
Abstract
Background:
Bone marrow (BM) Angiotensin II (Ang II) type 1 (AT1) receptor plays a crucial role in atherosclerosis development; however, the effect of BM Ang II type 2 (AT2) receptor on atherogenesis remains undefined.
Methods and results:
We generated BM chimera apoE-deficient (apoE−/−) mice whose BM cells were repopulated with AT2-deficient (Agtr2−/−) or wild-type (Agtr2+/+) cells. After 2 months of a high-cholesterol diet, the atherosclerotic lesion area was significantly increased in the apoE−/−/BM-Agtr2−/− mice compared with the apoE−/−/BM-Agtr2+/+ mice (51%, P < 0.05), accompanied by an augmented accumulation of lesion macrophages. Although phenotypic polarization in BM-derived macrophages and lipopolysaccharide-induced expression of proinflammatory cytokines in thioglycollate-induced peritoneal macrophages (TGPMs) were not affected by AT2-deficiency, mRNA and protein expression levels of macrophage liver X receptor β (LXRβ) were significantly decreased in Agtr2−/− TGPMs compared with Agtr2+/+ TGPMs. Anti-inflammatory effects of LXR agonist (GW3965) were markedly inhibited in Agtr2−/− TGPMs. Furthermore, the expression levels of ATP-binding cassette transporter ABCA1 and CCR7 were much lower in Agtr2−/− TGPMs than Agtr2+/+ TGPMs, accompanied by a significantly reduced cholesterol efflux.
Conclusions:
Our findings demonstrate that BM-AT2 deficiency aggravates atherosclerosis, at least in part, by eliminating the anti-atherogenic properties of macrophages elicited by LXRβ activation.
Introduction
Angiotensin II (Ang II) promotes atherosclerosis development through type 1 (AT1) receptor,1–3 which is present in a variety of cells, including bone marrow (BM) cells.4,5 Previous studies have shown that atherosclerosis development was significantly attenuated in atherosclerosis-prone mice whose BM cells were repopulated with AT1-deficient (Agtr1−/−) cells. 6 We have also reported that BM AT1 regulates macrophage-colony-stimulating factor (M-CSF)-mediated differentiation/proliferation of BM monocyte-lineage cells followed by the mobilization of monocytes, which contribute to AT1-mediated pro-atherogenic actions. 7 These findings indicate that BM AT1 plays a crucial role in the development of atherosclerosis; however, the effect of BM Ang II type 2 (AT2) receptor on atherogenesis remains to be defined.
Macrophages play a central role in the atherogenic process by showing phenotypic diversity and plasticity in response to different environmental signals present in atherosclerosis lesions.8,9 Macrophage phenotypes are conventionally divided into two broad types, known as classically activated macrophages (M1), induced by Th1 cytokines or lipopolysaccharide (LPS), and alternatively activated macrophages (M2), induced by Th2 cytokines.10–12 In atherosclerotic plaque, M1 and M2 macrophages are located in a lesion-specific manner, in which distribution and dominance of M1 and M2 macrophages are closely implicated with lesion progression and instability.11,12 Recently, the AT1 receptor has been reported to modulate macrophage polarization into M1 macrophages and to exaggerate renal injury and atherosclerosis.13,14 These findings led us to examine the effect of AT2 on macrophage phenotypic polarization and its functional relevance to the development of atherosclerosis.
In this study, we focused on the effect of the BM-AT2 receptor on macrophage phenotypic alteration and studied whether (1) BM-AT2 deficiency exaggerates the development of hypercholesterolemia-associated atherosclerosis and (2) BM-AT2 deficiency affects macrophage polarization and inflammatory response. We also investigated the underlying mechanism of AT2-mediated atheroprotective action. Our results demonstrated for the first time that BM-AT2 deficiency accelerates the development of atherosclerosis, and liver X receptor β (LXRβ)-mediated anti-atherogenic actions such as anti-inflammatory response, cholesterol efflux, and emigration-related gene expression were attenuated in AT2-deficient macrophages. Our findings support the notion that direct AT2 activation could be a potential therapeutic option for preventing the development of atherosclerotic cardiovascular diseases.
