose of pentobarbital (pentobarbital natrium; Abbott Laboratories, Solna, Sweden) as previously described.
22
The experiments were approved by the local animal care committee and conducted in accordance with national animal welfare guidelines.
Schematic outline of portal and systemic blood samples collection of the pigs during IS. Samples were taken before induction of ischemia (baseline), 3 h after induction of ischemia, 6 h after induction of ischemia or before reperfusion, and 5, 10 and 30 min after re-establishment of intestinal circulation.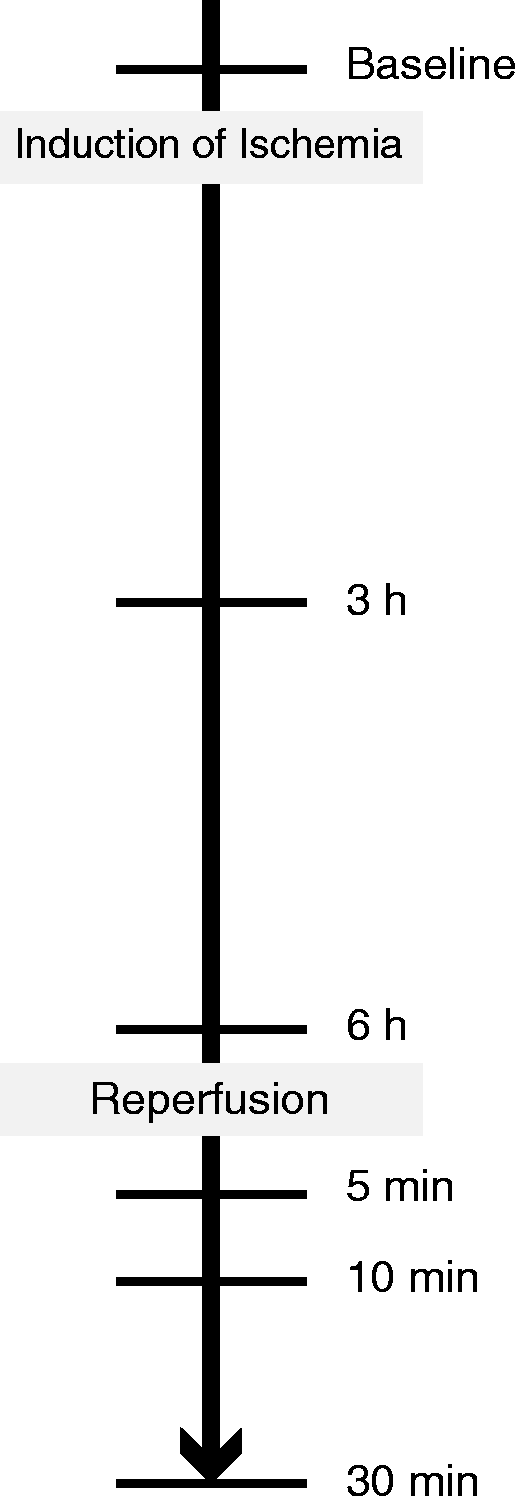
Measurement of CCN1 and TNF-α
CCN1 protein and TNF-α were measured in the blood samples with a commercially available ELISA [CCN1: EIA-5108 (DRG Diagnostics, Marburg, Germany); porcine TNF-α: DuoSet (R&D Systems, Minneapolis, MN, USA)] according to the manufacturer’s instructions. Acquired absorbance data were analyzed by the VictorTM X5 2030 Multilabel Reader (Perkin Elmer, Waltham, MA, USA).
Quantitative RT-PCR for CCN1 mRNA
Total RNA was isolated from snap-frozen tissues by means of RNeasy mini kit (Qiagen, Düsseldorf, Germany) according to the manufacturer’s instructions. Samples were treated with an RNase-free DNase set (Qiagen) to remove genomic contamination. RNA quality was controlled at random using the Experion automated electrophoresis system (BioRad Laboratories, Hercules, CA, USA). Reverse transcription was done using a TaqMan RT kit (Applied Biosystems, Foster City, CA, USA). RT-qPCR was run in triplicate using a relative standard curve approach. CCN1-TFRC (CD71/transferrin receptor) was analyzed using pre-inventoried TaqMan assays according to the manufacturer’s instructions (Applied Biosystems). CCN1 expression was analyzed by SYBR Green technology, using 2 × Universal master mix (Applied Biosystems), cDNA template, and 30 µM sense and anti-sense primers. SYBR green primers were designed to span intron junctions. Specificity of the SYBR green primers was assessed by melting point analysis. All assays were optimized to a threshold cycle (CT) value between 20 and 30 cycles. The primers utilized to amplify target genes were as follows: CCN1 forward 5′-TCG GCA GCC TGA AAA AGG GCA, reverse 5′-TCG CAG CGG AAG CGC ATC TT, TFRC, forward 5′-CTT TGG AGT TAT TAA GGG CTT TG, reverse 5′-GGC AAG GTT CAA TAG GAG AC.
