
Abstract
A hallmark of Francisella tularensis, a highly virulent Gram-negative bacterium, is an unusual LPS that possesses both structural heterogeneity and characteristics that may contribute to innate immune evasion. However, none of the methods yet employed has been sufficient to determine the overall LPS composition of Francisella. We now demonstrate that metabolic labeling of francisellae with [14C]acetate, combined with fractionation of [14C]acetate-labeled lipids by ethanol precipitation rather than hot phenol–water extraction, permits a more sensitive and quantitative appraisal of overall compositional heterogeneity in lipid A and LPS. The majority of lipid A of different francisellae strains grown in diverse bacteriologic media and within human phagocytes accumulated as very hydrophobic species, including free lipid A, with <10% of the lipid A molecules substituted with O-Ag polysaccharides. The spectrum of lipid A and LPS species varied in a medium- and strain-dependent fashion, and growth in THP-1 cells yielded lipid A species that were not present in the same bacteria grown in brain heart infusion broth. In summary, metabolic labeling with [14C]acetate greatly facilitates assessment of the effect of genotypic and/or environmental variables on the synthesis and accumulation of lipid A and LPS by Francisella, including during growth within the cytosol of infected host cells.
Introduction
Recognition of LPS by the innate immune system is typically extremely efficient, with picomolar concentrations of many LPS species sufficient to induce robust inflammation. 1 LPS consists of a glycophospholipid ‘lipid A’ anchor attached to a core oligosaccharide that is attached to a characteristic repeating polysaccharide (O–Ag). Each component of LPS—the lipid A, the core and the O–Ag—can interact with the host to stimulate immune responses, but, generally, the lipid A region is the most potent. The potency of the lipid A of many bacteria derives from its interaction with an array of host LPS-sensing molecules, including LPS-binding protein (LBP), CD14 and MD-2, with subsequent signaling via TLR4. 2 The core and O–Ag polysaccharides and their substituents (e.g. phosphates) can modulate interactions of lipid A with the aforementioned proteins, and can also interact with other host receptors and soluble molecules, such as antimicrobial peptides and complement proteins. 3 Each of these structural components of LPS varies among strains and species of Gram-negative bacteria, contributing to distinct immune responses and pathogenicity.3,4
Francisella tularensis is the extremely infectious Gram-negative bacterium that causes tularemia. Fewer than 10 bacteria can cause life-threatening disseminated infection after inoculation on mucosa or in the skin. These properties stimulated use of Francisella as a potential agent of biowarfare during the Cold War. 5 As suggested by its remarkable infectiousness, Francisella possesses multiple virulence factors that frustrate the host immune response. Together, these render the bacterium a ‘stealth’ pathogen able to establish its replicative niche in the cytosol of host cells. 6 Among its virulence factors is an unusually inert LPS, which likely blunts early inflammatory responses to infection that are usually instrumental in host control of infection.7,8
The inert properties of Francisella LPS are manifest by the failure both to stimulate TLR4 activation and to antagonize the action of potently stimulatory LPS.7,8 These properties suggest that Francisella LPS does not interact with host LPS-binding proteins important in TLR4 activation. In support of this notion, we have shown that Francisella LPS does not bind to LBP or to the closely-related host protein, bactericidal permeability-increasing protein. 9 This failure to engage prominent host innate immune LPS-sensing molecules likely results from the unusual structure of Francisella LPS. In comparison to the structural hallmarks of LPS species recognized efficiently by innate immunity, atypical features of Francisella LPS structure include the number and chain length of 3-OH and non-hydroxylated fatty acids (FA) on the lipid A, the relative paucity of anionic moieties within the lipid A backbone and neighboring inner core sugars, the unusual sugars of F. tularensis-LPS O–Ag and, apparently, the relatively high percentage of LPS that accumulates as free lipid A without substituted core ± O–Ag polysaccharide.10–14 The latter properties, because of the much greater lipophilic nature of free lipid A (vs LPS), have complicated assessment of the overall Francisella envelope LPS composition. Namely, isolation protocols that rely upon the relative hydrophilicity of LPS may lose a substantial fraction of lipid A-containing material if free lipid A partitions to lipophilic solvents. 13 Consequently, the structural and functional analyses of such preparations may fail to include significant sub-populations of free lipid A and LPS molecules. 15
In order to facilitate the study of heterogeneous free lipid A and LPS structures accumulating in Francisella under a variety of growth conditions, we sought to develop methods to metabolically label Francisella LPS. All molecules of LPS, irrespective of precise chemical structure, contain 1 mol of lipid A/mol of LPS. Defining features of lipid A are its 3-OH fatty acyl substituents, which provide a molecular signature of nearly all lipid A.1,3,16,17 Each of the fatty acyl chains within lipid A derive from fatty acids generated by the bacteria by de novo synthesis, utilizing acetyl derivatives to provide the two-carbon building blocks needed in fatty acid synthesis. Thus, bacterial lipid A can be efficiently and uniformly radiolabeled during growth in medium supplemented with radiolabeled acetate, provided the bacteria express cytosolic enzymes needed for conversion of the passively accumulated acetate to the acetyl precursors utilized in de novo fatty acid synthesis. 18 The annotated genomes of both F. tularensis and of Francisella novicida contain orfs predicted to encode these enzymes (acetate kinase and phosphate acetyltransferase—FTL_0015 and FTL_0016, respectively, NCBI accession no. NC_007880.1), suggesting that such metabolic labeling of Francisella lipid A should be possible.
We now report metabolic radiolabeling of F. tularensis and of F. novicida with [14C]acetate to achieve a better definition of the overall composition of the lipid A and LPS of these bacteria. In contrast to other methods that have been used previously, metabolic labeling with [14C]acetate during several generations of bacterial growth permitted representative labeling of all bacterial lipid-containing molecules, irrespective of the extent to which charged substituents or sugar moieties are covalently linked. We applied this experimental approach not only to bacteria growing in bacteriologic media but also to bacteria growing within the cytosol of infected human phagocytes. The latter studies revealed infection-induced alterations of the lipid A species produced and accumulated during intracellular growth of F. tularensis LVS. These new experimental approaches provide sensitive and quantitative methods for lipid A and LPS analysis that can be readily applied to different francisellae strains and species, and to lipid A and LPS that are synthesized in specific growth environments.
Materials and methods
Materials
Monoclonal Ab to F. tularensis LPS (clone FB11) was purchased from QED Bioscience, Ltd (San Diego, CA, USA). [1,2-14C] Acetic acid sodium salt (112 mCi/mmol) was purchased from Moravek Biochemicals Inc. (Brea, CA, USA). 14C-labeled Neisseria meningitidis lipooligosaccharide (LOS) was isolated as described previously. 18 Sheep blood was purchased from Colorado Serum Co. (Denver, CO, USA). Bacto brain–heart infusion broth (BHI), Difco Mueller–Hinton broth (MMH), cysteine heart agar and Isovitalex were from Becton Dickinson (Sparks, MD, USA). Micrococcal DNAse was from New England BioLabs, Inc. (Ipswich, MA, USA). Crystalline phenol was obtained from Sigma-Aldrich Co. (St. Louis, MO, USA). One-hundred percent ethanol (EtOH) was purchased from Deon Labs, Inc. (King of Prussia, PA, USA). PBS was from Mediatech, Inc. (Herndon, VA, USA). EconoSafe scintillation fluid was purchased from Research Products International (Mount Prospect, IL, USA). Granulocyte-macrophage colony-stimulating factor and IL-4 were from R& D Systems, Inc. (Minneapolis, MN, USA).