Materials and methods
Animals
ApoE−/− mice (C57BL/6) were obtained from Taconic Co. Ltd (Germantown, NY, USA) and AT 2 receptor-deficient (Agtr2−/−) mice (C57BL/6) were kindly provided by Dr. M. Horiuchi (Ehime University). Agtr2−/− mice were crossed with apoE−/− mice. Heterozygous animals were crossed until homozygous double knockout (Agtr2−/−/apoE−/−) mice were obtained (backcrossed more than 10 times). Two-month-old female apoE−/− recipient mice were lethally irradiated at 9 Gy using an X-ray source. BM cells were harvested from the femurs and tibias of donor male mice (Agtr2−/−/apoE −/− or Agtr2+/+/apoE−/−) by flushing with RPMI-1640 medium (Gibco BRL), and then recipients received 5 × 106 BM cells per mouse in 0.2 ml of medium by tail-vein injection. Two months after BM transplantation, all mice started to receive a high-cholesterol diet (36% fat, 1.25% cholesterol; Oriental Yeast Co., Tokyo, Japan) for an additional 2 months. The animals were housed in a room that was maintained at 22°C under a 12-h light/dark cycle and were provided with drinking water ad libitum. Before the tissues (aortas) were harvested, mice were euthanized by trans-cardiac perfusion under terminal anesthesia induced by intraperitoneal administration of pentobarbital (200 mg/kg). All investigations conformed to the Guidelines for Animal Experiments of the Kyoto Prefectural University of Medicine, following approval by a local university ethics review board.
Hemodynamic analysis
Mean blood pressure and heart rate were measured under conscious and unrestrained conditions using a programmable sphygmomanometer (BP-98A; Softron, Tokyo, Japan).
Plasma lipid analysis
Total cholesterol and triglyceride concentrations were determined using an automated chemistry analyzer (Wako Chemicals Co., Tokyo, Japan). Low density lipoprotein (LDL)-cholesterol levels were quantified by an enzymatic reaction using a commercially available kit (Daiichi Pure Chemicals Co., Tokyo, Japan).
Quantitative measurement of atherosclerotic lesions
The mice were euthanized at 24 weeks and the atherosclerotic lesions were analyzed. Image analysis was performed on Oil Red O-stained aortas using Scion Imaging software (Informer Technologies, Inc. Shingle Springs, USA). The aortic lesion area in each animal was measured as a percentage of the total aortic area.
Immunohistochemistry
For immunohistochemical analysis, the aortic root was rapidly removed after phosphate buffered saline (PBS) perfusion, embedded in optimal cutting temperature compound, and quickly frozen in liquid nitrogen. The atherosclerotic lesions in the aortic root region were examined at five locations at 100-µm intervals, and stained immunohistochemically with fluorescein isothiocyanate (FITC)-conjugated antibodies against monocyte/macrophage antibody-2 (MOMA-2; Serotec, Oxford, UK), and then observed using a confocal microscope (FLUOVIEW FV300; Olympus, Tokyo, Japan). As a negative control, non-immune immunoglobulin and FITC-conjugated secondary antibodies were used. Positive MOMA-2 staining was evaluated using image-analysis software. Coverage of the MOMA-2-stained area as a percentage of the total plaque area was assessed in five sections from six animals in each group.
Isolation of bone marrow-derived macrophages (BMDMs) and polarization assay
Primary cultures of bone marrow-derived macrophages were obtained from femurs and tibias of 8–12-week-old Agtr2+/+ or Agtr2−/− mice and cultured in complete medium (Minimum Essential Medium (MEM) supplemented with 10% fetal calf serum (FCS), 800 pg/ml macrophage-colony stimulating factor-1 (20% L-929-conditioned medium)). Non-adherent cells were collected after 24 h and were differentiated for seven days, yielding 98% F4/80+ cells (data not shown). Differentiated bone marrow-derived macrophages were polarized with either 100 ng/ml LPS+100 U/ml interferon gamma (IFNγ) (M1) or 5 ng/ml interleukin-4 (IL-4) (M2). 12
Isolation of thioglycollate-elicited peritoneal macrophages (TGPMs) and lipopolysaccharide (LPS)-induced inflammatory response
Peritoneal exudate cells were harvested from the peritoneal cavities of wild-type (Agtr2+/+) and Agtr2−/− mice four days after an intra-peritoneal injection of 2.0 ml of 3% sterile thioglycollate medium. After 6 h of incubation, non-adherent cells were removed. Flow cytometric analysis showed that more than 90% of the adherent cells were positive for F4/80. After 18 h of incubation, isolated TGPMs were stimulated by 100 ng/ml of LPS for 6 h. LXR agonist (GW3965, 2 μM) was added 18 h prior to LPS stimulation.
