
Abstract
There is now convincing evidence that liver X receptor (LXR) is an important modulator of the inflammatory response; however, its mechanism of action remains unclear. This study aimed to examine the effect of LXR on the IL-12 family of cytokines and examined the mechanism by which LXR exerted this effect. We first demonstrated that activation of murine-derived dendritic cells (DC) with a specific agonist to LXR enhanced expression of LXR following activation with LPS, suggesting a role in inflammation. Furthermore, we showed LXR expression to be increased in vivo in dextrane sulphate sodium-induced colitis. LXR activation also suppressed production of IL-12p40, IL-12p70, IL-27 and IL-23 in murine-derived DC following stimulation with LPS, and specifically targeted the p35, p40 and EBI3 subunits of the IL-12 cytokine family, which are under the control of the NF-κB subunit p50 (NF-κBp50). Finally, we demonstrated that LXR can associate with NF-κBp50 in DC and that LXR activation prevents translocation of the p50 subunit into the nucleus. In summary, our study indicates that LXR can specifically suppress the IL-12 family of cytokines though its association with NF-κBp50 and highlights its potential as a therapeutic target for chronic inflammatory diseases.
Introduction
Liver X receptors (LXRs) are members of the nuclear receptor superfamily, which act as ligand-activated transcription factors. Once activated, LXR can heterodimerise with other members of this superfamily in order to function. LXR consists of two subtypes, LXRα and LXRβ, which are 77% structurally identical to each other; 1 however, the expression pattern between these two receptors differs greatly. While LXRβ is expressed ubiquitously, the expression of LXRα is primarily detected in liver, intestine, kidney, adipose tissue and certain immune cells, such as macrophages and dendritic cells (DCs). 2 Endogenous LXR ligands consist of oxidised cholesterol derivatives, such as oxysterols, although synthetic ligands, such as GW3965 and T0901317, have also been described as potent LXR agonists. 3 Numerous studies to date have highlighted an important role for LXR in cholesterol, fatty acid and glucose metabolism and homeostasis. Recently an important role for LXR in immunobiology has been highlighted. LXR activation promotes potent anti-inflammatory effects in B cells, T cells, neutrophils, macrophages and DC.4–8 Furthermore, LXR expression has been linked to inflammatory bowel disease (IBD) where polymorphisms in the LXR gene have been strongly associated with an individual’s susceptibility to ulcerative colitis. 9 Previous studies have also highlighted a reciprocal relationship between the nuclear receptor pathway and the TLR pathway, where activation of one leads to the inhibition of the other. 10 These reports all provide convincing evidence that LXR is an important modulator of the inflammatory response; however, the exact mechanism by which it does this remain unclear.
DCs are essential APCs that efficiently link the innate immune system with the adaptive immune system. Following a DC encountering a pathogen and subsequent PRR engagement, a program of DC maturation is initiated. Only after this DC maturation process is complete can the mature effector DC drive the development and differentiation of Th cells from naive precursors.11,12 A key family of cytokines secreted by DCs is the IL-12 family, which includes IL-12, IL-23 and IL-27. These cytokines are structurally related to one another and share cytokine subunits among family members. Given that these cytokines are important in the differentiation and maintenance of Th1 and Th17 cells, respectively, targeting their production has been explored as a means to treat Th-mediated autoimmune disease.
In this study we show that LXR is regulated during inflammation and we report, for the first time, that LXR can significantly decrease IL-12, IL-23 and IL-27 production by inhibiting NF-κB activation. Specifically, we show that LXR targets the IL-12p40, IL-12p35 and EBI3 subunits of these cytokines through a direct interaction with NF-κB subunit p50 (NF-κBp50). This interaction physically sequesters NF-κBp50 in the cytoplasm and prevents its translocation to the nucleus.
Materials and methods
Animals and materials
Balb/c mice were purchased from Charles River (Margate, UK) and were used at 6–8 wks of age. Animals were maintained according to the regulations of the European Union and the Irish Department of Health. For the colitis experiments, specific pathogen-free female C57BL/6OlaHsD mice, 7–12 wks old, were obtained from Harlan (Bicester, UK). Mice were housed with sterile bedding, at a temperature of 21℃, with 12 h light:12 h darkness and a humidity of 50% in a dedicated animal holding facility. They were fed a sterilized pellet diet and tap water ad libitum. Mice were allowed ≥ 2 wks to acclimatize before entering the study. All animal procedures were performed according to national ethical guidelines following approval by University College Cork Animal Experimentation Ethics Committee. Escherichia coli LPS (serotype R515) was purchased from Alexis Biochemicals (Exeter, UK). The LXR agonist T0901317 was purchased from Sigma-Aldrich (St Louis, MO, USA), dissolved in sterile DMSO and stored at −20℃.
Isolation and culture of bone marrow-derived DC
Bone marrow-derived immature DC (BMDC) were prepared by culturing bone marrow cells obtained from the femurs and tibia of mice in RPMI 1640 medium with 5% FCS supplemented with 10% supernatant from a granulocyte macrophage (GM)-CSF-expressing cell line (J558-GM-CSF), as previously described. 13 The cells were cultured at 37℃ for 3 d and the supernatant was carefully removed without disturbing the cell monolayer and replaced with fresh medium with 10% GM-CSF cell supernatant. On d 7 of culture, cells were collected, counted and used for assays. For experiments, either DMSO (vehicle control) or T0901317 were added to cells on d 1, 5 and 7 of culture.
Addition of T0901317 from d 1 of BMDC harvest was necessary in order to alter the differentiation process of the DC, and concurs with previous published literature.14–16 Dose–response experiments were carried out to determine the dose with maximum effect and minimal cytotoxicity. For all subsequent experiments a dose of 2 μM was used, which correlates with previous literature. 8
Effect of LXR activation on DC cytokine mRNA and protein
On d 7 of culture, DMSO or T0901317 treated cells were cultured in 24-well plates with LPS (100 ng/ml) or medium alone for 0–24 h. At the end of the relevant incubation periods, supernatants were removed and IL-10, IL-1β, TNF-α, IL-6, IL-12p70, IL-12p40, IL-23 and IL-27 were measured using DuoSet ELISA kits from R&D Systems according to the manufacturer’s instructions. For RT-PCR experiments, total RNA was extracted from the cells using Nucleospin (Macherey Nagel, Duren, Germany) RNA II spin columns, according to the manufacturer’s instructions. Two micrograms of RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s instructions. Quantitative real-time PCR was carried out on an ABI 7500 (Applied Biosystems), and TaqMan Gene Expression Assays were purchased from Applied Biosystems and used according to manufacturers instructions. The relative levels of each target transcript were calculated using the ΔΔCt method, after normalising with 18S rRNA as the endogenous control. The relative level of mRNA in untreated control cells was adjusted to 1 and served as the basal reference value thoughout experiments.