Growth of bacteria
Francisella tularensis subsp. holarctica strain LVS (ATCC strain 29684) was obtained from Dr Michael Apicella, University of Iowa and F. tularensis subsp. novicida U112, henceforth referred to as F. novicida (Ftn), was obtained from Dr Lee-Ann H. Allen at the University of Iowa. An LVS Himar transposon mutant lacking functional wbtA was a generous gift from Dara Frank (Medical College of Wisconsin, Milwaukee, WI, USA). 19 An flmK mutant of Ftn was a kind gift from Dr Robert K. Ernst (University of Maryland, MD, USA). 20 A fevR mutant of F. tularensis LVS was obtained from Dr Bradley Jones at the University of Iowa. 21 Stocks were frozen at −80℃ and grown overnight (18 h) on cysteine heart agar with sheep blood at 37℃ in 5% CO2. We supplemented BHI with [1,2-14C] sodium acetate, to a final concentration of 5–20 µCi/ml (acetate concentration, 45–180 µM). 22 Escherichia coli CL99 lacks uridine diphosphate galactose epimerase; to allow synthesis of core and O–Ag, the bacterial growth medium was supplemented with 2 mM galactose. Bacteria were suspended to an OD600 of 0.025–0.050 and incubated at 37℃ with shaking at 200 rpm until the OD600 reached ∼1.0 (6 h for Ftn, 9 h for F. tularensis LVS, 6 h for E. coli). Bacteria were washed three times with PBS before subsequent manipulation. For some experiments, modified MMH [supplemented with 0.1% (wt/vol) Glc, 0.025% ferric pyrophosphate and 2% IsoVitaleX] was used.
Isolation of LPS
After growth and harvesting, the bacteria (1/4 of a 7 ml culture) were re-suspended to their original volume with 2% SDS, 10 mM EDTA, 60 mM Tris base (pH 6.8) and incubated at 95℃ for 5 min. The lysate was brought to 37℃ and incubated overnight with proteinase K (Sigma-Aldrich), at a final concentration of 50 μg/ml. The treated lysate was diluted with three volumes of 100% EtOH and 1/10 volume of 3 M sodium acetate (pH 5.4) followed by incubation at −20℃ for at least 4 h. LPS and nucleic acids were pelleted at 12,000 g at 4℃ and washed twice in −20℃ EtOH/sodium acetate as above. Recovered pellets were re-suspended in 500 µl of 50 mM Tris with 5 mM CaCl2 (pH 7.9) containing micrococcal nuclease (2000 Kunitz units/ml) and incubated overnight at 37℃. Hot (65℃) phenol (450 µl) was then added followed by incubation at 65℃ for 30 min, with occasional vortexing. The mix was chilled on ice and then centrifuged for 10 min at 3000 g. The upper aqueous phase was removed and the phenol phase was washed twice more with water. The combined aqueous fractions were washed three times in EtOH/sodium acetate as above. The resulting EtOH-insoluble pellet was washed twice in HPLC-grade H2O by centrifugation at 147,000 g for 75 min at 4℃. Total 14C Cpm in aliquots of various fractions obtained during this procedure was measured in a Beckman LS 5000TD liquid scintillation counter (Beckman Instruments, Inc., Fullerton, CA, USA).
Isolation of LPS by the conventional hot phenol–water procedure was performed as follows. Bacteria were disrupted by overnight incubation with shaking (100 rpm) at 37℃ in 0.2% lysozyme in 10 mM Tris with 50 mM MgCl2 (pH 8.0), followed by incubation with micrococcal nuclease for 3 h at 37℃ shaking at 200 rpm, and precipitation and washing of extracted LPS in acidic EtOH as above. In this article, the term ‘lipid A’ refers to the acylated diglucosamine moiety that may either be bound to core ± O–Ag or may be found free. ‘Free lipid A’ refers specifically to lipid A molecules that lack core and O–Ag polysaccharides. ‘Lipid A/LPS’ is used to refer to the entirety of lipid A-containing molecules expressed by Francisella, given that a large proportion of these are free lipid A.
TLC
FA of intact bacteria and of various derived fractions were prepared for analysis by sequential treatment with 4 N HCl and 4 N NaOH at 90℃ to release ester- and amide-linked FA from the parent lipids, followed by neutralization of the solution and Bligh–Dyer lipid extraction. 22 The released free fatty acids (FFA) were recovered in the lower (chloroform) phase. Normal-phase TLC was performed on 0.25-mm silica gel G HPTLC (Analtech, Newark, DE, USA) using hexane/ethyl acetate/glacial acetic acid (50:50:1 v/v/v) as the solvent system. Reverse-phase TLC used 0.2 mm HPTLC (RP-18; Merck, Whitehouse Station, NJ, USA) with acetonitrile/acetic acid (1:1 v/v) as the solvent system. 18 Purified 14C-FA, obtained either from commercial sources ([14C]14:0 and [14C]16:0) or purified from metabolically-labeled [14C] meningococcal LOS {[14C]12:0(3-OH), 14:0(3-OH), and 12:018} and Francisella LPS {[14C]16:0(3-OH) and 18:0(3-OH); this study} were run in parallel as standards. The relative amounts of resolved 14C-labeled FFA were determined by image analysis (PhosphorImager; Molecular Dynamics, Sunnyvale, CA, USA) using a tritium screen that permitted quantitation of as little as 200 Cpm of 14C-labeled FFA after 24 h of exposure. Enrichment of highly hydrophobic LPS species, including free lipid A, was achieved using a modified Bligh–Dyer extraction as described by Wang et al. 13 Intact bacterial pellets and lipid A/LPS-enriched precipitates were re-suspended in chloroform/methanol/water (1:2:0.8 v/v/v) to create a single phase suspension. Insoluble debris was removed by sedimentation and the recovered supernatant was converted to a two-phase lipid extraction (chloroform/methanol/water, 2:2:1.8 v/v/v). Aliquots of the recovered pellet (re-suspended in water), aqueous and interface, and chloroform phases were measured by liquid scintillation spectroscopy to determine the partitioning of the total [14C] species. The [14C] species recovered in the chloroform phase were further resolved by silica gel HPTLC using chloroform/pyridine/88% formic acid/methanol/water (65:35:10:5:2 (v/v/v/v/v) as the solvent system (modified from Wang et al. 13 ). Resolved [14C] FFA and intact lipids were eluted from normal phase (NP)-TLC and HPTLC with chloroform/methanol (1:1) and either directly analyzed by reverse phase (RP)-TLC or first subjected to chemical hydrolysis and Bligh–Dyer extraction before further analysis by both NP- and RP-TLC. In some instances, in order to better resolve individual slow-migrating bands prior to elution, chloroform/pyridine/formic acid/methanol/water TLC was performed twice upon the same plate.
Reverse-phase HPLC
Chemically hydrolyzed samples containing FFA were dried under nitrogen, dissolved in 90% methanol/0.05% acetic acid and applied to a 5 µ Grace Prevail organic acid 10 mm × 250 mm column. Applied samples were eluted in 90% methanol/0.05% acetic acid at a flow rate of 1 ml/min on a Beckman System Gold HPL chromatograph (Beckman Coulter, Inc., Indianapolis, IN, USA) and collected in 0.5–1.0 ml fractions. Elution of 14C-labeled FFA was monitored by liquid scintillation spectroscopy and analyzed by NP- and RP-TLC (see above). Selected fractions were further analyzed by liquid chromatography (LC)–MS by the High Resolution Mass Spectrometry Facility at the University of Iowa using a Waters Q-TOF Premier with an Acquity ultra performance liquid chromatography system.