RNA isolation and real-time polymerase chain reaction (RT-PCR)
RNA isolation was performed using the RNeasy mini kit or RNeasy micro kit (Qiagen, Hilden, Germany). The obtained RNA was reversely transcribed with SuperScriptR III First-Strand kit (Invitrogen, Carlsbad, CA). Quantitative real time RT-PCR was carried out using a LightCycler ST300 (Roche Applied Science, Indianapolis, IN) with LightCycler FastStart DNA Masterplus SYBR Green I kit (Roche Applied Science, Mannheim, Germany). The dissociation curves were monitored to check for aberrant primer dimer formation. PCR-amplified products were electrophoresed on 2% agarose gels to confirm the presence of a single band. Data are expressed as levels relative to the Agtr2+/+ mice.
Western blotting
TGPMs were harvested and homogenized in Eppendorf tubes in 100 μl of lysis buffer (62.5 mM Tris-HCl, pH 6.8 containing 2% sodium dodecyl sulfate (SDS) (w/v), 10% glycerol, 50 mM dithiothreitol (DTT) and 0.1% bromophenol blue (w/v)). The cell lysates were sonicated for 2 s, heated at 95°C for 5 min and then centrifuged at 12,000 × g for 2 min at 4°C. Small aliquots (20 μl) of the supernatants were subjected to polyacrylamide gel (10%) electrophoresis containing sodium dodecyl sulfate (SDS-PAGE) and then transferred onto a nitrocellulose membrane (Hybond-P, Amersham-Pharmacia Biotech, Inc., Piscataway, USA). The membrane was then blotted using the LXRβ antibodies (LXRβ: sc-1001; Santa Cruz Biotechnology, Inc., Santa Cruz, USA). Immunoreactive proteins were visualized with an enhanced chemiluminescence (ECL) detection system (Amersham-Pharmacia Biotech, Inc.) followed by exposure to Kodak X-ray film.
Flow cytometric analysis
TGPMs were detached from culture plates using cell dissociation buffer (GIBCO BRL, San Francisco, USA) and analyzed by FACSCalibur (Becton, Dickinson and Company, Franklin Lakes, USA). The antibodies used for analysis were PE-conjugated anti-F4/80 (eBioscience, Inc. San Diego, USA) and PerCP-CyTM5.5-conjugated anti-CCR7 (Becton, Dickinson and Company).
Cholesterol efflux assay
Peritoneal macrophages were obtained as described above and cholesterol efflux was evaluated as described previously. 15 After washing, 5 × 105 cells/well were plated on an eight-well μ-slide in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% FBS, 2 mM glutamine, and 1 U/ml penicillin/streptomycin and were grown overnight. Cells were then incubated with NBD-cholesterol (0.35 μM) in the above medium containing 2.5% FBS for 1 h at 37°C. Cells were washed twice with Puck’s buffer (1 mM Na2HPO4, 0.9 mM H2PO4, 5 mM KCl, 1.8 mM CaCl2, 0.6 mM MgSO4, 6 mM glucose, 138 mM NaCl, and 10 mM HEPES) and placed in serum-free medium. The fluorescence-labeled cholesterol released from the cells into the medium was measured with a multi-label counter. Cholesterol efflux was evaluated as a percentage of fluorescence in the medium relative to the total amount of fluorescence (cells and medium), and expressed as levels relative to the Agtr2+/+ mice.
Statistical analysis
All data are expressed as means ± standard error (SE). Mean values were compared using analysis of variance. If a statistically significant effect was found, Scheffe’s F test was performed to detect the difference between groups. Values of P < 0.05 were considered statistically significant.