Western blotting
Snap-frozen tissue samples were gently homogenized in RIPA buffer with protease and phosphatase inhibitors (Thermo Fisher Scientific, Waltham, MA, USA) prior to 30 min incubation on ice. The lysates were centrifuged twice at 10,000 g for 10 min to remove insoluble material, and the total protein concentration was determined by Bradford assay (BioRad). Proteins (70 µg) were separated by 10% SDS-PAGE and transferred to a Hybond-P membrane (Amersham Bioscience, Amersham, UK). Blocking was carried out by incubating membranes with 5% non-fat milk in Tris-buffered saline with 0.05% Tween-20 (TBST) for 1 h at room temperature. Membranes were incubated with primary Abs against mouse-CCN1 (AF 4055; R&D Systems) and goat anti-human GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight (18–24 h) at 4℃. GAPDH was used as an internal control to ensure that equal amounts of sample were applied for electrophoresis and immunoblotting. After washing in TBST three times, the membranes were incubated with a secondary anti-sheep (sc-2473) or anti-rabbit Ab (sc-2004) (both from Santa Cruz) for 1 h at room temperature, and immunoreactive bands were visualized by LumiGlo Chemiluminescent Substrate system (KPL, Gaithersburg, MD, USA). Displayed images and semi-quantitative densitometry analysis was performed using ImageJ software (Image Processing and Analysis in Java; NIH, Bethesda, MD, USA).
Immunohistochemistry
Paraffin-imbedded intestinal tissues were cut in 6 -µm sections, deparaffinized and rehydrated. For immunofluorescence staining, Ag-retrieval was performed in Dako retrieval solution (Dako, Ely, UK) for 30 min at 95℃. The sections were blocked in PBS containing 0.1% Tween-20 PBS, 5% BSA and 5% donkey serum for 30 min at room temperature prior to overnight incubation at 4℃ with purified primary sheep anti-mouse CCN1 (AF4055; R&D Systems) in combination with NK cells: mouse anti-Pig CD11R1 (MCA1220GA; AbD Serotec, Kidlington, UK). For detection of primary Abs, the sections were incubated at room temperature in a mixture of donkey anti-Sheep IgG AlexA 488 (A11015; Molecular Probes, Eugene, OR, USA) and donkey anti-mouse IgG