SDS-PAGE
Radiolabeled material was dissolved in sample buffer and applied to a 4–12% Bis-Tris gradient gel in 2-(N-morpholino)ethanesulfonic acid buffer (Invitrogen, Carlsbad, CA, USA), after which samples were transferred to polyvinylidene difluoride (PVDF) membranes (Perkin Elmer, Boston, MA, USA). For immunoblotting, the membrane was blocked in 5% dried-milk in Tris-buffered saline with 0.1% Tween-20 (pH 7.5) prior to incubation with mouse anti-F. tularensis LPS monoclonal Ab (clone FB11; Biolegend, San Diego, CA). Bound Ab was detected using goat anti-mouse HRP-conjugated Ab and West Pico enhanced chemiluminescent substrate (Thermo Fisher Scientific, Inc., Rockford, IL, USA). For imaging by autoradiography, the membrane was dried and subjected to image analysis as for TLC. To align the immunoblot and autoradiogram images, two small (∼5 µl) spots on the membrane were dabbed with labeled Francisella LPS, and were thus visible both by immunoblot and autoradiography. For extraction of individual bands from SDS-PAGE, an unfixed gel was washed in hot water and stained with zinc and imidazole as described previously. 23 The desired bands were cut out and sliced into slivers, and zinc was removed from the gel by chelation in 10 mM Tris with 100 mM EDTA (pH 8). The resulting gel slices were disrupted in a tissue grinder, and lipid A/LPS was extracted three times from the resulting slurry with 5% triethylamine. The recovered sample was dried under air and subjected to chemical hydrolysis and lipid extraction of FFA as above.
Isolation and infection of human monocyte-derived dendritic cells
Venous blood was obtained from healthy volunteers in accordance with a protocol approved by the Institutional Review Board for Human Subjects at the University of Iowa. Pooled human serum (PHS) was obtained from approximately 10 healthy donors without a history of tularemia and stored in single-use aliquots at −80℃. Dendritic cells (DC) were differentiated from PBMC that had been obtained from venous blood by Ficoll–Paque density gradient centrifugation.
24
Monocytes were isolated by allowing PBMC to adhere to tissue culture flask for 2 h at 37℃ in RPMI + 10% FBS. Non-adherent cells were washed away, and adherent cells were incubated for 5 d at 37℃ in 5% CO2 in RPMI+ (RPMI 1640 with
DC were infected with unlabeled F. tularensis LVS in the presence of 10% PHS for 2 h at a multiplicity of infection of 500 to 1. DC were washed twice to remove free bacteria and suspended at a concentration of 5 × 105/ml in the same media indicated above (without cytokines). Cell suspensions were spiked with [1,2-14C] sodium acetate to a final concentration of 20 µCi/ml and incubated for 48 h, at which time cells were washed twice to remove free bacteria. DC were pelleted and frozen at −20℃ until subsequent analysis. Growth of LVS in this cell culture medium was scant (< 0.5 log growth), whereas bacterial growth within phagocytes DC was >10-fold (Ben Nasr et al. 25 and data not shown).
Infection of THP-1 cells
THP-1 cells from Dr Lee-Ann H. Allen (University of Iowa) were placed in 12-well cell culture dishes (300,000 per well) and matured to macrophage-like cells with phorbol myristate acetate (100 nM) for 48 h. Adherent THP-1 were washed and then infected by incubating LVS (1 × 108 CFU/ml) in each well and centrifuging 600 g for 10 min. After 2 h, THP-1 cells were washed, treated with 50 µg/ml gentamicin for 45 min, and then incubated with cell culture media supplemented with gentamicin (0.5 µg/ml) and sodium [14C]acetate (10-20 µCi/ml). Cells were harvested 24 h later using trypsin/EDTA solution, washed and stored at −20℃ for subsequent analysis.
Statistical analysis and presentation
Indicated statistical tests were performed using Prism 5 (GraphPad Software, San Diego, CA, USA). Graphs were created with DataGraph 3 (Visual Data Tools, Inc., Chapel Hill, NC, USA), and compiled and annotated with OmniGraffle Pro 5 (Omni Group, Seattle, WA, USA).
Results
Incorporation of 14C acetate into Francisella
We first determined whether [14C]acetate was incorporated by Francisella and, more specifically, into 3-OH fatty acids (3-OH-FA) unique to lipid A. We chose to use two Francisella strains, F. tularensis LVS and F. novicida U112, as their LPSs have been studied most extensively.10,11,15,26 We included E. coli CL99 grown in the presence of Gal to extend our analyses to another Gram-negative bacterial species that produces a well-characterized smooth LPS. 27 A variety of media have been used to cultivate francisellae. Unless indicated otherwise, we chose BHI broth, as this growth medium has been used in previous studies of francisellae, 28 and both francisellae and E. coli grow readily in this medium. [14C]acetate was incorporated into E. coli and a variety of Francisella strains to varied degrees, both in BHI and MMH broth, with all strains incorporating ∼15% of the Cpm in the initial broth inoculum (data not shown). The filtered (0.2 µm) conditioned medium of harvested Ftn and LVS grown in BHI contained <0.1% of the added [14C] acetate in the form of lipid A-containing membrane ‘blebs’ as judged by methods used previously for collection and characterization of blebs from N. meningitidis. 29 In contrast, the medium from harvested E. coli contained LPS-rich membrane blebs that represented 2–3% of the total radiolabeled material in the medium (data not shown).
To measure more specifically the incorporation of [14C] acetate into lipid A, we made use of methods previously established to recover, isolate, and characterize the 3-OH-FA that are specifically present in lipid A (Figure 1A).16–18,22 As expected, little or no [14C]3-OH-FA were present in the bacteria as FFA without pre-treatment of the bacteria or extracted lipid fractions with, sequentially, 4 N HCl and 4 N NaOH at 90℃ to release these fatty acids from ester and amide links to the lipid A backbone (data not shown).
22
After this chemical hydrolysis, the released [14C]FFA—both 3-OH-FA from lipid A and various non-hydroxylated fatty acids (NFA) from phospholipids, as well as lipid A and lipoproteins—were recovered following Bligh/Dyer extraction in the chloroform phase (Figure 1B), representing >80% of the total incorporated bacterial radioactivity. The more hydrophilic properties of the 3-OH-FA (vs NFA) made possible complete separation of the 3-OH-FA from the NFA using NP-TLC (silica gel G HPTLC) and hexane/ethyl acetate/glacial acetic acid (50:50:1, v/v/v) as the solvent system (Figure 1C). The identity of the [14C]lipid (FFA) species of LVS, Ftn and E. coli co-migrating with 3-OH-FA standards was determined by elution of these lipid species from the silica gel, and further resolution and characterization by RP-TLC (Figure 1C) and by HPLC (see below), with various [14C]3-OH-FA standards run in parallel. The major recovered [14C] species were 18:0(3-OH) and 16:0(3-OH) from LVS and Ftn, and, exclusively, 14:0(3-OH) from E. coli. After correcting for the differences in the molar amounts of [14C]acetate incorporated into 18:0(3-OH) vs 16:0(3-OH) owing to differences in fatty acyl chain length, these results indicate a molar ratio of metabolically-labeled [14C]18:0(3-OH)/[14C]16:0(3-OH) of 5–7:1, similar to previous estimates of the composition of 3-OH-FA in Francisella.26,30–32 These findings demonstrate that representative metabolic labeling and recovery of the 3-OH-FA of Francisella lipid A were achieved by incorporation of [14C]acetate during several generations of bacterial growth followed by chemical hydrolysis of the bacteria and quantitative analysis of the 3-OH-FA by TLC/[14C] image analysis. These procedures were used, as described below, to monitor the overall lipid A/LPS composition of the bacteria and the recovery of lipid A/LPS of francisellae during various LPS isolation protocols. RP-TLC of [14C]FA species from LVS, Ftn and E. coli co-migrating with NFA standards (Figure 1C, right panel, rightmost three lanes) showed virtually identical NFA composition of LVS and Ftn. This includes prominent slow-migrating species absent in E. coli that likely correspond to the long-chain FAs (i.e. 22:0/1 and 24:0/1) that have been previously reported in fatty acid analyses of francisellae phospholipids.30–33
Incorporation of [14C]acetate into fatty acids of Francisella and E. coli. (A) Schematic diagram of a predominant species of Francisella lipid A. The ester- and amide-linked 3-OH-FA are indicated.
