
Abstract
Upon virus infection, the host innate immune response is initiated through the activation of IFN regulatory factor 3 (IRF3) and NF-κB signaling pathways to induce IFN production. Previously, we demonstrated EBV BGLF4 kinase suppresses IRF3 function in a kinase activity-dependent manner. The replacement of Ser123, Ser173 and Thr180 into alanines at the proline-rich linker region of IRF3 abolishes BGLF4-mediated suppression. In this study, we show that BGLF4 phosphorylates glutathione-S-transferase (GST)–IRF3(110-202), but not GST–IRF3(110-202)3A mutant (S123/S173/T180A) in vitro. Compared with activation mimicking mutant IRF3(5D), the phosphorylation-defective IRF3(5D)3A shows a higher transactivation activity in reporter assays, whereas the phosphorylation-mimicking IRF3(5D)2D1E, with Ser123 and Ser173 mutated to aspartate and Thr180 to glutamate, has a much lower activity. To explore whether similar cellular regulation also exists in the absence of virus infection, candidate cellular kinases were predicted and the transactivation activity of IRF3 was examined with various kinase inhibitors. Glycogen synthase kinase 3 (GSK3) inhibitor LiCl specifically enhanced both IRF3(5D) and wild type IRF3 activity, even without stimulation. Expression of constitutive active GSK3β(S9A) represses LiCl-mediated enhancement of IRF3 transactivation activity. In vitro, both GSK3α and GSK3β phosphorylate IRF3 at the linker region. Collectively, data here suggest GSK3 phosphorylates IRF3 linker region in a way similar to viral kinase BGLF4.
Introduction
IFN regulatory factor 3 (IRF3) is a major transcription factor of host innate immune response. IRF3 is composed of three domains and is activated through multiple tank-binding kinase 1 (TBK1)- or IκB kinase (IKK) ɛ-mediated phosphorylation at its C-terminus to release the auto-inhibition effect. The phosphorylated IRF3 then forms a dimer, translocates into the nucleus and binds to its responsive elements. With the cooperation of NF-κB and activating transcription factor 2 (ATF-2)/C-Jun, IRF3 can efficiently activate the expression of IFNβ, which subsequently binds to IFN receptors and induces the phosphorylation and activation of the Janus kinases JAK1 and Tyk2, followed by the phosphorylation of signal transducer and activator of transcription (STAT)1 and STAT2, and further antiviral gene expression. 1 To turn off IRF3 signaling, the active IRF3 may be phosphorylated at Ser339 and subsequently targeted for proteasome dependent degradation. 2 Because IRF3 signaling can also lead to the apoptosis pathway through activation of p53,3,4 it is important to keep a low background of intracellular IRF3 activity in the absence of pathogen challenge.
To avoid the broad antiviral effects of IFN signaling, viruses have evolved various strategies to block the IRF3 signaling pathway to benefit virus replication. In the case of RNA viruses, the leader protein of mengovirus prevents IRF3 from forming dimers, 5 and the leader protein of Theiler’s virus reduces the amount of IRF3 translocated into nucleus. 6 Herpesviruses have evolved different strategies to block IRF3 function. K-bZIP of Kaposi’s sarcoma-associated herpesvirus (KHSV) binds to and competes for IRF3 DNA target sites, and therefore prevents IRF3 binding to DNA. 7 KHSV also encodes vIRF1 to interfere with the interaction between IRF3 and CREB-binding protein (CBP). 8
Previously, we demonstrated that EBV BGLF4 kinase down-regulates IRF3 transactivation through linker region phosphorylation. 9 Using mutagenesis and reporter assays we identified that phosphorylation at Ser123, Ser173 and Thr180 of IRF3 contributed additively to BGLF4-mediated suppression of IRF3 function. The phosphorylation did not interfere with IRF3 dimerization and nuclear translocation, but the DNA binding ability of IRF3 to its responsive element was down-regulated. Interestingly, the BGLF4 homolog of murine gammaherpesvirus 68 (MHV68), ORF36, was also observed to block IRF3 transactivation function through inhibiting the interaction between CBP and IRF3, leading to the suppression of RNA polymerase II to be recruited to the IFN-β promoter. 10 These findings indicate that the viral kinase-mediated down-regulation of IRF3 activity is a conserved mechanism for γ-herpes viruses to counteract the innate antiviral responses. It leads us to hypothesize that in the absence of viral infection, there may be cellular kinase(s) that also negatively regulate IRF3 activity.