AlexA 594 (A-21203; Life Technologies, Renfrew, UK) according to the manufacturer's instructions. Slides were mounted with Prolong Gold antifade (Life Technologies) and left in the dark for at least 24 h at 4℃. Three overlapping single-color images (red and green) and differential interference contract were captured using AxioVision software (Zeiss Axiocam; Carl Zeiss, Jena, Germany).
Statistical analysis
Statistical analyses were performed by one-way ANOVA with Bonferroni correction or by Students t-test, using Prism software (GraphPad, La Jolla, CA, USA). Differences were considered significant at the level of P < 0.05.
Results
Animals subjected to JI/R suffer tissue injury
As assessed from mean arterial pressure, mean airway pressure and systemic oxygenation (among others), the cardiopulmonary function of the pigs remained stable throughout the study (data not shown). Histological examination of the intestinal biopsies revealed mucosal damage with a reduction in mucosal thickness in the IS and, in accordance with other reports, this reduction was mainly attributable to the loss in villous height, with a relative sparing of the depth of the crypts.
1
Edema and hemorrhage were prominent in the ischemic segment (IS), while less pronounced in edge zone segment (EZS). Furthermore, these analyses showed that the NIS were weakly inflamed as recruitment of the immune cells were also observed in these segments. Representative data are shown in Figure 2.
Representative photomicrographs stained with hematoxylin and eosin are shown, which includes the area of the jejunal intestine biopsies taken from NIS, EZS and IS. Section magnification is 1.25 × with an additional 5 × magnification of IS (below right). The IS segment demonstrates a marked villous damage, edema and hemorrhage, while the EZ segment shows less intense findings.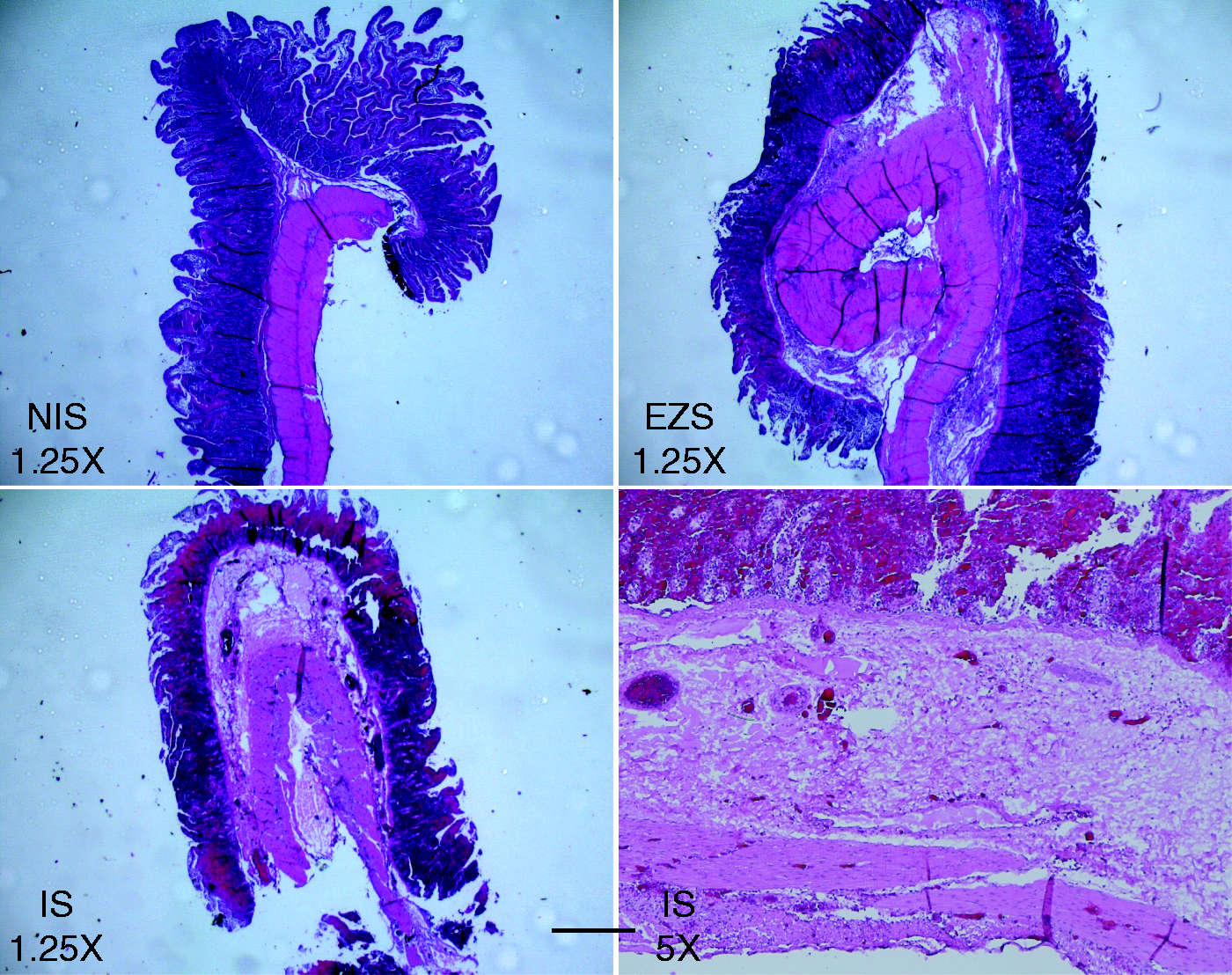
CCN1 expression in the ischemic intestine