12
(B) Schematic overview of the extraction and analysis of metabolically-labeled [14C]FA. (C) NP-TLC of recovered chloroform phase after lipid extraction of the indicated metabolically-labeled bacteria. Separated [14C]3-OH-FA and NFA were eluted and further resolved by RP-TLC. Migration of 3-OH-FA and NFA standards are indicated on the left and right of the RP-TLC respectively.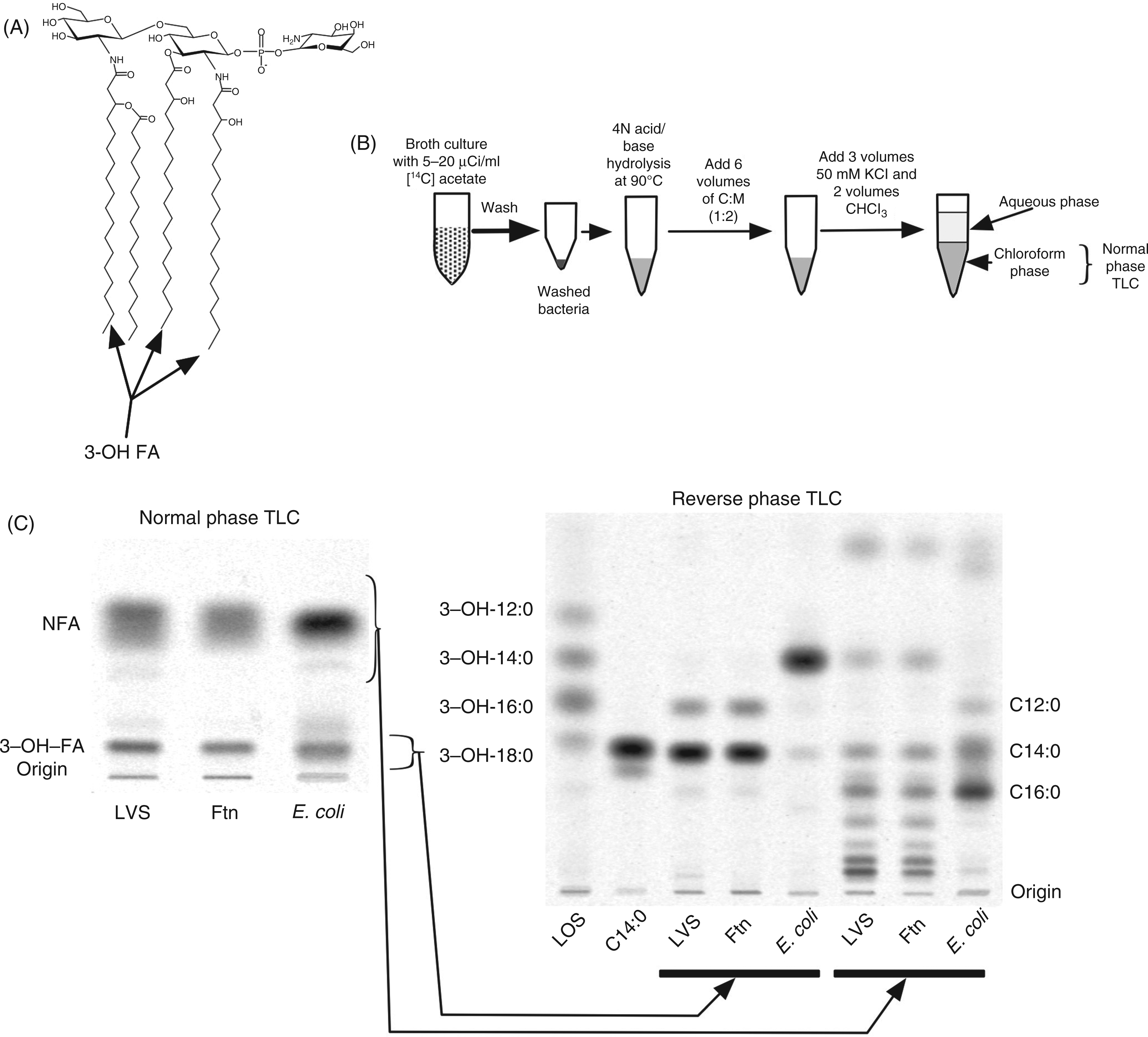
Re-evaluation of methods for isolation of francisellae lipid A/LPS
On the basis of analysis of Ftn metabolically labeled with [32Pi], Wang et al.
13
observed that ∼90% of Francisella LPS is present as free lipid A rather than as LPS containing lipid A with attached 3-deoxy- Comparison of methods to isolate Francisella lipid A/LPS. (A) Schematic representation of conventional phenol–water extraction of LPS. (B) RP-TLC of FA of LPS purified by conventional phenol–water extraction and hydrolyzed to release FA. (C) Fraction of total bacterial 3-OH-FA recovered in the aqueous phase of a conventional phenol–water extraction procedure. (D) Schematic representation of modified LPS extraction protocol. (E) Fractionation of total [14C] cpm by the modified LPS extraction. Total recovery is <100%, reflecting losses occurring during manipulation of samples. (F) RP-TLC of selected fractions recovered during the modified extraction after chemical hydrolysis; the migration of the predominant lipid A FA of LVS is indicated. (G) SDS-PAGE/autoradiogram of LVS LPS preparations purified by conventional phenol–water method versus DNase-treated ethanol precipitate (EtOHp) of SDS/proteinase K-treated bacteria. All results shown are representative of two or more independent experiments.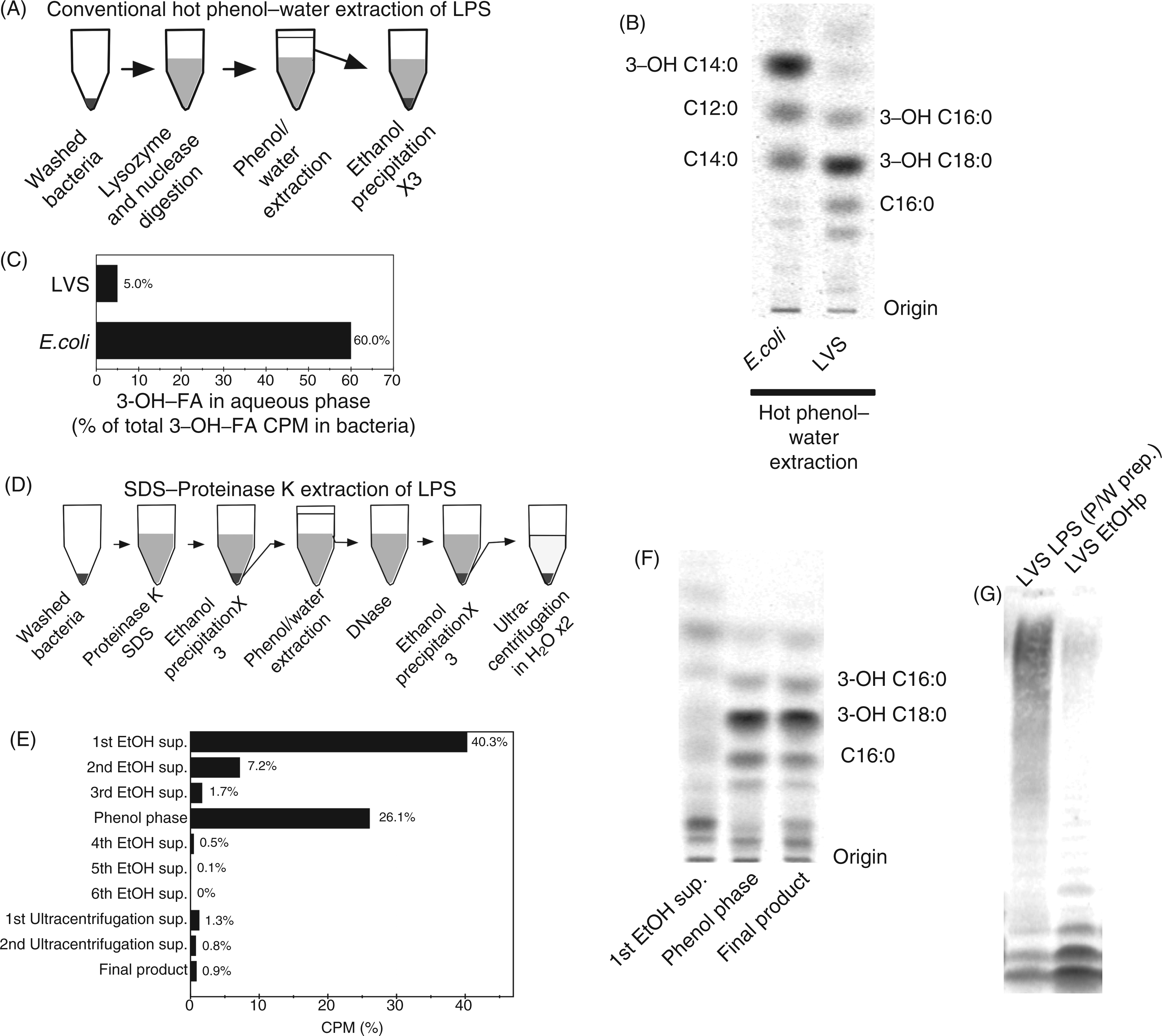
Further characterization of the spectrum of lipid A/LPS species accumulating in Francisella growing in bacteriologic medium (BHI)
Migration of LPS during SDS-PAGE varies inversely with the length of the LPS polysaccharide chain34,35 Free lipid A migrates most rapidly whereas LPS species containing long polysaccharide chains with multiple O–Ag repeats migrate during SDS-PAGE much more slowly. To further define the composition of the [14C]-labeled species recovered in the EtOHp, samples resolved by SDS-PAGE were analyzed in parallel by [14C]-imaging and by immunoblotting, using a monoclonal Ab reactive with the terminal O–Ag repeat of LVS.
36
The immunoblot revealed a ladder of many bands, reflecting the presence of diverse LPS species containing progressively increasing numbers of O–Ag repeats. However, as judged by [14C]-imaging, the most abundant (∼80% of total) [14C]-labeled species in the EtOHp corresponded to the two species migrating fastest during SDS-PAGE that did not react with mAb FB11 and thus apparently lack O–Ag (Figure 3A). Comparison of samples derived from wbtA mutant LVS (which lacks O–Ag
19