Results
BM-AT2 deficiency exaggerates atherosclerosis development in apoE−/− mice
Eight weeks after BM transplantation, 16-week-old chimeric mice were fed a high-cholesterol diet for 2 months. The atherosclerotic lesion area was significantly larger in apoE−/−/BM-Agtr2−/− mice than that in apoE−/−/BM-Agtr2+/+ mice (51%, P < 0.05) (Figure 1(a) and (b)), accompanied by an augmented accumulation of lesion macrophages (Figure 1(c) and (d)). Hemodynamic data (Supplemental Tables 1 and 2) did not differ between the two groups before and after the high-cholesterol diet. Lipid profile (Supplemental Table 3) and circulating blood cell counts (Supplemental Table 4) after 8 weeks of a high-cholesterol diet were also equivalent between the two groups, suggesting that the BM-AT2 receptor plays a crucial role in the pathogenesis of atherosclerosis development.

Bone marrow AT2 deficiency exaggerates atherosclerotic lesion formation.
AT2 deficiency has no effect on phenotypic polarization of BM-derived macrophages
Macrophage phenotype has been conventionally divided into two classes: inflammatory M1 macrophages and resident M2 macrophages, induced by IFN-γ or IL-4, respectively. 12 First, we examined the effect of BM-AT2 on BMDM polarization. The expression levels of M1 markers, such as TNF-α, CXCL10, iNOS, and ICAM-1, were equivalent between the two groups before polarization. Stimulation with IFN-γ significantly increased the expression of M1 markers in both Agtr2+/+ and Agtr2−/− BMDMs, with no discernible difference between the two groups except for iNOS gene expression (Figure 2(a)). Likewise, the expression of M2 markers, such as CD206 and Arg1, was comparable between Agtr2+/+ and Agtr2−/− BMDMs before and after stimulation with IL-4 (Figure 2(b)), suggesting that BM-AT2 did not affect BMDM polarization elicited by IFN-γ or IL-4. We further compared LPS-induced M1 polarization between Agtr2+/+ and Agtr2 −/− TGPMs. Flow cytometric analysis showed no difference in F4/80 expression between Agtr2+/+ and Agtr2 −/− TGPMs (Supplemental Figures 1A and 1B). AT1 receptor mRNA expression levels, as well as F4/80 and CD68, did not differ between the two groups (Supplemental Figure 1C). LPS-induced expressions of inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, were significantly elevated in both Agtr2+/+ and Agtr2 −/− TGPMs; however, no significant difference could be observed between the two groups (Figure 2(c)), suggesting that LPS-induced macrophage polarization was not affected by AT2 deficiency.

AT2 receptor deficiency has no effect on macrophage polarization.
AT2 deficiency remarkably attenuates LXRβ expression in TGPMs
Next, we examined the mRNA expression levels of LXRα and LXRβ, which are a nuclear receptor superfamily of ligand-dependent transcriptional factors exerting anti-atherogenic properties.16–20 LXRα mRNA expression in TGPMs was comparable between Agtr2+/+ and Agtr2 −/− TGPMs, irrespective of LPS stimulation (Figure 3(a)). In contrast, LXRβ mRNA expression was modestly, but significantly, reduced in Agtr2−/− TGPMs compared with Agtr2+/+ TGPMs before LPS stimulation (Figure 3(b)). LXRβ expression was remarkably increased by LPS treatment in Agtr2+/+ TGPMs; however, it was remarkably inhibited in Agtr2−/− TGPMs. Consistently, the protein expression levels of LXRβ also showed a modest, but significant, decrease in Agtr2−/− TGPMs irrespective of LPS stimulation (Figure 3(c) and (d)), suggesting that the LXRβ-associated signaling pathway was impaired in Agtr2−/− TGPMs.