Effect of LXR activation on NF-κB activation
HEK-293 cells stably transfected with TLR4, CD14 and MD-2 17 were grown in DMEM supplemented with 10% FCS and 1% penicillin/streptomycin solution (v/v) with the addition of 50 µg/ml Hygrogold (Invitrogen; Life Technologies Carlsbad, CA, USA) and 1 µg/ml Blasticidin (Invitrogen) to maintain expression of TLR4, CD14 and MD-2. Some cells were cultured in the presence of T0901317. Cells were then seeded in 24-well plates and incubated overnight (18 h) prior to transfection using geneJuice transfection reagent (Novagen, Madison, WI, USA). For NF-κB luciferase assays, 75 ng of NF-κB luciferase plasmid, 30 ng of Renilla luciferase and empty pcDNA3.1 vector made up to a total of 220 ng of DNA were transfected into each well. Cells were left to rest overnight and then stimulated with LPS (100 ng/ml). Cells were lysed in 100 μl of passive lysis buffer (Promega, Southampton, UK) for 15 min. Firefly luciferase activity was assayed by the addition of 40 μl of luciferase assay mix [20 mm Tricine, 1.07 mm (MgCO3)4, Mg(OH)2·5H2O, 2.67 MgSO4, 0.1 m EDTA, 33.3 mM dithiotheitol, 270 mM coenzyme A, 470 mM luciferin, 530 mm ATP] to 20 μl of the lysed sample. Renilla luciferase was read by the addition of 40 μl of a 1:1000 dilution of Coelentrazine (Argus Fine Chemicals; Vernio, Italy) in PBS. Luminescence was read using the reporter microplate luminometer (Turner Designs; Sunnyvale, CA, USA). The Renilla luciferase plasmid was used to normalise for transfection efficiency in all experiments.
Expression of LXRα in DSS-induced colitis
Mice were exposed to DSS as previously described.18,19 Three percent DSS (45 ku; TdB Consultancy, Uppsala, Sweden) was provided ad libitum in the drinking water to mice for 6 d followed by water. The different stages of colitis development were defined as 6 d of DSS and 1 d of water = early colitis (d 7); 6 d of DSS and 6 d of water = late acute colitis (d 12); and 6 d of DSS and 20 d of water = transition into chonic colitis (d 26). In the control healthy group (d 0), animals received only water. Mice were sacrificed at the specified days and the colon was dissected and a longitudinal section from the distal colon was transferred to RNA later, kept at 4℃ overnight, frozen and kept at −80℃ until RNA extraction. The number of mice per group was five. The mouse distal colon tissue was transferred to mirVana lysis buffer from the mirVana miRNA Isolation Kit (Ambion; Life Technologies, Carlsbad, CA, USA). Samples were homogenized (×3) at 6500 g for 15 s using a Magnalyser Instrument (Roche; Basel, Switzerland). RNA was extracted using mirVana miRNA Isolation Kit with acid-phenol:chloroform, as per the manufacturer’s instructions.
Effect of LXR activation on NF-κB subunit expression
After T0901317 or DMSO treatment, BMDCs were washed and lysed with pre-chilled lysis buffer [50 mM Tris-HCl, pH 7.5, containing 150 mM NaCl, 0.5% (w/v) igepal and 50 mM NaF, with 1 mM Na3VO4, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride (PMSF) and protease inhibitor mixture (leupeptin (25 µg/ml), aprotinin (25 µg/ml), benzamidine (1 mM), trypsin inhibitor (10 µg/ml)] for 30 min on a rocker at 4℃. Lysates were centrifuged at 12,000 g for 10 min at 4℃. After centrifugation the supernatant was removed and stored at −80℃. The concentration of protein in each sample was quantified by the Bradford assay (Bio-Rad; Hertfordshire, UK) and used for subsequent Western blot analysis, as previously described. 20 Pre-stained protein molecular mass marker (Bio-Rad) and protein samples were resolved on 12% SDS-PAGE gels and transferred onto nitrocellulose membranes. Membranes were blocked in 5% (w/v) nonfat dried milk in Tris buffered saline with Tween and incubated overnight at 4℃ with either anti-NF-κBp105/p50 (Merck Millipore; Darmstadt, Germany), anti-NF-κBp65 (Cell Signaling Technology; Boston, MA, USA), anti-total NF-κBp65 (Cell Signaling Technology) or anti-β-actin (Sigma; St Louis, MO, USA) Abs. Membranes were washed and incubated for 2 h at room temperature (RT, 15℃) with peroxidase-conjugated anti-mouse or anti-rabbit IgG (Sigma-Aldrich) before being developed by enhanced chemiluminescence (Immobilin; Millipore; Darmstadt, Germany). Protein bands were quantified using the GeneSnap acquisition and GeneTools analysis software (GeneGenius Gel Documentation and Analysis System; Syngene; Cambridge, UK).
Effect of LXR activation on NF-κB subunit translocation and co-localisation with LXR
Following treatment with either DMSO or T0901317, DC were cultured in six-well plates on coverslips (1 × 106 cells/ml), left overnight to adhere and stimulated with LPS (100 ng/ml) for 15 min, and fixed in paraformaldehyde on ice for 30 min. Following rinsing 3 × 5 min in PBS baths, cover slips were incubated with 100 mM glycine PBS-1.2% fish gelatin blocking buffer and incubated with a goat polyclonal anti-LXR Ab (Sigma Aldrich) and rabbit polyclonal anti NF-κBp105/p50 Ab (Merck Millipore) or rabbit polyclonal anti NF-κBp65 (Cell Signaling) overnight at 4℃. Cell preparations were washed and incubated with an AlexaFluor 488 anti-goat secondary Ab or AlexaFluor 546 anti-rabbit secondary Ab (Molecular Probes, Invitrogen; Carlsbad, CA, USA) for 1 h at 37℃. For investigation of NF-κBp50 and NF-κBp65 translocation, propidium iodide (Miltenyi; Teterow, Germany) was added to slides for 10 min in order to stain the nucleus. Finally, cover slips were washed and mounted on slides with antifade medium (Dako; Stockport, UK). Slide preparations were observed using a Zeiss Axio Observer Z1 equipped with a Zeiss 710 and ConfoCor 3 laser scanning confocal head (Carl Zeiss; Jena, Germany). Images were analysed using ZEN 2008 software.
For co-immunoprecipitation experiments, cell extracts were generated at 4℃, as above. Lysates were scraped into pre-chilled 1.5-ml Eppendorf tubes and centrifuged at 4℃. Supernatants were removed to fresh tubes (a sample retained for whole cell lysate analysis) and incubated overnight with an LXR primary Ab (2 µg; Abcam). The following day protein A/G agarose beads (Santa Cruz) were added to each sample and incubated overnight. The subsequent day samples were centrifuged, washed with CoIP lysis buffer [50 mM Tris-HCl, pH 7.5, containing 150 mM NaCl, 0.5% (w/v) igepal and 50 mM NaF, with 1 mM Na3VO4, 1 mM DTT, 1 mM PMSF and protease inhibitor mixture (leupeptin (25 µg/ml), aprotinin (25 µg/ml), benzamidine (1 mM), trypsin inhibitor (10 µg/ml)] and subject to re-centrifugation. This step was repeated four times. The 2 × sample buffer [0.125 M Tris-HCl, pH 6.8, containing 20% (w/v) glycerol, 4% (w/v) SDS, 1.4 M β-mercaptoethanol and 0.0025% (w/v) bromophenol blue] was added to the beads for 30 min at RT. Samples were boiled at 100℃ for 10 min and analysed for NF-κBp50 using SDS-PAGE and Western blotting.