In the adult, CCN proteins are generally expressed at a low level in most tissues but become elevated again in sites of inflammation and injury repair.8,23 We investigated first the regulation of CCN1 in biopsies from jejunum subjected to 6 h of ischemia followed by 30 min of reperfusion. Gene expression analysis revealed that CCN1 was induced by an average of threefold in the IS biopsies and twofold in the EZS compared with the distant NIS segment (Figure 3A). Western blot analysis confirmed CCN1 regulation at the protein level. The protein expression pattern of CCN1 in all pigs correlated to their transcript levels (Figure 3A, C). We observed that the sex of the pigs did not influence the expression of CCN1.
Regulation of CCN1 in the I/R intestine. (A) CCN1 mRNA was analyzed with RT-PCR using TFRC as a loading control. CCN1 expression was, on average, threefold higher in the IS (P < 0.005) and twofold in the edge zone segment (EZS) (P < 0.005) compared with the NIS. (B) Western blot analysis shows a CCN1 protein profile match with CCN1 gene expression where the IS and EZS band appeared stronger compared with the NIS band. GAPDH, used as an internal control, remained unchanged. (C) Histogram shows the result of the densitometric analysis of immunoreactivity bands in (B). Values are means ± SEM (n = 2 males and 2 females). All statistical analyses were analyzed by one-way ANOVA with Bonferroni or Students t-test.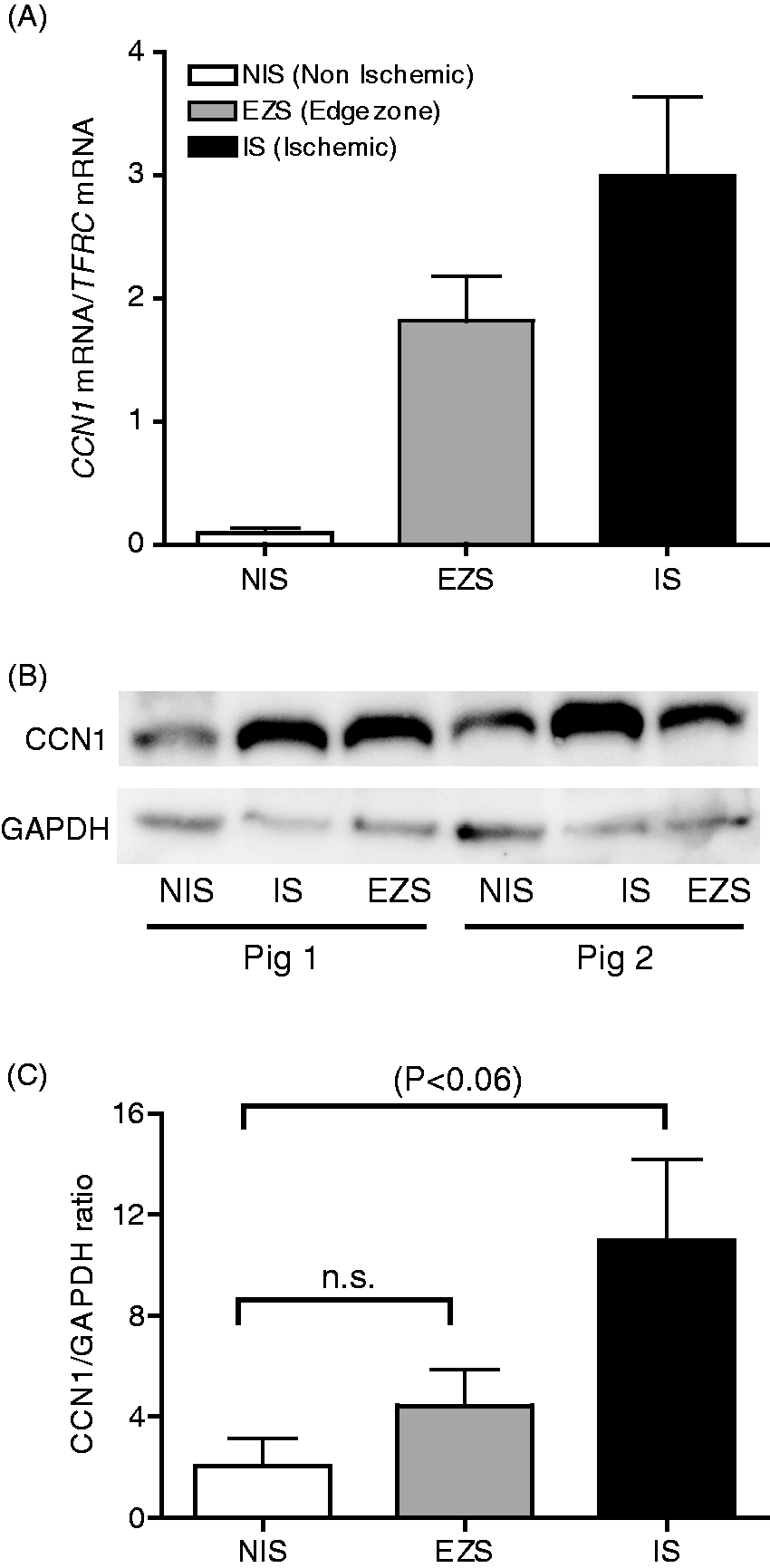
Localization of CCN1 in the JI/R-tissue and identifying CCN1-producing cells by immunohistochemistry
Immunofluorecense staining was performed to identify the tissue localization and cellular source of CCN1 in ischemic intestine. CCN1 immunoreactivity was concentrated in mucosa and around blood vessels. The staining intensity in the IS and EZS appeared similar, whereas very few positive cells were identified in the NIS biopsies (Figure 4A). The latter observation may be in accordance with weak inflammation that was observed in the NIS (Figure 2). Morphologic and anatomic evaluation showed that most of the epithelial cells on the surface of microvilli and vascular endothelial cells were highly positive for CCN1 in response to ischemia (Figure 4B). Unfortunately, it was not possible to tell whether the staining was due to binding of CCN1 to the cell surface or the cells producing CCN1, or a combination of both.