In this study, we set out to identify the candidate cellular kinase that can negatively regulate IRF3 transactivation function. Through phosphorylation site prediction and kinase inhibitor screening, glycogen synthase kinase 3 (GSK3) was identified as a candidate negative regulator of IRF3. GSK3 α and β are ubiquitous serine/threonine kinases first identified to phosphorylate and inactivate glycogen synthase. GSK3 was later demonstrated to be the mediator in multiple cellular signaling and physiological processes.11,12 Under normal physiological conditions, GSK3 is present as an active form. Upon pathogen infection or other stimulation, the phosphorylation of GSK3α (Ser21) or GSK3β (Ser9) inactivates GSK3 in immune cells. 13 GSK3 is also involved in the regulation of TLR-mediated cytokine production. Upon inhibition of GSK3, TLR4- and TLR2-mediated release of IL-10 is increased and the production of pro-inflammatory cytokines, including IL-12 and IL-1β, are decreased. 14 Additionally, GSK3β negatively regulates TLR4-mediated IFN-β production through down-regulating levels of the transcription factor c-Jun and its nuclear association of ATF-2 in LPS-stimulated macrophages. 15 Nevertheless, GSK3β was found to regulate the IRF3-mediated antiviral response through physical association to activate TBK1 in a GSK3 kinase activity-independent manner. 16 Our current study further identifies that GSK3 also down-regulates IRF3 activity through direct phosphorylation at its linker region, suggesting GSK3 as a fine-tuning factor that regulates IFN production through multiple pathways.
Materials and methods
Plasmid constructs
pCR3.1-based plasmids (Invitrogen, Carlsbad, CA, USA) expressing wild type IRF3 (pCR3.1-huIRF3) and IRF3(5D) [pCR3.1-huIRF3(5D)] were kindly provided by Dr Y. L. Lin.
17
pJTW74 [glutathione-S-transferase (GST)-huIRF3(110-202)] and pJTW75 [GST-huIRF3(110-202)3A] were generated by cloning a PCR-amplified DNA fragment encoding amino acids 110–202 of IRF3 from pCR3.1-huIRF3 or pJTW68 [pCR3.1-huIRF3(5D)3A]
9
into the BamHI site of pGEX-4X-1 (Amersham Biosciences, Fairfield, CT, USA) respectively. The primers used in the PCR reaction were sense primer 5′-CGGGATCCGGAGGCGTGAACTCAGGAGTTGGGG-3′ and anti-sense primer 5′-CGGGATCCTCACCACTCTTCCCCCGGCACCAACA-3′. A series of mutant plasmids were generated to map the phosphorylation site of IRF3, including pJTW65 [pCR3.1-huIRF3(5D)S123A], pJTW66 [pCR3.1-huIRF3(5D)S173A], pJTW67 [pCR3.1-huIRF3(5D)T180A], pJTW69 [pCR3.1-huIRF3(5D)S123/173A], pJTW71 [pCR3.1-huIRF3(5D)S173/T180A], and pJTW70 [pCR3.1-huIRF3(5D)S123/T180A], which were generated by a single primer-based in vitro mutagenesis strategy,
18
and the primers used were 5′-CCCAGCCAGACACC
Cell culture and transfection
HEK293 cells were maintained in DMEM supplemented with 10% FCS, and cultured in the presence of penicillin (100 U/ml) and streptomycin (100 mg/ml) at 37℃ with 5% CO2. Plasmid DNA was transfected into HEK293 cells using the calcium phosphate-N,N-bis(2-hydroxyethyl)-2-amino-ethanesulfonic acid method. 22
Protein expression and purification from recombinant baculovirus and in vitro kinase assay
Recombinant baculovirus expressing His-BGLF4 or His-K102I protein was generated using Bac-to-Bac system (Invitrogen, Carlsbad, CA, USA) as described previously. 23 The expression and purification of GST fusion proteins were carried out as described previously. 9 Purified GST, GST–IRF3(110–202) and GST–IRF3(110–202)3 A were used as substrates for the kinase assay. For the in vitro kinase assay, purified His-BGLF4 or His-K102I from the recombinant baculovirus system were incubated with 0.5 µg GST fusion protein or GST in the presence of 10 μCi [γ-32P]ATP and kinase buffer A (20 mM Tris HCl, pH 7.5, 1 mM EDTA, 1 mM DTT, 10 mM MgCl2, 0.2 mM Na3VO4, 100 µM ATP) for 1 h at 37℃. For the GSK3α or GSK3β in vitro kinase assay, recombinant active human GSK3α or GSK3β (Upstate Biotechnology, Billerica, MA, USA) was incubated with 0.5 µg GST fusion protein or GST in the presence of 10 μCi [γ-32P]ATP and kinase buffer B (50 mM HEPES, pH 7.4, 0.5 mM DTT, 5% glycerol, 800 mM MgCl2 and 10 µM ATP) for 1 h at 30℃. To stop the reaction, 2× SDS sample buffer was added, and the reaction mixtures were boiled for 5 min before electrophoresis. The gel was then processed for autoradiography and Coomassie Blue staining.