Statistics
One-way ANOVA was used to determine significant differences between conditions. When this indicated significance (P < 0.05), post-hoc Student–Newmann–Keul or Dunnett’s multiple comparison test analysis was used to determine which conditions were significantly different from each other. GraphPad software was used for the statistical tests. For confocal experiments a one-tailed unpaired t-test was used to check the percentage translocation of p50 to the nucleus.
Results
LXRα expression is regulated during inflammation
Little is currently known about the regulation of nuclear receptors during inflammation and, indeed, how the expression of one nuclear receptor is regulated in response to activation of another. We therefore examined the expression of LXRα, and one other nuclear receptor with which it can dimerise, RXRα, in response to LXR activation over a 24-h course of LPS stimulation in BMDC. LXRα has been shown to be the dominant isoform in DCs.
8
The expression of LXRα was significantly enhanced at 6, 12 and 24 h (P < 0.001) post-LPS stimulation (Figure 1a). The LPS-induced expression of LXR was further enhanced when LXR was activated 4 h (P < 0.001), 6 h (P < 0.001) and 12 h (P < 0.01) prior to addition of LPS. In contrast, the expression of RXR decreased following LPS stimulation in DMSO control cells, except at 24 h. However, we observed an increase in RXRα expression upon LXR activation (4, 6 and 12 h; P < 0.001).
The expression of LXRα is regulated during inflammation. (a) BMDC were differentiated in GM-CSF in the presence of 2 μM T0901317 or DMSO for 7 d and stimulated over the course of 24 h with 100 ng/ml LPS. Total RNA was isolated, converted to cDNA and used for subsequent RT-PCR experiments. Results are expressed as fold-change after normalising to the endogenous control 18S. Results are ± SEM of triplicate assays and represent three independent experiments. ***P < 0.001, **P < 0.01, *P < 0.05 comparing control versus LXR treated groups and ###P < 0.001 comparing control versus LPS-stimulated cells as determined by one-way ANOVA. (b) Mice were treated with 3% DSS for 6 d followed by up to 20 d of water. Total RNA was isolated from colonic tissue of mice at various stages of disease after induction of colitis with DSS. One µg of RNA was converted to cDNA and used for subsequent RT-PCR experiments. Results are expressed as fold change after normalising to the endogenous control S18. Results are ± SEM of triplicate assays and represent five mice per group (DSS) or four mice per group (Citrobacter rodentium) ***P < 0.001, **P < 0.01, *comparing control vs. disease groups as determined by one-way ANOVA.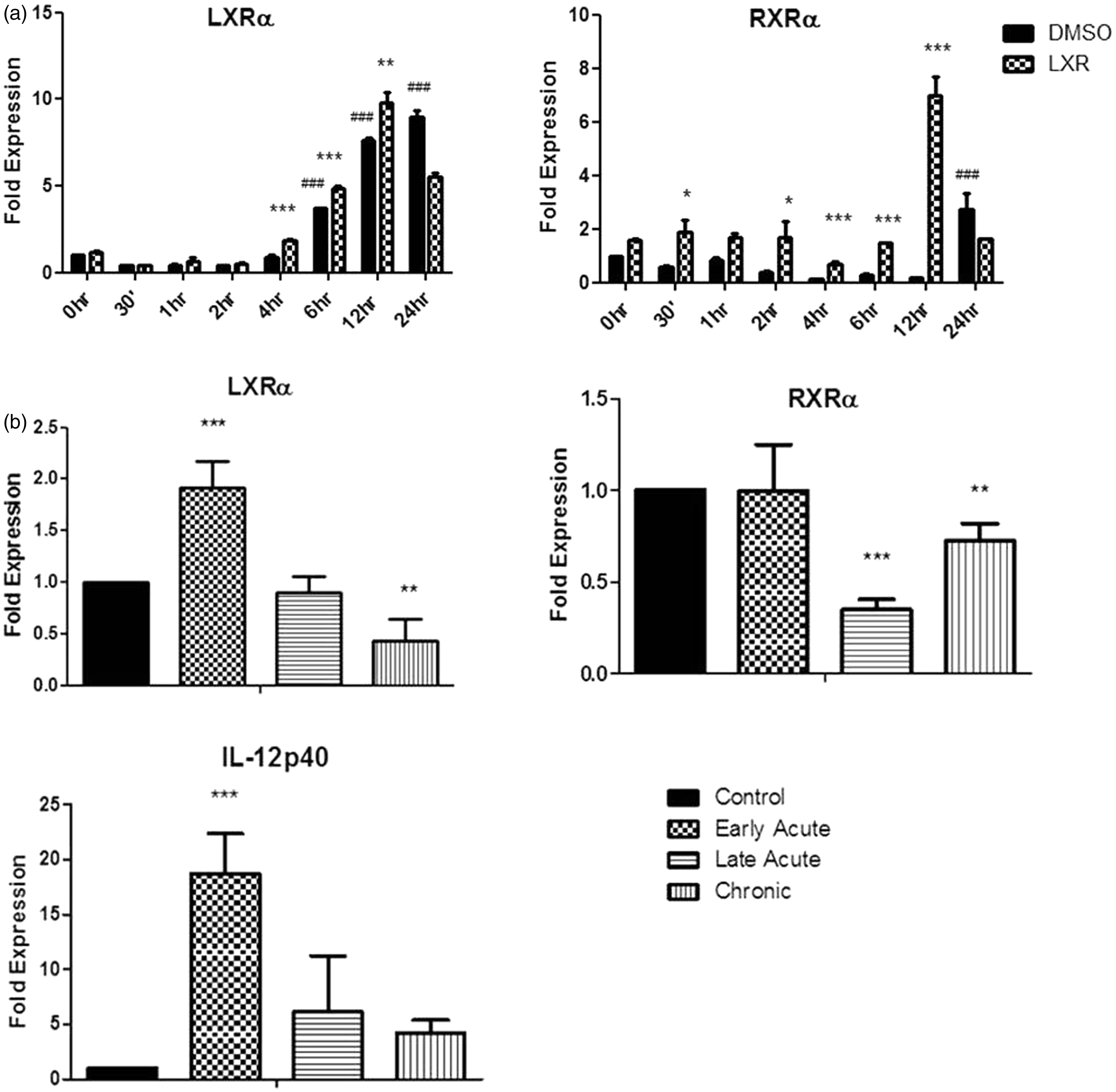
Given that the expression of LXR is regulated in response to an inflammatory signal we next examined the expression of this receptor during inflammation in vivo in DSS-induced colitis. Mice treated with DSS developed acute colitis that progressed into chronicity, which is characterised by body mass loss in the acute phase followed by its recovery in the chronic phase and increase in colon mass in both phases of disease. IL-12p40 is significantly increased in the early acute stage of DSS treatment and acts as a marker of inflammation in this model (P < 0.001). The expression of LXRα was significantly increased during the initial onset of disease, that is the early acute stage (P < 0.001; Figure 1B), before peak of body mass loss. However, its expression was reduced to normal levels by the late acute phase and significantly decreased in mice with chronic colitis compared to healthy controls (P < 0.01). There was no increase in RXR expression during any phase of disease; indeed, it was significantly decreased (Figure 1b).