) and the parent LVS strain confirmed that the two fastest migrating [14C]-species lack O–Ag whereas the other, slower migrating [14C]-species represent LPS containing O–Ag. Both the immunoblot and [14C]-imaging suggested greater molar abundance of the fastest migrating O–Ag-containing species (‘a’ in Figure 3A) in comparison to the other larger O–Ag-bearing species. The intensity of the [14C] signal within the slower-migrating group was likely enhanced by incomplete resolution of multiple LPS species containing longer polysaccharide chains within this group and increased radiolabeling of the long polysaccharide chain itself, which contains acetylated sugars (see Discussion).
The vast majority of lipid A-containing material in Francisella lacks O–Ag. (A) EtOHp of radiolabeled LVS and a wbtA mutant of LVS were subjected to SDS-PAGE and transfer to a PVDF membrane. The membrane was probed with an Ab to Francisella O–Ag (clone FB11, left) and subjected to autoradiography (right). The images are aligned so that identical portions of the membrane are shown. (B) The fastest-migrating band of [14C]-labeled LVS EtOHp was located using a zinc stain, extracted and subjected to hydrolysis and RP-TLC. (C) Schematic representation of a modified Bligh–Dyer extraction. (D) Immunoblot and SDS-PAGE/[14C]-imaging of molecular species partitioning in chloroform phase after modified Bligh–Dyer extraction of EtOHp or of whole bacteria. (E) (left) Autoradiogram after TLC (chloroform/pyridine/formic acid/methanol/water) of [14C]-labeled LVS EtOHp species recovered in chloroform phase after Bligh–Dyer extraction; (right) NP-TLC of [14C] fatty acids recovered from each of four major resolved [14C]-species after elution and chemical hydrolysis. Migration of [14C] 3-OH-FA and NFA standards is indicated. (F) Percent of total bacterial [14C] 3-OH-FA that partition to the chloroform phase after modified Bligh–Dyer extraction of LVS, FtN and E. coli. Results shown are either from one experiment representative of two or more experiments (A, B, D, E) or the mean ± SD of three experiments.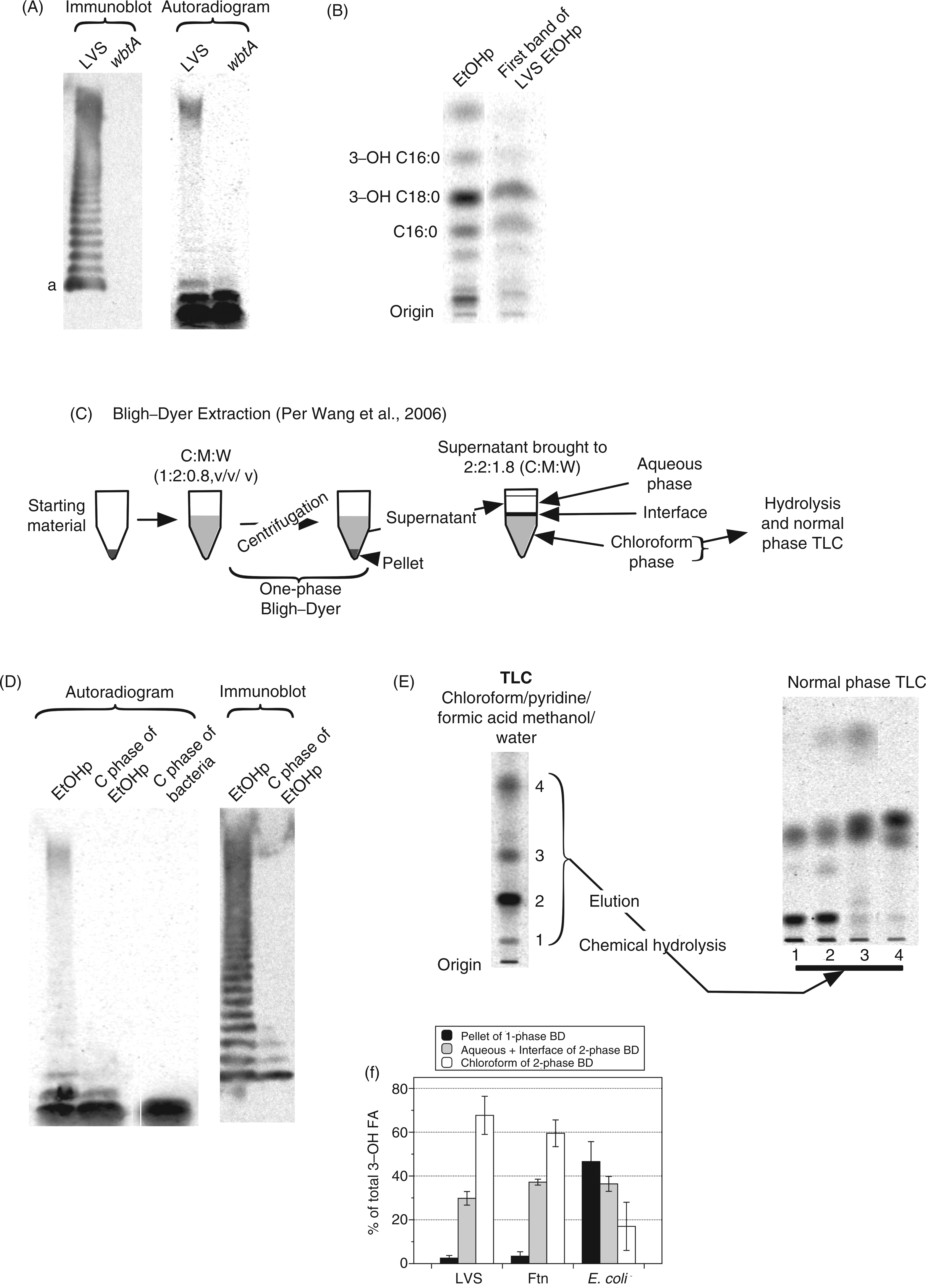
The two fastest migrating [14C]-species could represent, respectively, free lipid A and lipid A containing core, but not O–Ag polysaccharide. It should be noted, however, that the sample analyzed (EtOHp before hot phenol–water extraction) contained a higher fraction of [14C]NFA than expected if this fraction only included lipid-A-containing material, raising the possibility that the fastest migrating [14C]-species on SDS-PAGE included other [14C]-lipids (e.g. phospholipids). To more clearly assess the composition of the fastest-migrating species, the first band was extracted from the gel and subjected to chemical hydrolysis and [14C]FA RP-TLC analysis (Figure 3B). The most prominent [14C] FA recovered co-migrated with 18:0(3-OH) and 16:0(3-OH), confirming that the major [14C]-labeled species present in the fastest migrating band represented lipid A-containing molecules, quite possibly free lipid A. This band represented ∼60% of the total cpm present in the EtOHp, thus suggesting that the majority of lipid A-containing material accumulating during several generations of growth of Francisella is free lipid A.