Immunofluorescence staining of CCN1 in the intestine biopsies. (A) Immunohistochemical analysis of intestine sections from microvillus area is shown. Sections from NIS, EZS and IS were stained with anti-CCN1 (AlexA488, green color) and DAPI (blue color) (left panel, 40×; right panel, 40×). CCN1 protein is observed in few areas in NIS, while many areas in the IS and EZS showed expression of CCN1. (B) CCN1-positive vascular endothelial cells (white circles) in serosal (left, 20×) and mucosal side (right, 40×) of IS are shown.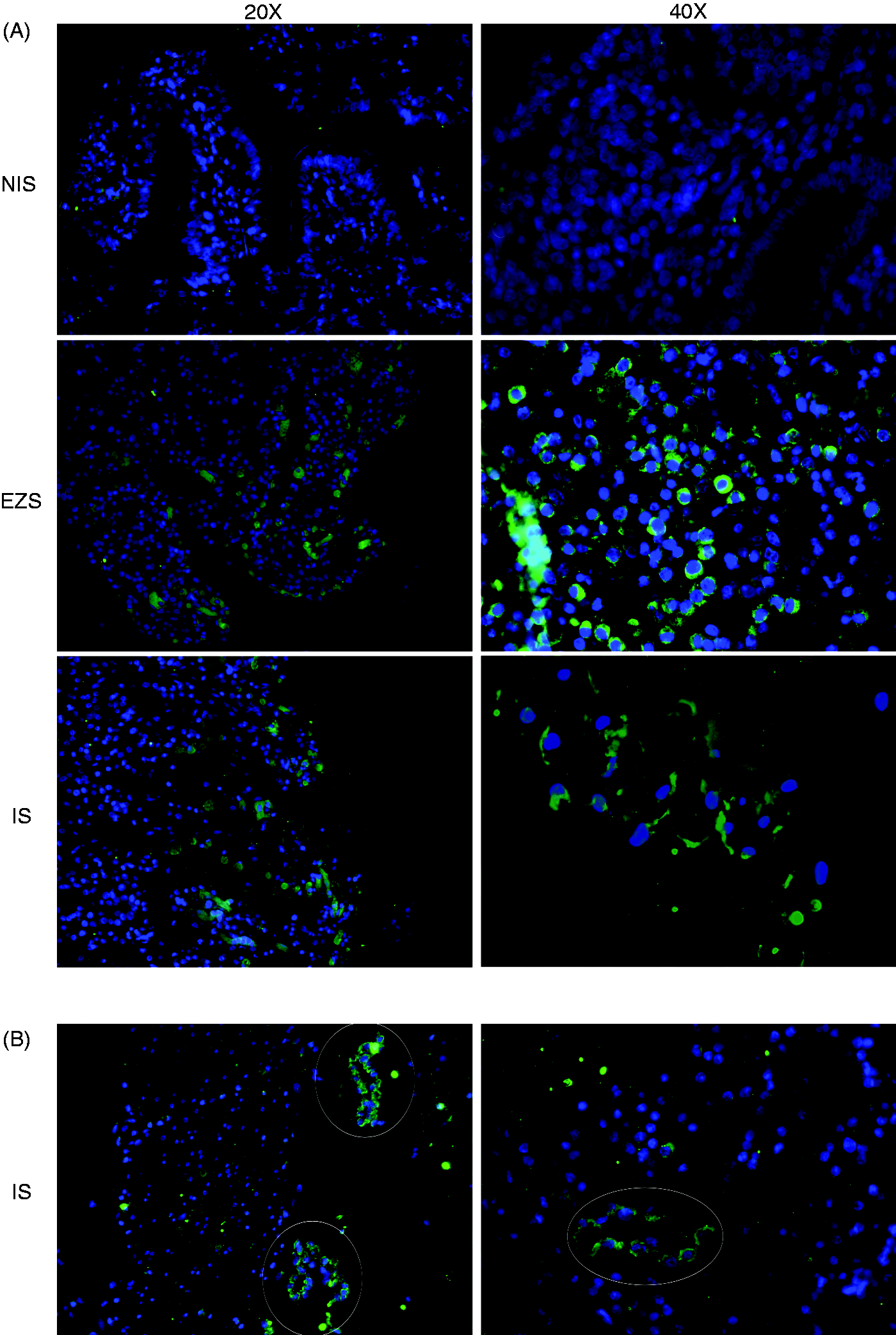
Immunofluorescence analyses with anti-CCN1 were limited to reveal the distribution and expression of CCN1 in the intestine based on morphological and anatomical evaluation. Hence, to characterize other cell types that may produce CCN1 we used double-label immunofluorescence staining of the biopsies. It has been shown that T cells and NK cells bind to CCN1, and may also up-regulate CCN1 in response to different stimuli such as pro-inflammatory cytokines.9,15,24 In order to find whether NK cells produce (or bind to) CCN1 in response to JI/R, NK cells were co-stained with anti-CCN1 for immunofluorecense staining. We found no NK cells (CD11R1+ cells) positive for CCN1 in the NIS (Figure 5A). However, we found some NK cells positive for CCN1 and in the brush border in both ischemic (IS) and edge zone (EZS) intestinal biopsies (Figure 5B, C). Altogether, these findings suggest that epithelial and endothelial cells are the main producers of CCN1 protein in ischemic intestines, while NK cells only produce modest amounts. We should mention that the Ab we used against NK cells also stains a subset of neutrophils (see ‘Discussion’).