Activation of LXR decreases the IL-12 family of cytokines in BMDC
Given that LXR is clearly regulated during inflammation, we next examined how activation of this nuclear receptor affected cytokine secretion. We focused, in particular, on the IL-12 family of cytokines, as it has been widely implicated in the generation and maintenance of specific Th cell responses, which are implicated in autoimmune disease. LXR activation by T0901317 significantly decreased production of IL-12p40, IL-23 (P < 0.001), IL-12p70 (P < 0.05) and IL-27 (P < 0.01; Figure 2) by BMDC following LPS stimulation. There was no significant effect on TNF-α, IL-6 or IL-10 production after 24 h.
Activation of LXR specifically suppresses the production of the IL-12 family of cytokines. BMDC were differentiated in GM-CSF in the presence of 2 μM T0901317 for 7 d and stimulated for 24 h with 100 ng/ml LPS. Supernatants were then harvested and assessed for levels of IL-12p40, IL-12p70, IL-23, IL-27, TNF-α, IL-6, IL-1β and IL-10 using specific immunoassays. Results are ± SEM of triplicate assays and represent three independent experiments. ***P < 0.001, **P < 0.01, comparing DMSO/LPS versus T0901317/LPS groups as determined by one-way ANOVA.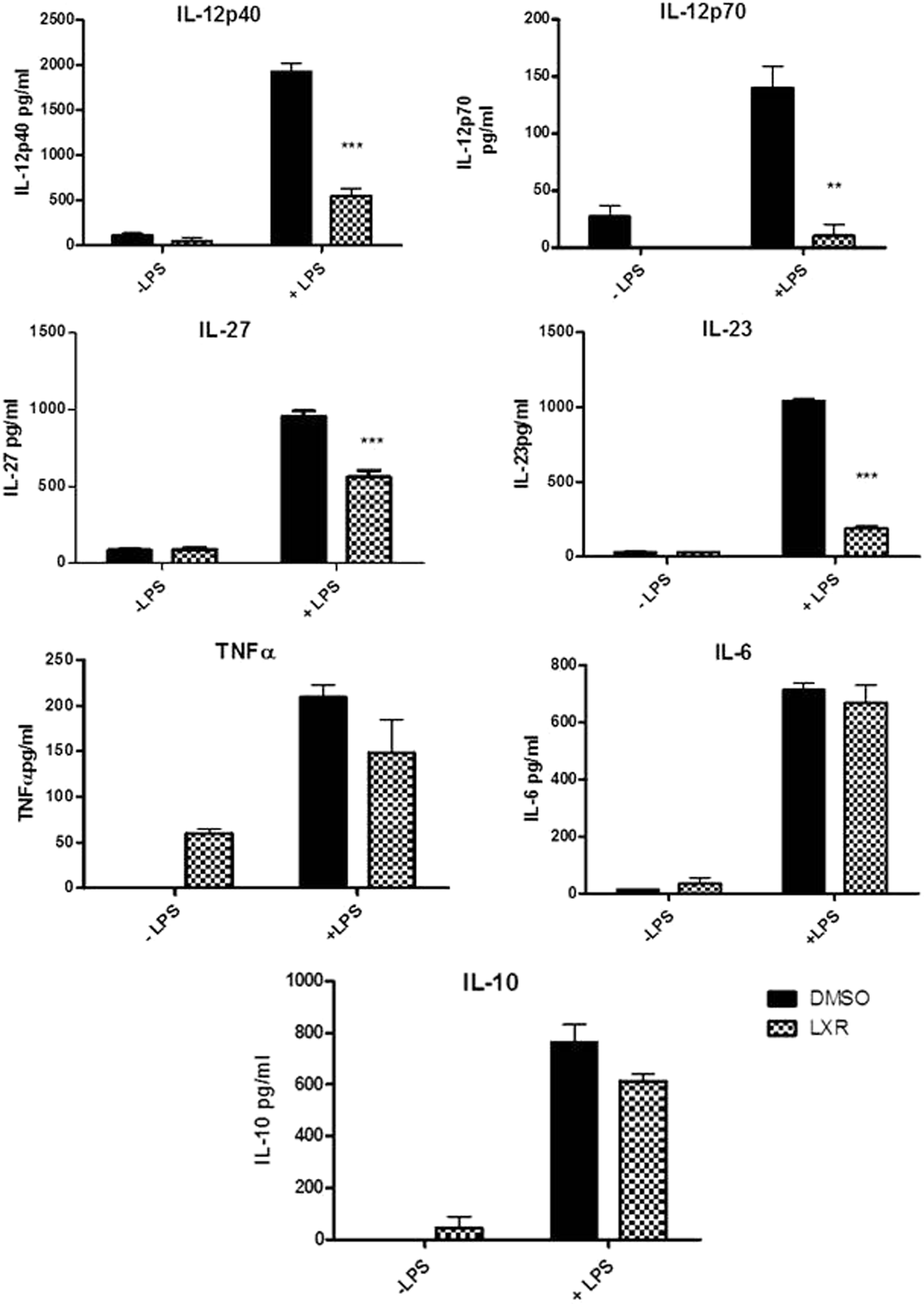
LXR activation in BMDC specifically inhibits expression of the EBI3, IL-12p40 and IL-12p35 subunits of the IL-12 family
Given that the IL-12 family of cytokines are made up of distinct subunits, and we have shown that LXR activation can inhibit the production of these cytokines, we next determined the effects of LXR activation on the individual subunits that form the cytokines IL-12, IL-23 and IL-27. The expression of IL-12p35 was significantly increased after 2 h LPS stimulation and continued to increase over this 24 h stimulation period. LXR activation in these cells significantly inhibited IL-12p35 expression after 2 (P < 0.001; Figure 3), 4, 6 and 12 h LPS stimulation (P < 0.05). The expression of the IL-27 subunit EBI3 was significantly increased after 6 h and 12 h of LPS stimulation (P < 0.001, P < 0.05 respectively; Figure 3). LXR activation in these cells significantly decreased the expression of EBI3 in both unstimulated cells and cells stimulated with LPS for 30 min to 4 (P < 0.001), 6 (P < 0.01) and 12 h (P < 0.05; Figure 3). The expression of IL-12p40 was also increased following LPS stimulation, with changes in expression detected as early as 1 h (P < 0.01). LXR activation in these cells significantly decreased the expression of IL-12p40 in cells stimulated for 30 min (P < 0.01) and from 1 h to 24 h (P < 0.001). While IL-23p19 and IL-27p28 expression increased significantly following LPS stimulation at 30 min and 4 h, respectively (P < 0.001), LXR activation did not significantly decrease their expression. Although there was a decrease in IL-27p28 at the later time points of 12 h and 24 h post-LPS stimulation. Indeed, the expression of IL-23p19 is significantly increased following LXR activation 30 min post-LPS stimulation (P < 0.001).