To confirm that a substantial fraction of the fastest migrating [14C]-species represents free lipid A, we took advantage of methods described by Wang et al. 13 to enrich and resolve the more hydrophobic lipid A-containing species (e.g. free lipid A) away from both the more hydrophilic LPS and the more hydrophobic phospholipids (Figure 3C). SDS-PAGE/[14C]-imaging and immunoblot of a modified Bligh–Dyer extraction of both whole LVS and LVS EtOHp demonstrated a marked enrichment of the fastest migrating [14C]-band, consistent with enrichment of free lipid A in the chloroform phase, away from the more hydrophilic core and O–Ag-polysaccharide-containing LPS species that partition mainly in the pellet and interface/aqueous phases during this extraction (Figure 3D). 22 Resolution of the more polar lipid A-containing species from more hydrophobic bacterial lipids (e.g. phospholipids) was achieved by TLC using a solvent system using chloroform/pyridine/formic acid/water (50:50:16:5, v/v/v/v/) (Figure 3E, left panel). 13 Each of the most prominent [14C]-species was eluted and, after chemical hydrolysis and extraction, analyzed by NP-TLC to identify those species containing [14C]3-OH-FA (i.e. lipid A). As expected, the two slower-migrating species contained lipid A (Figure 3E, right panel), confirming the abundant presence of free lipid A in the metabolically-labeled LVS.
To test for the presence of free lipid A in other francisellae, we performed Bligh–Dyer extraction on whole LVS, Ftn and E. coli, reasoning that the majority of Francisella lipid A-specific 3-OH-FA would partition to the chloroform phase. In fact, ∼60% of the total 3-OH-FA of both LVS and Ftn were recovered in this fraction, whereas E. coli 3-OH-FA partitioned mainly to the interface and aqueous fractions, consistent with a predominance of more hydrophilic LPS species containing core ± O–Ag polysaccharide in these bacteria (Figure 3F). Thus, SDS-PAGE/[14C]-imaging and lipid-extraction of radiolabeled francisellae indicated that approximately 60% of Francisella LVS lipid A produced during bacterial growth in bacteriologic medium (BHI) accumulates as free lipid A lacking core and O-Ag polysaccharide.
Comparison of lipid A/LPS heterogeneity in different strains/subspecies of Francisella or after growth in different broth media
Expression of surface polysaccharides of various Gram-negative bacteria, including the O–Ag of LPS, can vary when bacteria grow in different media.37–39 Our ability to assess the overall lipid A/LPS composition of francisellae by the methods described above prompted us to use these methods to compare the overall lipid A/LPS composition of different Francisella strains/subspecies after growth of each in BHI. Effects of growth of LVS in BHI verus MMH on lipid A/LPS synthesis and steady state accumulation were compared because of previous evidence that expression of virulence-associated genes and a high molecular mass polysaccharide (that is distinct from the O–Ag capsule) were different in these two media.
40
After growth of LVS in BHI, the relative abundance in the EtOHp of the fastest migrating [14C]-species during SDS-PAGE, was slightly greater than the corresponding fraction from bacteria grown in MMH (Figure 4A). Conversely, a slightly higher percentage of the [14C]-species derived from the EtOHp of bacteria grown in MMH co-migrated with immunoreactive species (i.e. contained O–Ag) (Figure 4A). FevR is a transcription factor involved in stress responses and is required for virulence.41,42 fevR LVS accumulated a somewhat higher percentage of [14C]-species in the EtOHp that migrated within the fastest band during SDS-PAGE, and a lower percentage that migrated as very long chain LPS. Finally, SDS-PAGE/[14C]- imaging analysis of [14C]acetate-labeled Ftn revealed a much higher percent of lipid A/LPS containing one, two or an intermediate number of O–Ag repeats, but little of the very long chain LPS that is characteristic of LPS from LVS (Figure 4A).
Comparison of metabolic labeling of francisellae with [14C]acetate under different growth conditions and in different strains and subspecies. (A) SDS-PAGE/autoradiography of [14C]-labeled EtOHp from the indicated bacteria. Quantitation of the [14C] within the fastest migrating band and within all bands migrating slower than the two faster migrating bands was determined by [14C]-imaging. Results are representative of two independent experiments. (B) Human DC were incubated either alone or after infection with LVS in culture medium containing [14C]acetate. After 48 h incubation, harvested DC were subjected to hydrolysis and NP-TLC, along with the indicated controls. (C) Uninfected DC, LVS-infected DC, and BHI-grown LVS were subjected to SDS-proteinase K digestion followed by precipitation in acidic EtOH. The recovered EtOHp were subjected to SDS-PAGE followed by autoradiography and immunoblot for LVS O-Ag. Equal cpm from EtOHp of extracellular LVS and infected DC were loaded; the much lower yield of Cpm in the recovered EtOHp from the uninfected DC resulted in loading of eightfold fewer cpm from this sample. Results are representative of two experiments of identical design. (D) Equal quantities EtOHp derived from either BHI- or THP-1-grown LVS [as determined by LC-MS quantitation of 18:0(3-OH)] were subjected to SDS-PAGE/immunoblot for O–Ag. (E) Similar cpm of the same samples were subjected to SDS-PAGE/[14C]autoradiography. (F) (left) Free lipid A isolated from EtOHp of the indicated samples was extracted by modified Bligh–Dyer extraction and resolved by chloroform/pyridine/formic acid/methanol/water TLC. * and **, bands appearing only in LVS grown within THP-1 and not in BHI-grown LVS. (right) The band indicated by ** was extracted from the TLC plate and subjected to chemical hydrolysis and NP-TLC to reveal 3-OH-FA.