Characterization of CCN1-producing cells in ischemic intestine. (A) Sections (40×) from non-ischemic sections were stained anti-CCN1 (green) and anti-CD11R1 (red color, NK cells and/or neutrophils) merged with DAPI (blue). Sections (20×) for (B) EZS and (C) IS are either stained alone as in (A) or additionally double-stained with anti-CCN1 and anti-CD11R1 (merged).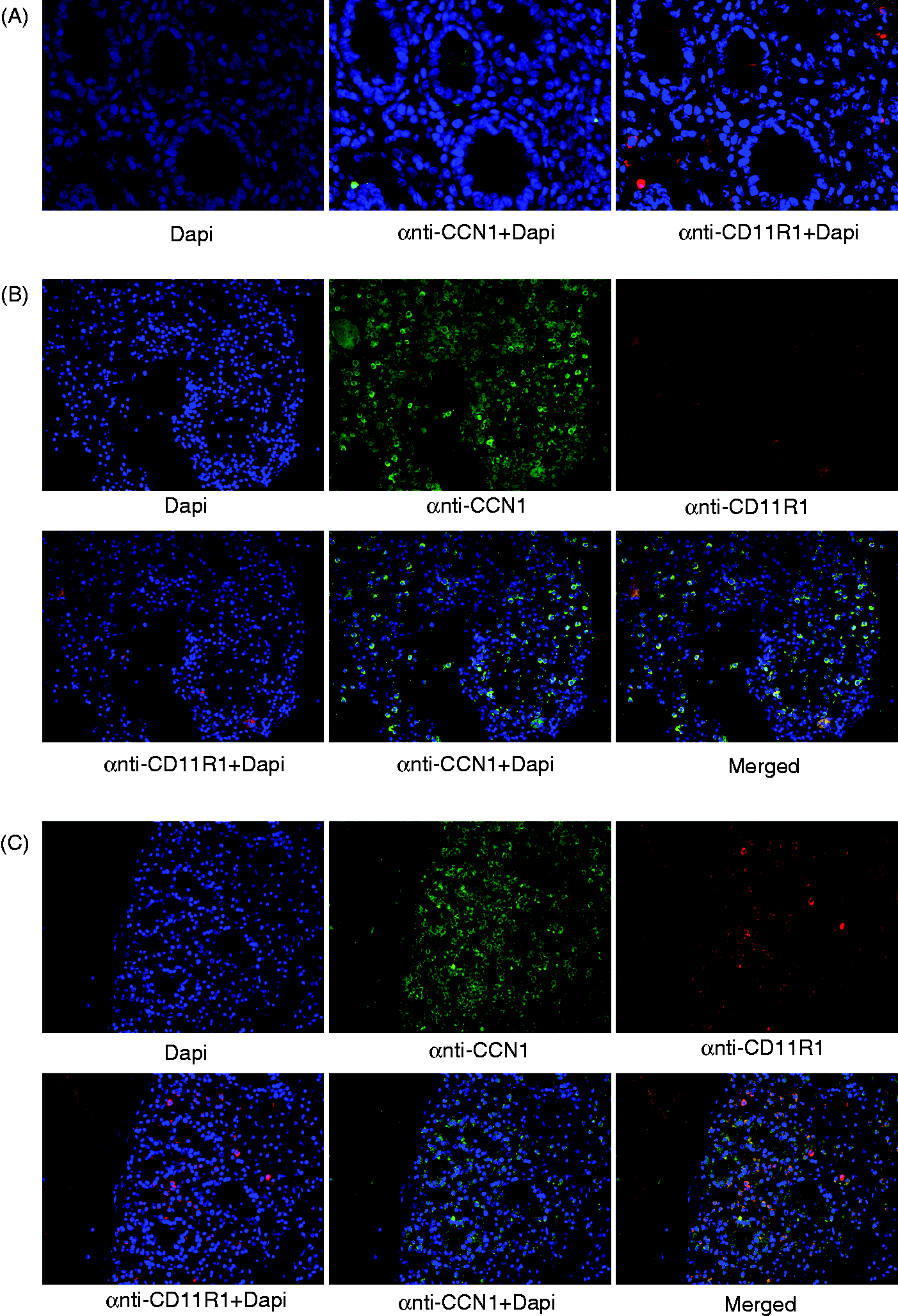
Measurement of CCN1 in the blood
To investigate if CCN1 is released concomitantly with inflammatory mediators from ischemic intestine after reperfusion, CCN1 and TNF-α levels were measured in portal and caval blood. However, both CCN1 (Figure 6) and TNF-α (data not shown) remained unchanged in the collected blood samples. These experiments indicate that CCN1 and TNF-α did not change in the circulation during local intestinal I/R.