LXR activation specifically targets the EBI3, IL-12p40 and IL-12p35 subunits of the IL-12 family of cytokines. BMDC were differentiated in GM-CSF in the presence of 2 μM T0901317 or DMSO for 7 d and stimulated over the course of 24 h with 100 ng/ml LPS. Total RNA was isolated, converted to cDNA and used for subsequent RT-PCR experiments. Results are expressed as fold-change after normalising to the endogenous control 18S. Results are ± SEM of triplicate assays and represent three independent experiments ***P < 0.001, **P < 0.01, *P < 0.05 comparing control versus LXR-treated groups as determined by one-way ANOVA.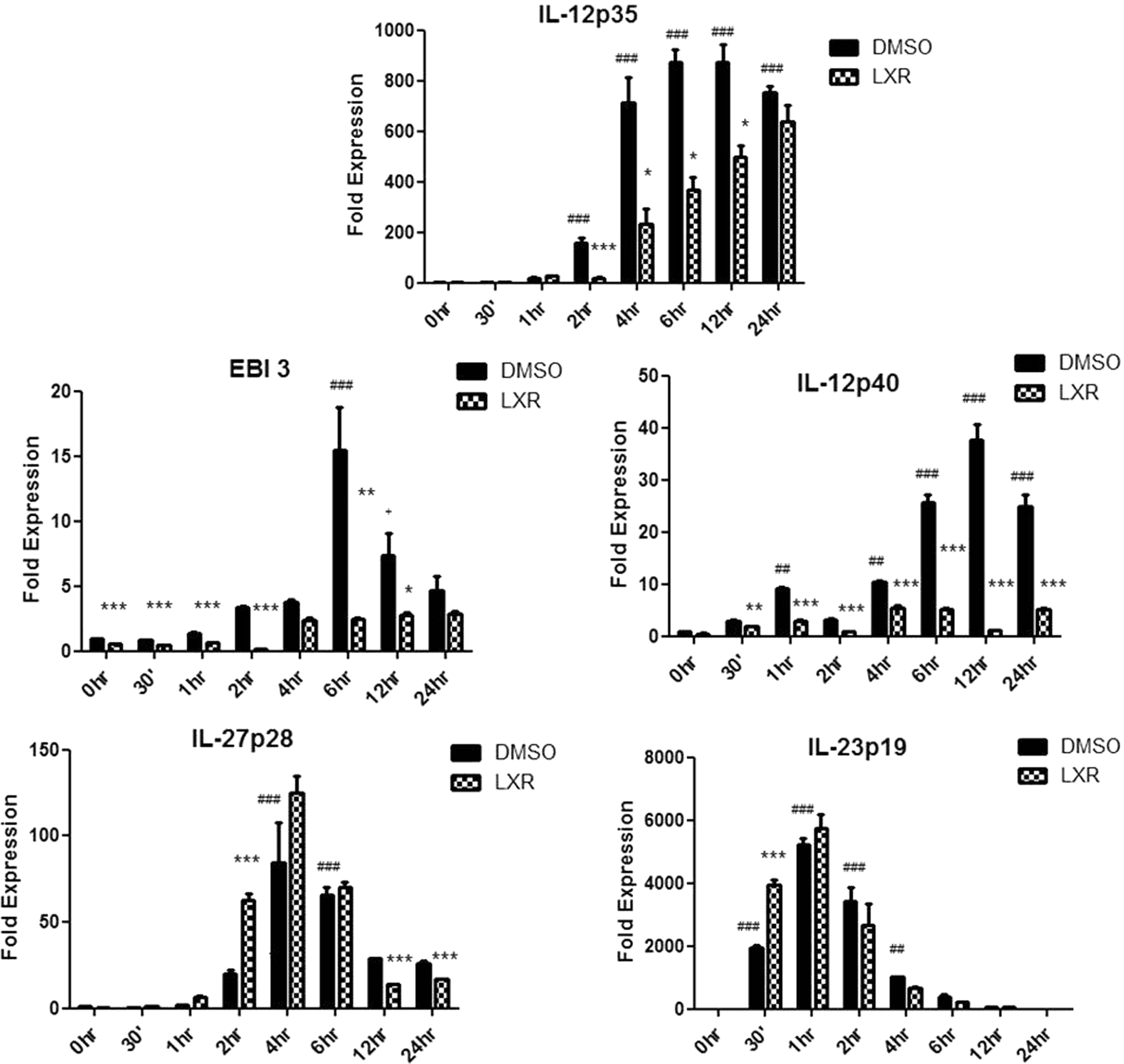
Activation of LXR suppresses NF-κB activation
It has previously been reported that nuclear receptors may regulate inflammatory processes through inhibition of key transcription factors that are involved in inflammation. Given the well documented relationship between IL-12 and NF-κB, we next examined if LXR activation could suppress the activation of NF-κB. Our results show that LXR activation does significantly inhibit NF-κB activation in HEK293-TLR4/CD14/MD-2 cells (P < 0.001; Figure 4a).
LXR inhibits NF-κB activation and the expression of NF-κBp50. (a) HEK293 cells stably expressing CD14, MD2 and TLR4 were cultured for 7 d in the presence of the specific LXR agonist (T0901317 2 μM) Cells were plated and transiently transfected for 24 h with NFκB luciferase constructs before being stimulated with LPS (100 ng/ml). Firefly luciferase activity was quantified 6 h after LPS stimulation and all samples were normalised to Renilla luciferase. Results are ± SEM of triplicate assays and represent three independent experiments. ***P < 0.001 comparing DMSO/LPS versus T0901317/LPS groups as determined by one-way ANOVA. (b) BMDC were differentiated in GM-CSF in the presence of 2 μM T0901317 or DMSO for 7 d and stimulated over the course of 30 min with 100 ng/ml LPS. After this time, cells were lysed and immunoblotted for p50, p105 and p65. Total cellular levels of β-actin and total p65 were used as a loading control. Representative immunoblots of three experiments are shown. *P < 0.05 comparing DMSO/LPS and T0901317/LPS. RLA: relative luciferase activity.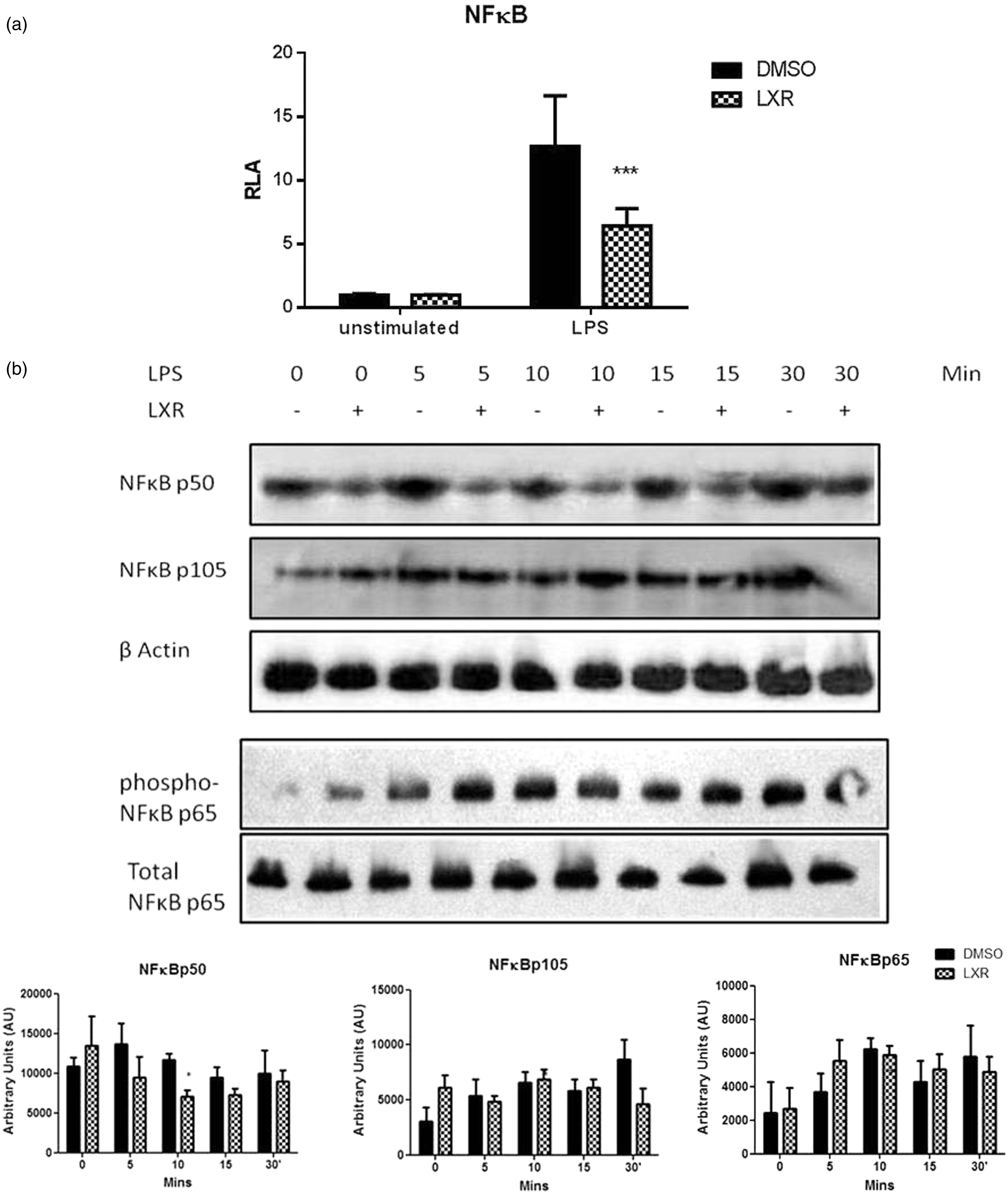
LXR physically associates with NF-κBp50 in BMDC and prevents its translocation to the nucleus
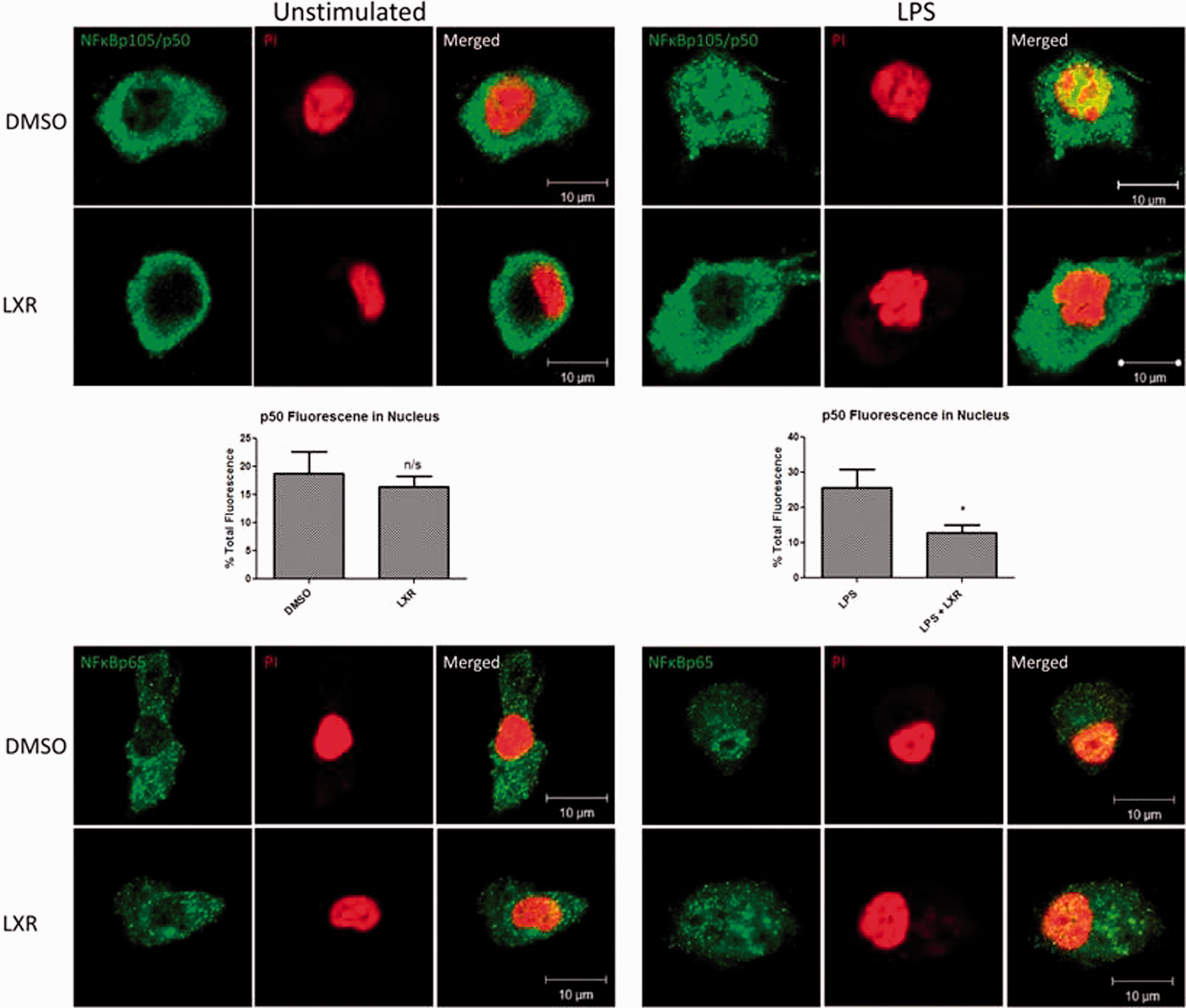