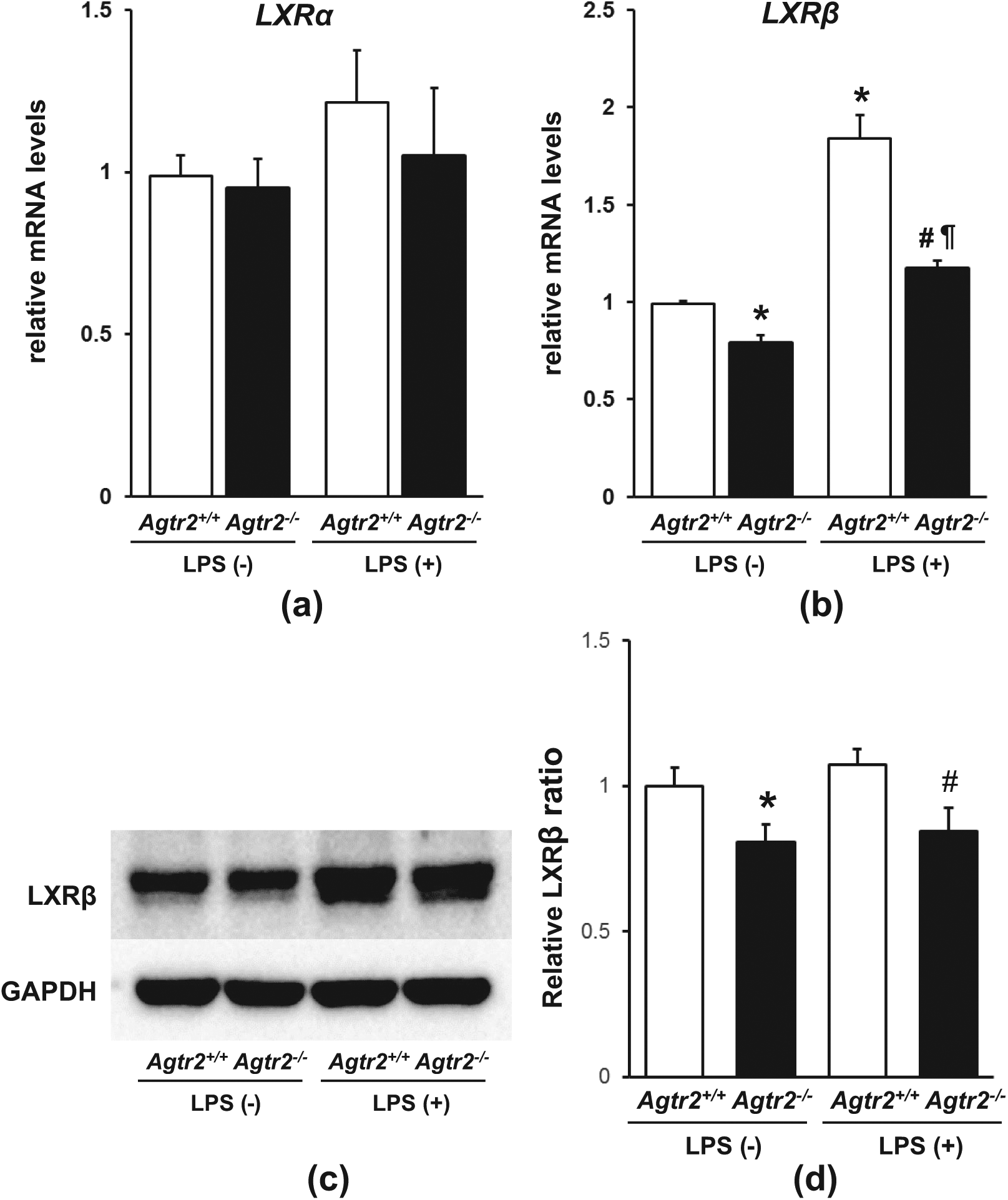
AT2 receptor deficiency attenuates LXRβ expression in thioglycollate-induced peritoneal macrophages.