Measurement of CCN1 protein level in the circulation. CCN1 protein level did not change in the circulation of pigs subjected to I/R. CCN1 protein was measured by an ELISA kit in the (A) systemic and (B) portal blood samples taken at the time points illustrated in Figure 1 (1 = 25.50 pg/ml).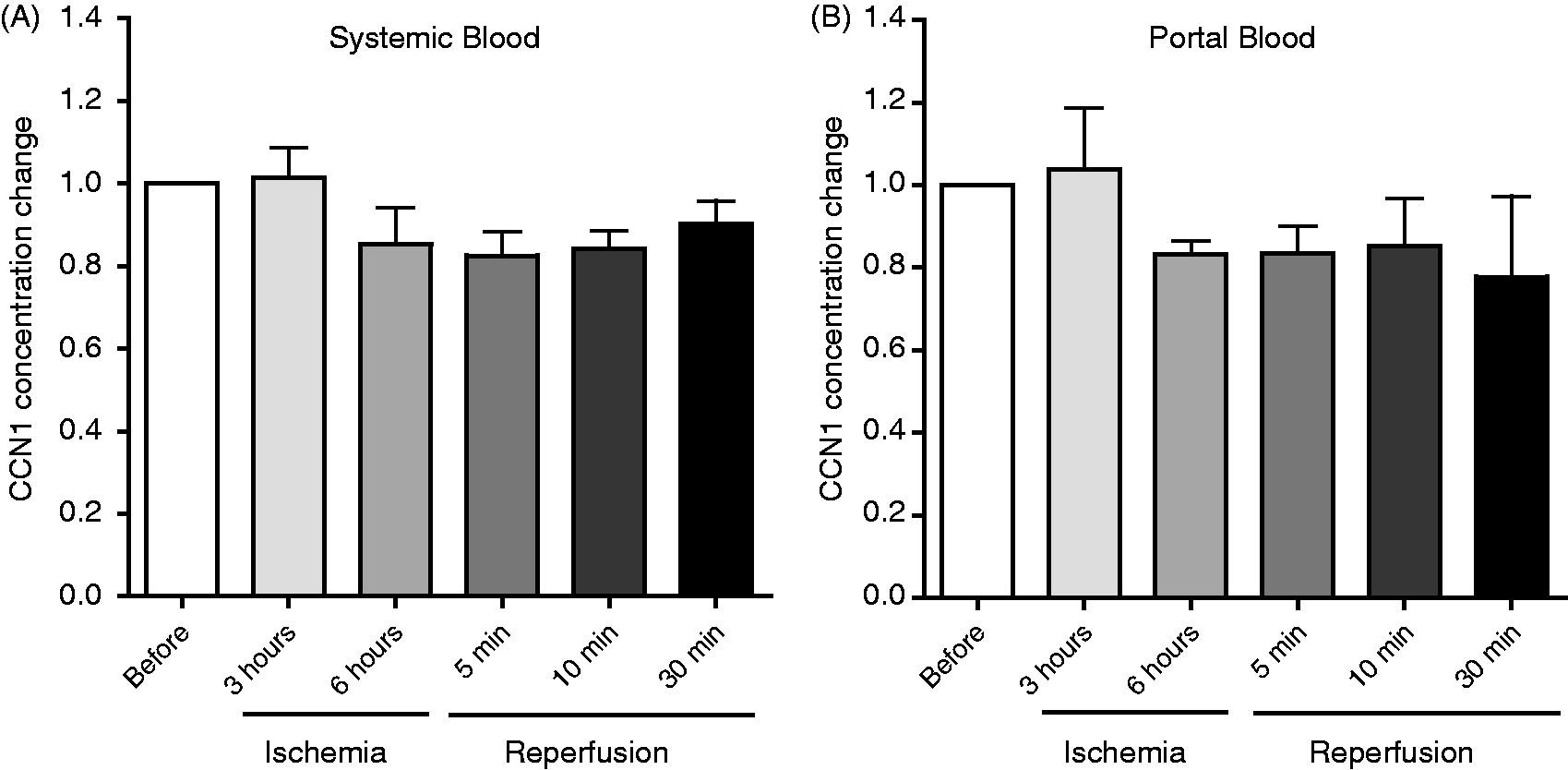
Discussion
The present study investigates CCN1 regulation in porcine intestine following I/R injury. It demonstrates that CCN1 is highly up-regulated, suggesting that the degree of regulation follows the level of prior oxygen deprivation. Despite the local CCN1 production in response to the I/R injury, CCN1 was not detectable in the portal or systemic circulation in the immediate phase after reperfusion, suggesting that the observed CCN1 up-regulation is a localized phenomenon in intestinal I/R injury.
CCN1 protein has been shown to exhibit diverse functions in various cell types, including cell adhesion, survival and apoptosis, and play important roles in inflammation, tissue repair and, recently, a protective role in I/R.10,13,20,25 Intestinal ischemia is a serious condition that can result in death due to development of intestinal gangrene. 26 Better methods of treatment and diagnosis may be achieved by understanding the impact of ischemia on intestinal cells and ECM proteins in more detail. To our knowledge, regulation of CCN1 in mesenteric ischemia has never been investigated. However, it has been shown that hypoxia transcriptionally controls the regulation of CCN1. 27 The authors suggested that CCN1 might play a significant role in ischemic diseases. Early detection of CCN1 in urine following renal ischemic reperfusion injury has been investigated, and CCN1 as a potential disease marker was raised. 28
In this study, we used an intestinal I/R porcine model to study the expression of CCN1 in mesenteric ischemia. We also wanted to find out where and which cell types produce (or bind to) CCN1 in response to I/R, and determine whether this response is locally or systemically regulated. We used two male and two female pigs in our experiments. In the experimental analysis the sex of the animals may alter the results. However, in the current study we did not observe any difference in CCN1 regulation between the sexes throughout our analysis. We concluded that the sex of the pigs does not influence the CCN1 response to I/R. The 6h jejunal ischemia period was chosen owing to surgical experience from human patients and the fact that transplantation surgeons allows up to 12 h cold ischemia during intestinal transplantations (personal communication). Accordingly, we observed after 6 h ischemia advanced ischemic changes but the intestines recovered after 30 min reperfusion in all four animals.