While the IL-12 family of cytokines is regulated by NF-κB, each subunit is under the control of a distinct pairing of NF-κB subunits. The three IL-12 subunits whose expression we report to be suppressed by activation of LXR are IL-12p40, IL-12p35 and EBI3, which are under the control of NF-κBp65/p50, NF-κBp50/c-Rel and NF-κBp50/p65 respectively.21,22 Given that NF-κBp50 is the common subunit, we hypothesised that LXR may exert its effects though this subunit. We first examined the effect of LXR activation on the expression of this subunit. NF-κBp50 was expressed in both unstimulated and LPS-stimulated BMDC (Figure 4b). Interestingly, LXR activation resulted in a significant decrease in the expression of this protein after 10 min LPS stimulation (P < 0.05). NF-κBp105, the precursor protein of p50, is also present in unstimulated and LPS-stimulated BMDC, and its expression does not significantly change following LXR activation. To examine whether the effect of LXR was specific to the p50 subunit, we also examined the effect of LXR activation on NF-κBp65. LXR activation did not significantly alter the expression of the p65 subunit nor did it affect phosphorylation of the p65 subunit (Figure 4b). We next examined if LXR could also physically associate with NF-κBp50 using confocal microscopy. Co-localisation was observed between LXR and NF-κBp50 in both LXR-activated and control DMSO cells following 15 min of LPS stimulation (Figure 5a). We also examined potential co-localisation between LXR and NF-κBp65, and showed that while they are abundantly expressed in the cell they do not co-localise with each other in either LXR-activated or control DMSO cells following LPS stimulation (Figure 5a). We confirmed a direct interaction between LXR and p50 using immunoprecipitation, and densitometric analysis showed a significant interaction between LXR and p50 following LXR activation (P < 0.01, P < 0.001 respectively). Finally, we examined if the association between LXR and NF-κBp50 could interfere with the translocation of NF-κBp50 to the nucleus. In control cells NF-κBp50 translocates to the nucleus following exposure to LPS for 15 min, as indicated by the yellow colour (Figure 6). However, following activation of LXR, NF-κBp50 does not translocate to the nucleus and remains cytoplasmic following LPS stimulation (Figure 6). We also show that NF-κBp65 translocates to the nucleus following LPS stimulation and that this is not affected by activation of LXR (Figure 6), confirming that LXR specifically targets the NF-κBp50 subunit.