AT2 deficiency eliminates LXRβ-mediated anti-inflammatory response
To examine the effect of AT2 deficiency on LXRβ-mediated anti-inflammatory response, TGPMs were stimulated by LPS in combination with or without the LXRβ agonist GW3965. Treatment with GW3965 significantly reduced the LPS-induced mRNA expression levels of TNF-α, IL-1β, and IL-6 in Agtr2+/+ TGPMs (Figure 4(a)). In contrast, the anti-inflammatory effect of GW3965 was attenuated in Agtr2−/− TGPMs (Figure 4(b)). We also examined the anti-inflammatory effect of GW3965 treatment on protein expression levels in the culture supernatant fluids. Tumor necrosis factor alpha (TNF-α) protein levels were significantly decreased in Agtr2+/+ TGPMs after treatment with GW3965 (Figure 4(c)), whereas GW3965 treatment had no effect on TNF-α protein levels in Agtr2−/− TGPMs (Figure 4(d)). These findings suggest that the LXRβ-mediated anti-inflammatory response was impaired in Agtr2−/− TGPMs.

AT2 deficiency eliminates LXRβ-mediated anti-inflammatory response.
AT2 deficiency reduces ABCA1 expression and cholesterol efflux in TGPMs
Next, we examined the effect of AT2 deficiency on ATP-binding cassette transporter 1 (ABCA1) expression, which is transcriptionally regulated by LXRβ. 21 ABCA1 mRNA expression was significantly reduced in Agtr2−/− TGPMs by 31% compared with Agtr2+/+ TGPMs (Figure 5(a)). Consistently with this finding, cholesterol efflux was substantially attenuated in Agtr2−/− TGPMs by 35% (P < 0.05) (Figure 5(b)), suggesting that the LXRβ-mediated increase in ABCA1 expression and its anti-atherogenic properties were impaired in apoE−/−/BM-Agtr2−/− mice.

AT2 deficiency attenuates ABCA1 gene expression and cholesterol efflux.
AT2 deficiency inhibits CCR7 expression in TGPMs
We further examined the effect of AT2 deficiency on CCR7 expression, which is up-regulated by LXRβ activation and contributes to macrophage emigration from the atherosclerotic lesions. 22 CCR7 mRNA expression was substantially reduced by 58% in Agtr2−/− TGPMs compared with Agtr2+/+ TGPMs (Figure 6(a)). Flow cytometric analysis also showed that CCR7 expression levels were significantly attenuated in Agtr2−/− TGPMs compared with Agtr2+/+ TGPMs (Figure 6(b)). These findings suggest that the AT2 receptor plays a crucial role in macrophage emigration from atherosclerotic plaque lesions.

AT2 deficiency attenuates mRNA and protein expression of CCR7.
Discussion
In this study, we demonstrated for the first time that BM-AT2 deficiency markedly exaggerated atherosclerosis development with an augmented accumulation of lesion macrophages. Although phenotypic polarization of BM-derived macrophages was not affected by AT2 deficiency, mRNA and protein expression levels of LXRβ, which belongs to a nuclear receptor family of ligand-dependent transcriptional factors exerting anti-atherogenic properties,16–20 were significantly decreased in AT2-deficient TGPMs. While LXR agonist treatment significantly inhibited LPS-induced proinflammatory response in wild-type TGPMs, it was markedly ameliorated in AT2-deficient TGPMs. Moreover, mRNA expression levels of ABCA1 and CCR7, transcriptionally up-regulated by LXRβ activation,21,22 showed significant decreases in AT2-deficient TGPMs, accompanied by markedly reduced cholesterol efflux. These findings suggest that LXRβ-mediated anti-atherogenic action was substantially impaired in BM-AT2-deficient chimeric mice and support the notion that direct AT2 activation could be a potential therapeutic strategy to prevent the development of atherosclerotic diseases.