We found that the 6h ischemic period caused severe damage to the intestine (Figure 2), and CCN1 was highly up-regulated at both the gene and protein level (Figure 3). To investigate the localization of CCN1 up-regulation in IS and CCN1-producing cells we explored immunofluorescence staining. These analyses revealed that CCN1-up-regulation was mainly located in mucosal area (Figure 4), and microvillus epithelial cells and vascular endothelial cells were identified to be the main cells positive for CCN1 as judged by microscopic evaluations (Figures 3 and 4). Co-staining revealed that NK cells were modestly positive for CCN1, especially in the mucosal area of ischemic intestine. By excluding the possibility that being positive for CCN1 in the immunofluorescence analysis is not due to the binding of CCN1 to cells we suggest that epithelial, vascular endothelial cells and NK cells may be the source of CCN1 in the reperfused tissue (Figure 5). There was no change in CCN1 protein in the systemic or portal blood samples taken during ischemia or after reperfusion, indicating that CCN1 was locally regulated (Figure 6).
Cell adhesion to ECM molecules promotes cell survival, whereas detachment from the ECM induces rapid cell death, commonly termed anoikis. For instance, normal epithelial cells require matrix attachment for survival. 6 CCN1 can promote the proliferation, migration and adhesion of endothelial cells and fibroblasts. 29 CCN1 stimulates integrin-dependent recruitment of CD34+ progenitor cells to endothelial cells, thereby enhancing endothelial proliferation and neovascularization. 30 Furthermore, CCN1 binds to αvβ3 integrin on endothelial cells to promote angiogenesis and regulate the expression and activities of angiogenic factors.10,31 Hence, up-regulation of CCN1 in endothelial cells may recruit other cells from blood and repair the I/R injury.
After epithelial layer damage intestinal bacteria may cross this barrier, and the activation of the immune cells is therefore important. Although these speculations cannot be formally tested in the present experiments, production of CCN1 in epithelial cells can be explained by the heavily damaged mucosal layer due to ischemia and the urgent need for repairing this vital intestinal barrier. In this context, CCN1 may also act as a danger signal that could initiate the immune responses. Furthermore, it has been shown that T cells and NK cells bind to CCN1 and may also up-regulate CCN1 in response to different stimuli such as pro-inflammatory cytokines.9,15 CCN1 can bind to decidual NK cells and modulate vascular endothelial growth factor C (VEGF-C), 24 and play a role in NK cell-mediated pathogen clearance. 16 The protective role of NK cells in hepatic I/R injury has been reported. 32 We found NK cells positive for CCN1 staining in the brush border in both IS and EZ intenstinal biopsies (Figure 5). Of note, according to the manufacturer, the anti-CD11R1 we used as the NK cell marker also stains a subset of neutrophils. Therefore, we cannot exclude that some of the positive cells may be neutrophils. As a first line of defense, production of CCN1 by NK cells and/or binding of CCN1 to NK cells in IS may be a novel function for this cell type to participate in the repairing process. In line with this notion, we observed weaker CCN1 production by NK cells in EZ segments. Assuming that CCN1 binds to NK cells rather than being produced by NK cells in IS, it could be suggested that NK cells are modulated to produce growth factors such as VEGF-C. 24 Assessing whether other lymphocyte subsets (T and B cells) produce or bind to CCN1 in response to ischemia deserves further investigation. Limitations in available commercial antibodies precluded the identification of other CCN1-producing cells in IS tissue. It therefore remains an interesting subject for future studies to identify other CCN1-producing cell types in intestinal I/R. It should be remembered that CCN1 positivity on immunohistochemistry is not the same as these cells being producers. They could have engulfed the protein or have it bound to their membrane.
We expected to detect CCN1 and TNF-α release from the intestine to the portal circulation, or at least from the liver macrophages into the caval circulation. However, none of them was detectable in the circulation. This may be owing to the model, and CCN1 might have been released if the reperfusion time had been prolonged. This should be addressed in future studies. As a pro-inflammatory mediator, it is possible that CCN1, when acting together with other signal proteins, can elicit responses that it does not elicit alone.12,33,34 The engagement of CCN1 may induce genes encoding chemokines and molecules involved in the repairing response such as angiogenesis, epithelial regeneration and immune regulation. CCN1 production has been found in models of inflammatory bowel disease, which can be considered models of chronic inflammation. 21 We have previously connected CCN1 regulation with acute inflammatory conditions (sepsis).35,36 This study even places CCN1 in acute intestinal inflammation where NK cells may play an inflammatory role.
In conclusion, we report the first data on CCN1 regulation in intestinal I/R. We provide evidence that CCN1 is locally up-regulated in response to ischemia. Although it remains to be proven, CCN1 may (amongst other mediators), presumably, act as a danger or survival signal through its binding to surface receptors on different cell types eliciting responses to eliminate and rebuild the ischemic damage.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Conflict of interest
The authors do not have any potential conflicts of interest to declare.
Acknowledgments
The authors sincerely thank Håvard Attramadal for critically reading the manuscript, and Mohammad Shakil Ahmed for his expert assistance and comments to this article.