Metabolic labeling of F. tularensis LVS lipid A/LPS during intracellular growth in human phagocytes: Synthesis/accumulation of novel lipid A species
The efficiency and specificity of metabolic labeling of Francisella 3-OH-FA with [14C]acetate prompted us to apply this approach to Francisella growing in human cells in cell culture. For this purpose we incubated LVS + 10% PHS with human monocyte-derived DC at 37℃ for 2 h to allow phagocytosis. After washing the DC to remove remaining extracellular bacteria, we spiked the tissue culture medium with [14C]acetate (20 µCi/ml, final concentration) and incubated the infected cells for 48 h to permit intracellular bacterial growth and metabolic labeling. After the 48 h incubation, DC and DC-associated bacteria were separated from non-adherent extracellular bacteria by differential centrifugation, and the recovered cell pellets were washed and subjected to either (i) chemical hydrolysis (to release [14C]FFA) and [14C]FA/TLC analyses (Figure 4B); or (ii) SDS-proteinase K extraction and EtOH precipitations followed by SDS-PAGE/immunoblot and [14C]-imaging (Figure 4C). Normal-phase TLC revealed the prominent presence in LVS-infected DC, but not uninfected DC of Francisella-derived 3-OH-FA (Figure 4B). Application of the same multi-step SDS-proteinase K/ethanol–sodium acetate extraction procedure used to quantitatively recover highly enriched lipid A/LPS from extracellular francisellae also yielded [14C]-labeled species from infected DC, but not from uninfected DC, that closely resembled the [14C] species in EtOHp from metabolically-labeled LVS grown in BHI, as judged by SDS-PAGE/immunoblot-[14C]-imaging (Figure 4C). These results clearly demonstrate that these novel methods can be used to quantitatively characterize the lipid A/LPS species produced by Francisella both during extracellular growth in culture media and within host cells.
To facilitate comparison of the overall composition of F. tularensis lipid A/LPS accumulated during growth in BHI versus intracellular growth in infected human cell cytosol, we scaled-up experiments with infected human cells. For this purpose, we adapted the conditions of the experiments with primary human monocyte-derived DC to larger cell cultures using cultured THP-1 cells pre-treated with PMA to express a macrophage-like phenotype. Conditions were optimized to achieve infection of ∼60% of the THP-1 cells with 1–5 bacteria per cell (not shown), resulting in ∼100-fold growth in 24 h of culture. The larger quantities of EtOHp that we could isolate from these experiments permitted a more detailed analysis of the lipid A/LPS produced and accumulated by the intracellular replicating LVS. We first compared O–Ag expression by LVS grown either in BHI or in THP-1. In order to quantitate the amount of lipid A/LPS in samples of EtOHp, we isolated [14C]18:0(3-OH) by chemical hydrolysis, Bligh–Dyer extraction and HPLC of the recovered chloroform phase (Supplementary Figure 2A). LC–MS of the recovered 18:0(3-OH) was used to estimate the molar amounts of lipid A-containing material recovered in corresponding EtOHp fractions (Supplementary Figure 2B), permitting comparison of equal quantities of lipid A/LPS from intracellular LVS versus BHI-grown LVS by SDS-PAGE/immunoblot imaging. This comparison (Figure 4D) revealed a distribution of O–Ag containing LPS molecules that was grossly similar, though there may have been fewer of the largest (i.e. slowest-migrating) LPS molecules in bacteria grown in THP-1 cells. Comparison of the same samples by SDS-PAGE/[14C] autoradiography revealed a similar relative abundance of free lipid A verus lipid A linked to core polysaccharide ± O–Ag in LVS grown in BHI and LVS multiplying in the cytosol of THP-1 cells (Figure 4E). Further resolution of the free lipid A species was achieved by extraction and enrichment of these species with a modified Bligh–Dyer procedure followed by TLC of the recovered chloroform phase in a solvent system containing chloroform/pyridine/formic acid/methanol/water (65:35:10:5:2; v/v/v/v/v, modified from Wang et al. 13 ). Comparison of the corresponding fractions derived from bacteria grown in BHI versus LVS-infected THP-1 cells revealed two relatively slow migrating (i.e. relatively polar) [14C] species that were selectively present in the LVS-infected cells vs LVS grown in BHI (Figure 4F, left panel; bands are marked with asterisks) or uninfected THP-1 cells (Supplementary Figure 3). Resolution of the faster-migrating of these two species was sufficient to permit partial structural characterization of this species after elution from the silica gel. This was achieved by chemical hydrolysis, Bligh–Dyer extraction, and NP-TLC of the recovered chloroform phase. This analysis established that this [14C]-species contained 3-OH-FA (Figure 4F, right panel) and, hence, represented an altered form of F. tularensis lipid A. The slower migrating of the two novel [14C]-species from LVS-infected THP-1 cells (marked in Figure 4F with an asterisk) appeared to co-migrate with a lipid A species of wt Ftn grown in BHI that is absent in flmK Ftn, consistent with intracellular infection-induced polar modifications of Ft lipid A (see Discussion).
Discussion
Francisella lipid A/LPS differs from that of many Gram-negative bacteria in its structure and its ability to evade engagement of many innate immune recognition systems.7–12,43,44 Efforts to better characterize the structural basis for the unusual functional properties of Francisella lipid A/LPS and the possible genetic and environmental determinants of the synthesis and structure of lipid A/LPS have been hampered by the heterogeneous array of lipid A/LPS species produced by even a single strain under particular growth conditions.13,15,26,44 Adding to this analytical quandary has been the apparent inability of any of the conventionally used methods for LPS isolation to recover a substantial fraction of the lipid A-containing species from francisellae and of any of the routinely used detection methods (e.g. immunoblot with anti-O–Ag Abs, zinc or silver staining, metabolic labeling with 32Pi)13,36 to accurately reflect the relative molar abundance of the various lipid A-containing species. The data presented in this study show that metabolic labeling of francisellae with [14C]acetate combined with fractionation of [14C]-labeled lipids by EtOH precipitation permits sensitive and quantitative analysis LPS heterogeneity within multiple bacterial strains and bacterial culture/growth conditions. In contrast to the other methods previously used for detection of Francisella LPS, the characteristic FA composition of Francisella lipid A and the representative metabolic labeling achieved with [14C]acetate permit assessment by [14C]-imaging of the relative abundance of different lipid A/LPS species resolved by differential extraction, TLC or SDS-PAGE. With these new analytical tools in hand, we have been able to confirm, extend and make several new observations concerning the overall LPS composition of francisellae, grown both in broth and within human phagocytes.
Our data show clearly that in both F. tularensis LVS and F. novicida, only a small portion of the total lipid A/LPS pool contains O–Ag. By [14C]-imaging of samples resolved by SDS-PAGE, only ∼20% of the [14C]-species of LVS and Ftn recovered in the EtOHp after SDS/proteinase K treatment and several cycles of 70% EtOH/0.3 M sodium acetate (pH 5.4) washing migrated like LPS species containing O–Ag. Even these low percentages, however, represent an over-estimate of the relative molar abundance of the long chain LPS species in LVS and Ftn. In LVS and Ftn, three and all four of the sugars, respectively, of the O–Ag tetrasaccharide repeat are acetylated and thus are likely also metabolically labeled by [14C]acetate. Indeed, chemical hydrolysis of hot phenol–water-derived [14C]LPS from LVS highly enriched in long-chain LPS (Figure 2G) followed by Bligh–Dyer extraction yielded ∼60% of the recovered [14C] in the aqueous phase, where deacylated polysaccharides partition. By contrast, similar treatment of the EtOHp yielded about 90% of the recovered [14C] in the chloroform phase where the released FFA partition (data not shown). Given that 80% of the cpm in phenol–water-derived LPS migrated as O–Ag-containing species (Figure 2G), these findings indicate that ∼75% of the radiolabel in the overall O–Ag-containing LPS pool in LVS represented metabolic labeling of the O–Ag sugars and, thus, the mol fraction of this pool is only ∼5% of the total lipid A/LPS pool in LVS.