Liver X receptors (LXRα/β), members of the nuclear receptor superfamily of ligand-activated transcription factors, were originally thought to control lipid metabolism.23,24 The LXRα isoform is highly expressed in the liver and plays a dominant role in hepatic lipogenesis, thereby promoting triglyceride synthesis. 24 Considering that AT2 deficiency has no effect on LXRα expression (Figure 3(a)), the AT2-mediated antiatherogenic effect could be exerted without stimulating hepatic lipogenesis. On the other hand, LXRβ is ubiquitously expressed and has been found to exert anti-inflammatory and anti-proliferative effects. 25 Levin et al. demonstrated that the ability of the LXR agonist to inhibit atherosclerosis progression was lost in mice transplanted with BM lacking macrophage LXR expression, 26 suggesting that macrophage LXR may be a potential therapeutic target to prevent atherosclerosis development. Moreover, LXRβ activation has been reported to increase the expression of ABCA-1 and CCR7 in macrophages, which are closely implicated with cholesterol efflux and macrophage emigration from atherosclerotic plaque lesions.21,22 Systemic administration of the LXR agonist has been reported not only to inhibit atherosclerosis progression but also to cause regression of established atherosclerotic lesions.26,27 Since the mRNA and protein expression levels of CCR7 decreased in AT2-deficient TGPMs, AT2-mediated anti-atherogenic action is likely to be involved in the regression of atherosclerotic plaques by augmenting emigration of monocytes/macrophages from atherosclerotic lesions.
The underlying mechanism of AT2-mediated regulation of LXRβ gene expression remains to be elucidated. The AT2 receptor has been shown to be associated with the activation of a series of phosphatases, including protein tyrosine phosphatase SHP-1, mitogen-activated protein kinase phosphatase-1 (MKP-1), and serine/threonine phosphatase 2A (PP2A).28–32 In silico analysis of promoter sequences identified the binding sites of NFATC2, a transcriptional factor of the Nuclear Factor of Activated T cells (NFAT) superfamily, in the LXRβ promoter region (data not shown). While phosphorylated NFATC2 is located in the cytoplasm, dephosphorylated NFATC2 moves into the nucleus and binds to the promoter region to regulate gene expression. 33 Interestingly, AT2 activation has been demonstrated to induce diphosphorylation of NFATC2 and promote the binding of NFATC2 to the eNOS promoter region, resulting in increased eNOS expression in rat cardiomyocytes. 34 These findings suggest that AT2-mediated dephosphorylation of NFATC2 is likely to enhance its binding to the LXRβ promoter region, leading to increased expression of LXRβ.
LXR activation has been reported to lessen AT1 expression via dephosphorylation of Sp1. 35 Likewise, Takata et al. reported that ABCA1 gene expression was inhibited via AT1 activation. 36 To exclude the possibility that augmented inflammatory response and reduced gene expression of ABCA1 in AT2-deficient TGPMs can be attributed to increased AT1 activation, we examined AT1 receptor mRNA expression. AT1 receptor mRNA expression levels did not differ between AT2-deficient and wild-type TGPMs (Supplemental Figure 1(c)), suggesting that AT2-mediated anti-atherogenic actions are not dependent on AT1-mediated effects.
Recently, compound 21 (C21), a nonpeptide, orally active, and selective AT2 agonist has been developed. 37 Direct AT2 stimulation by C21 has been reported to exert beneficial effects on cardiac and vascular function in rodents through an increased production of NO and/or suppression of the inflammatory response.38,39 Rompe et al. reported that C21 treatment significantly reduced TNF-α-induced IL-6 mRNA levels in primary human and murine dermal fibroblasts. 40 They also showed that injection of bleomycin into murine skin resulted in significantly elevated mRNA expression levels of IL-6, MCP-1, and TNF-α, which were markedly reduced by C21 treatment. Our findings support the notion that AT2 stimulation exerts anti-atherogenic actions and could be a potential therapeutic strategy for producing a clinically beneficial effect on the cardiovascular system.
In conclusion, AT2 deficiency in macrophages markedly inhibited LXRβ-mediated anti-inflammatory response, ABCA1-mediated cholesterol efflux, and migration-related CCR7 expression. AT2-mediated regulation of macrophage LXRβ expression was mostly attributable to the atheroprotective effect of bone marrow AT2 receptor. These findings provide novel evidence for the bone marrow AT2-mediated anti-atherogenic actions and new insight into the mechanism for AT2-mediated effect on atherosclerosis.
Footnotes
Acknowledgements
Agtr2 −/− mice (C57BL/6) were kindly provided by Dr. M. Horiuchi (Ehime University).
Conflict of interest
None declared.
Funding
This work was supported by the Ministry of Education, Culture, Sports, Science and Technology of Japan (grant number C: KAKENHI-17590760).