LXR interacts with and co-localises with NF-κB p105/p50 in LXR-activated BMDC. BMDC were differentiated in GM-CSF in the presence of 2 μM T0901317 or DMSO for 7 d and stimulated for 15 min with 100 ng/ml LPS. Cells were subsequently stained for LXR and NF-κBp105/p50 or NF-κBp65, and co-localisation between these proteins was assessed by confocal microscopy. Co-localisation between LXR and NF-κBp105/p50 can be seen in the merged image (yellow), whereas no co-localisation is observed between LXR and NF-κBp65. Cells were also lysed, and co-immunoprecipitation experiments confirmed a physical association between LXR and NF-κBp50. (P < 0.01; 0.001). LXR activation in LPS-stimulated BMDC alters the translocation of NF-κBp50 to the nucleus. BMDC were differentiated in GM-CSF in the presence of 2 μM T0901317 or DMSO for 7 d and stimulated for 15 min with 100 ng/ml LPS. The nuclei of cells were subsequently stained using propidium iodide (red), and cells were also stained for either NF-κBp50 or NF-κBp65 (green). The translocation of NF-κBp50 and NF-κBp65 to the nucleus was assessed by confocal microscopy. The translocation of NF-κBp50 to the nucleus was quantified and expressed as nuclear fluorescence verses total cell fluorescence. *P < 0.05 as determined by a one tailed unpaired t-test.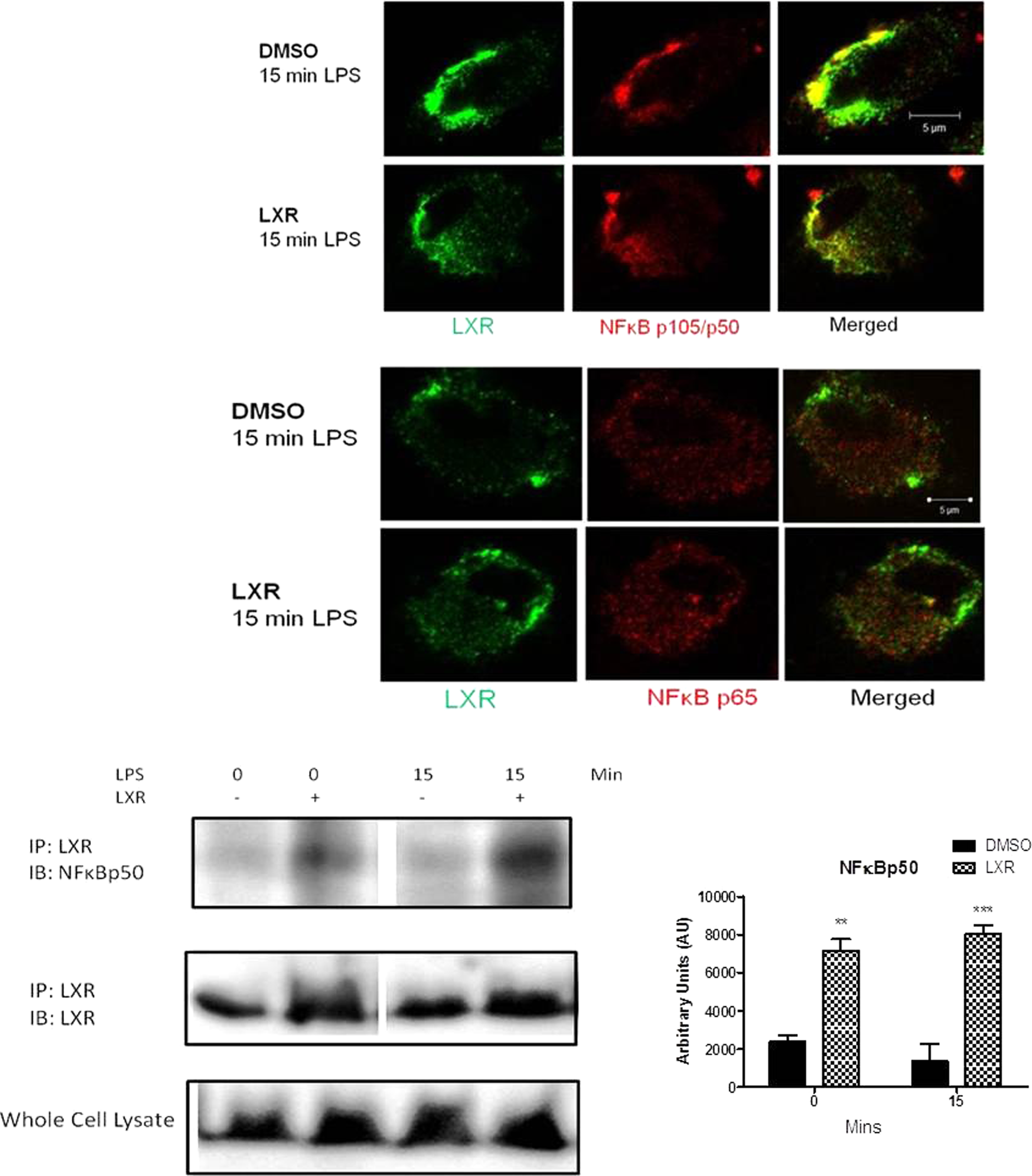
Discussion
LXR is an important regulator in lipid and cholesterol metabolism and therefore has been extensively studied in macrophages. Although previous studies have reported an anti-inflammatory role for LXR in a number of immune cells, the role of this receptor in DC and in intestinal inflammation is not well understood. We report an increased expression of LXR in DC in response to LPS. We also observed an increased expression of LXR in the early acute phase of inflammation in DSS-induced colitis. We report for the first time the specific effect of LXR on the IL-12 family of cytokines in BMDC with LXR activation specifically decreasing the IL-12p40, IL-12p35 and EBI3 subunits. Finally, we demonstrate that LXR activation in LPS-stimulated DC results in LXR interacting with NF-κBp50 and preventing its translocation to the nucleus, thereby leading to a reduction in the IL-12 family of cytokines.