The very low abundance of LPS containing O–Ag in LVS and Ftn is especially remarkable given that mutations in the wbt locus result in a profound diminution of virulence,45–48 an increase in sensitivity to complement, and reduced or delayed intracellular growth under complement-free conditions.45,49,50 Metabolic labeling with [14C]acetate made possible a comparison of the relative abundance and average polysaccharide chain length of O–Ag-bearing LPS from LVS and Ftn, and revealed that the most abundant long-chain LPS species in Ftn contain significantly shorter polysaccharide chains than those in LVS. In light of our recent findings that complement opsonization of Ftn is more efficient than that of LVS, it is tempting to speculate that the expression by LVS of LPS with longer polysaccharide chains, even in relatively low molar abundance, contributes to the greater complement resistance of LVS. 51 It should be noted that Francisella LVS also produces an O–Ag capsule that is not associated with lipid A. 52 Thus, bacterial resistance and virulence that is dependent on the wbt locus could reflect, at least in part, the effects of lipid A-free O–Ag capsule. Although the methods described in this study have been directed toward identification of lipid A-containing Francisella products, the demonstrated radiolabeling of O–Ag sugars achieved by metabolic labeling with [14C] acetate could assist future studies on the biogenesis and effects of lipid A-free O–Ag polysaccharide.
Typical isolation protocols for producing highly purified enterobacterial LPS have used phenol–water separation to separate the more hydrophilic LPS species from other, more hydrophobic acylated compounds, including phospholipids, and lipoproteins and peptides. Removal of contaminating lipopeptides can be especially difficult, sometimes requiring repeated phenol extractions. 53 Our work joins that of others who have noted that isolation protocols that depend upon the relative hydrophilicity of LPS molecules lose major subpopulations of Francisella lipid A-containing material. Furthermore, we confirm, in part, the findings of Wang et al. 13 that a large proportion of francisellae (both Ftn and LVS) lipid A-containing material accumulate in late log phase/early stationary phase bacteria as free lipid A. 13 In contrast to the observations of Wang et al., 13 which suggested that >90% of accumulated lipid A-containing material in Ftn and LVS was free lipid A, we estimate that this fraction was no more than 60%, as judged by the percentage of the metabolically-labeled [14C]-species that migrate fastest during SDS-PAGE and, following further purification by modified Bligh–Dyer extraction and TLC, have an FA composition indicative of purified lipid A. Because a portion of the core ± O–Ag-polysaccharide-containing molecules of Francisella LPS appears to lack phosphate groups altogether via action of a lipid A 1-phosphatase, 54 free lipid A is over-represented in radiographic analysis of 32Pi-labeled bacteria. Although differences in bacterial growth conditions used in our study vs that of Wang et al. 13 could contribute to differences in the amount of free lipid A that accumulates in francisellae, we believe that the more representative metabolic labeling of all lipid A/LPS molecules achieved with [14C]acetate vs 32Pi may be most important.
The very high mol fraction of the total lipid A/LPS pool of francisellae that is represented by carbohydrate-poor, hydrophobic molecules presumably explains the very low recovery of lipid A-containing material in the aqueous phase following hot phenol–water extraction by either the conventional (Figure 2A) or modified (Figure 2D) protocols. The partitioning of the vast majority of the [14C]3-OH-FA (lipid A)-containing material in the phenol phase (Figure 2E) presents a significant challenge to efforts to use purified francisellae lipid A/LPS to probe interactions (or the lack thereof) with defined host endotoxin-recognition systems. As our findings demonstrate, studies that have relied on Francisella LPS purified by conventional (i.e. hot phenol–water) methods, have analyzed interactions with LPS that represents only a small fraction of the total lipid A/LPS pool and have structural properties different from that pool as a whole. The binding of cationic antimicrobial proteins to the LPS of Gram-negative bacteria differs based upon the density and relative accessibility of anionic moieties (such as Kdo and phosphates) 55 within the inner-core-lipid A region and thus, in part, upon the length or composition of LPS polysaccharide chains. 56 Variation in the size distribution of LPS has also been associated with bacterial resistance to complement-mediated lysis and opsonization.57–59 Although a high proportion of free lipid A—and the corresponding lack of core and O–Ag sugars—might thus be expected to expose more bacterial surface structures to recognition by the host, the lack of negatively charged groups within free Francisella lipid A may simultaneously frustrate interactions with host cationic antimicrobial molecules that rely upon electrostatic attraction for high affinity interactions. 9
Finally, and perhaps most importantly, we have shown that the combined experimental approaches we developed in this study can also be applied to measuring and monitoring synthesis and accumulation of lipid A/LPS of bacteria replicating within the cytosol of infected host cells. We have demonstrated the feasibility of this approach in two different populations of human phagocytes: primary monocyte-derived DCs and cultured macrophage-like differentiated THP-1 cells. The use of the latter cells made it possible to scale-up our experiments sufficiently to permit comparison of the structural and compositional properties of F. tularensis LVS lipid A/LPS after intracellular (cytosolic) growth in THP-1 cells versus growth in BHI. No gross differences were seen in either the overall 3-OH-FA content of LVS lipid A/LPS or in the relative abundance of free lipid A versus lipid A substituted with core ± O–Ag polysaccharide. However, resolution by TLC of a fraction enriched in free lipid A species led to discovery of two metabolically-labeled species from infected cells that were not present in the corresponding fractions from LVS grown in BHI (Figure 4F) or from uninfected THP-1 cells. Although we have not yet recovered enough of either of these species for definitive structural analysis, several considerations suggest that these metabolically-labeled products represented polar modifications of LVS lipid A that were induced during intracellular/cytosolic replication of the bacteria in THP-1 cells, including (i) the more rapidly migrating of these two products (marked ** in Figure 4F) contained 3-OH-FA and, hence, was derived from Francisella lipid A; and (ii) the slower migrating of the two infection-induced [14C]-products appeared to co-migrate with a lipid A species formed during growth of wild type, but not flmK mutant Ftn (Figure 4F).
60
FlmK is a glycosyl transferase required for galactosamine and Man substitutions at the 1-phosphate group and the 4’ position (Figure 5), and we speculate that the slower migrating species is a lipid A substituted with one or more hexose residues not present in BHI-grown LVS. Further characterization of these molecules is required, particularly as lipid A modification of Ftn and other Francisella subspecies may be distinct,
44
but we hypothesize that alterations in free lipid A structure during adaptation to the host cytosol may result in bacteria more resistant to antimicrobial molecules or endowed with novel immunomodulatory properties.
Schematic representation of Francisella lipid A and the location of hexose substitutions dependent upon FlmK in Ftn.
60
In summary, we have described methods that permit, for the first time, sensitive and quantitative appraisal of the overall composition of lipid A/LPS produced by francisellae during bacterial growth in both laboratory media and within infected human cells. The latter has yielded preliminary data demonstrating—for the first time—infection-induced changes in synthesis and accumulation of Francisella lipid A/LPS. These observations add to recent findings demonstrating the capacity of francisellae to reprogram synthesis of lipid A/LPS in response to temperature-induced changes in the bacterial growth environment 61 and thus underscore the adaptive potential of these bacteria to modulate the structure of these major potentially immune-reactive bacterial surface components. The methods we have developed and tested in this study should be applicable to the study of other Gram-negative bacterial pathogens, including those that initially may depend on producing un-reactive lipid A/LPS and occupying intracellular niches to evade usually effective host defenses.
Footnotes
Funding
This work was supported in part by funds from the Public Health Service: P01AI044642-12 awarded to Lee-Ann H. Allen, and 5U54 AI057160-08 awarded to Jason H. Barker via the Midwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research.
Conflict of interest
The authors do not have any potential conflicts of interest to declare.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.