In this study, we first examined the expression of LXRα in DC following stimulation with LPS following activation of LXR. The expression of LXRα, but not RXR, increased in DC in response to LPS. The expression of LXRα and RXRα were significantly increased following activation of LXR. Our results are in agreement with a previous report that has shown LXR to regulate and induce its own expression. 10 We next confirmed that LXR could be regulated during inflammation in vivo using a model of colitis. We demonstrated that the expression of LXR was significantly increased in the early acute phase of inflammation, but decreased later during chronic inflammation compared with healthy controls. The data suggest that during the early onset of colitis LXR may be increased in order to regulate and suppress the production of pro-inflammatory cytokines that contribute to disease. During this early onset of inflammation there is also increased infiltration of immune cells, such as DC, macrophages and T-cells, to the colon.18,23 Given the receptor expression in these cells, this could also account for the increased LXRα expression in early acute colitis.
After confirming that LXR is, indeed, regulated during inflammation, we next examined the effects of LXR activation on cytokines secreted by DC, which are important in the pathogenesis of inflammatory diseases such as IBD. LXR activation in BMDC resulted in a significant decrease in IL-12 (p40 and p70), IL-23 and IL-27. While a previous report has shown that LXR activation in mature human DC results in a significant decrease in IL-12p40 and IL-12p70 secretion, 8 there was no indication of how LXR exerted these effects.
We also demonstrated that LXR can suppress NF-κB activation, which is important for the production of the IL-12 family of cytokines. The production of IL-12p40 has been linked to the formation of NF-κBp65/p50 heterodimers in response to LPS, 21 whereas the production of its cytokine partner, IL-12p35, is associated with NF-κBp50/c-Rel dimer complexes. 22 The expression of EBI3, one of the subunits of IL-27, is induced by p50/p65 heterodimers binding to the NF-κB response element in the promoter of the gene, 24 and, finally, IL-27p28 and IL-23p19 are produced in response to LPS by the formation of c-Rel/p65 heterodimers.25,26
Our results show that LXR can specifically inhibit the expression of the IL-12p40, IL-12p35 and EBI3 subunits, therefore accounting for an overall reduction in IL-12, IL-23 and IL-27 cytokine production. As IL-12p40, IL-12p35 and EBI3 are all under the transcriptional control of either p50:p65 or p50:c-Rel NF-κB complexes we therefore hypothesised that LXR may target the NF-κBp50 signalling subunit in order to exert its effects on the IL-12 family. We report that LXR activation can significantly decrease the expression of NF-κBp50 in LPS-stimulated DC, which would, consequently, reduce the level of p50 available to generate p50:p65 and p50:c-Rel dimers, which could account for a reduction in IL-12p40, IL-12p35 and EBI3 expression. We also report that LXR physically associates with NF-κBp50 in LXR-activated cells, and subsequently blocks its translocation to the nucleus.
Previous studies have also highlighted the anti-inflammatory potential of LXR activation in vitro. Geyeregger et al. 8 investigated the effects of LXR activation in human myeloid DCs and also reported a significant reduction in IL-12p40 levels. TO901317 treatment has also been shown to reduce atherosclerotic lesion numbers, size and severity, and to reduce both NF-κBp50 and p65 expression, as well as promoting lesion regression at several levels in apoE*3 Leiden mice. 27 Other studies have also reported that LXR expression is decreased in chronic inflammation. Zeng et al., 28 for example, highlighted decreased LXRα expression in monocyte-derived foam cells, suggesting that chronic atherosclerosis is mediated by loss of LXR.
A link between nuclear receptors and NF-κB has previously been described. Chen et al. 29 demonstrated a physical association between PPARγ and NF-κBp65 in colon cancer cells resulting in a decrease in NF-κB transcriptional activity. The glucocorticoid receptor, once activated, has been reported to increase the nuclear export rate of Rel A, therefore reducing the duration of an NF-κB transcriptional response. 30 The vitamin D receptor, upon activation, has been reported to reduce the phosphorylation of p65 and its subsequent translocation to the nucleus. 31
LXR-mediated effects on NF-κB have also been reported in other cell types. Cheng et al. 32 reported decreased p65 expression following ischaemic brain injury in rats treated with the LXR agonist GW3965, and Wu et al. 33 reported decreased NF-κB activity in cardiomyocytes as a result of LXR activation with T0901317. Studies have also shown that while nuclear receptor activation can inhibit NF-кB translocation, there is also a reciprocal relationship in which NF-кB can inhibit nuclear receptor translocation. Sticozzi et al. 34 reported that cigarette smoke could inhibit NF-кB activation, which prevented LXR translocation to its ABCA1 target gene. There have also been previous reports to suggest a possible LXR:p65-related mechanism in which LXR activation in DC prevented the association of p65 to a set of NF-κB target genes. 35 However, it is important to note that although LXR activation in this study inhibited a p65 response, no association between LXR and p65 was reported. Therefore, it is difficult to determine if LXR activation has a direct effect on NF-κBp65 or if the effect reported is a result of an interaction with p65’s heterodimerisation partner, that is p50 or c-Rel. We therefore propose a novel mechanism by which LXR can exert its anti-inflammatory effects in DC through its association with NF-κBp50 and prevention of its translocation to the nucleus. Inhibition of p50 translocation subsequently decreases the transcription of EBI3, IL-12p40 and IL-12p35 expression leading to a decrease in the production of the IL-12 family of cytokines.
It is well appreciated that the formation p50 homodimers can act as suppressors of cytokine genes; therefore, one could argue the biological relevance of targeting p50 in disease. However, it has previously been documented that p50 homodimers can activate transcription of the IL-10 gene by encouraging the recruitment of the co-activator CREB binding protein (CBP). 36 Indeed, it has also been proposed that depending on the relative amounts of each of the NF-κB family members, p50 may preferentially form heterodimers over repressive homodimers. It was suggested that the ratio of p50 relative to the other Rel family members is likely to be a determining factor for gene expression where high levels of p65 and c-Rel could compete for p50 binding to itself and thereby diminish p50 homodimer formation. Previous reports have also demonstrated that the use of terpenoids, small secondary metabolites released from plants, can suppress inflammation in adjuvant-induced arthitis through inhibition of p50:DNA binding, 37 further highlighting the biological significance in targeting this subunit.
The ability of LXR to directly target NF-κBp50 in DC and ultimately inhibit IL-12 production highlights the potential of targeting this receptor in disease. It has previously been shown that while p50 is essential in EBI3 transcription in DC, the expression of this IL-27 subunit is not controlled by p50 in B cells. 24 This finding suggests that activation of LXR could target the production of the IL-12 family of cytokines specifically released from the DC and not other immune cells. Therefore, activation of LXR could directly target cytokines important for the differentiation of Th cells without compromising the ability of other immune cells to respond normally to infections. Given that the current treatments for inflammatory diseases can leave patients immunosuppressed, targeting LXR in these diseases could improve on the therapies that are already available.
Footnotes
Funding
We acknowledge funding provided under the Programme for Research in Third Level Institutions (PRTLI) Cycle. The PRTLI is co-funded though the European Regional Development Fund (ERDF), part of the European Union Structural Funds Programme 2007–2013. The Immunomodulation Research Group at DCU was funded by Science Foundation Ireland for this work. The Alimentary Pharmabiotic Centre is a research centre funded by Science Foundation Ireland.