Reporter assay
HEK293 cells (2 × 105) were seeded into 12-well plates the day before transfection. Cells were then co-transfected with reporter plasmid pLuc-PRD(III-I)3, which contained the IRF3 responsive elements of IFNβ promoter, and wild type or mutant IRF3 expression plasmid as indicated. The total amount of DNA was kept constant by supplementing empty vector DNA. pEGFP-C1 was transfected at the same time for normalization of transfection efficiency. At 20 h after transfection, cells were treated with various kinase inhibitors for 24 h and luciferase activities were determined with Bright-Glo luciferase assay system (Promega, Madison, WI, USA). Inhibitors used were roscovitine (10 µM in DMSO; Calbiochem, Darmstadt, Germany) for Cdc2/ cyclin-dependent kinases (Cdk)5, U0126 (20 µM in DMSO, Calbiochem, Darmstadt, Germany) for ERK1 and LiCl (20 mM, Sigma-Aldrich, St. Louis, MO, USA) for GSK3. In all experiments, results are means ± SD from two separate transfections. Data are representative of two independent experiments.
Western blot
Western blotting analysis was performed as described previously. 24 The primary Abs used were IRF3 Ab (Santa Cruz Biotechnology, Santa Cruz, CA, USA), GAPDH Ab (Biodesign, Memphis, TN, USA), β-catenin Ab (Transduction Laboratories, Lexington, KY, USA) and HA Ab (Covance, Princeton, NJ, USA).
Results
The phosphorylation of IRF3 linker region suppresses IRF3 transactivation activity
Carboxyl-terminal phosphorylation at amino acids (aas) 396, 398, 402, 404 and 405 cause the activation of IRF3, and therefore phosphorylation mimicking mutant IRF3(5D) is considered as a constitutively active form of IRF3 (Figure 1A). In our previous study we found that Ser123, Ser173 and Thr180 within the linker region of IRF3 contribute additively to BGLF4-mediated suppression of IRF3, while an IRF3 mutant with alanine mutations at aas 123, 173 and 180 [IRF3(5D)3A] was resistant to BGLF4-mediated suppression.
9
To confirm whether Ser123, Ser173 and Thr180 of IRF3 are the target sites of BGLF4, in vitro kinase assay was performed with purified recombinant wild type BGLF4 or the kinase dead BGLF4 (K102I) as the kinase source, and GST–IRF3(110–202) or GST–IRF3(110–202)3A as the substrate. A phosphorylation signal was detected in the reaction containing IRF3(110–202) but not IRF3(110–202)3A, indicating that Ser123, Ser173 and Thr180 residues of IRF3 are possible BGLF4 target sites (Figure 1B). To further examine the influence of IRF3 phosphorylation on these three residues, a reporter assay was performed for the transactivation activity of IRF3(5D), phosphorylation-defective mutant IRF3(5D)3A or phosphorylation-mimicking mutant IRF3(5D)2D1E on the IRF3 responsive pLuc-PRD(III-I)3 reporter. The result showed that the transactivation activity of phosphorylation-defective IRF3(5D)3A was higher than that of IRF3(5D) on the reporter containing the IRF3 binding site, suggesting that some cellular kinase(s) may phosphorylate IRF3 at these three sites to regulate IRF3 activity. In contrast, the transactivation activity of IRF3(5D)2D1E was weaker than that of IRF3(5D) in IRF3-responsive reporter assay, especially at lower doses (Figure 1C). Collectively, the results hint that BGLF4-mediated phosphorylation of IRF3 on Ser123, Ser173 and Thr180 within the linker region may suppress IRF3 transactivation activity.
The phosphorylation of IRF3(5D) in proline-rich linker region negatively regulates IRF3 transactivation activity. (A) A summary of functional domains and currently identified phosphorylation sites of IRF3. Functional domains of IRF3 including the DNA binding domain (DBD), nuclear export signal (NES), proline-rich region (Pro), IRF associated domain (IAD), response domain (RD) and transactivation domain are indicated. IRF3(5D) is the constitutive active mutant of IRF3.19,36,37 The seven phosphorylation sites in RD can be phosphorylated by TBK1 or IKKɛ. The putative proline-dependent phosphorylation sites are Thr3, Ser123, Ser170, Thr180 and Ser339. Ser339 is phosphorylated by unidentified kinase(s) and contributes to Pin1-mediated proteasome dependent degradation of IRF3. The phosphorylation-defective mutant IRF3(5D)3 A with substitution of alanine residues for Ser123, Ser173 and Thr180 in the proline-rich linker region or the phosphorylation-mimicking mutant IRF3(5D)2D1E with two serine residues mutated to aspartate and one threonine mutated to glutamate were generated. (B) For the in vitro kinase assay, His-BGLF4 or His-K102I was purified from recombinant baculovirus-infected sf9 cells. Bacterially-expressed GST–IRF3(110–202) and GST–IRF3(110–202)3 A were purified as kinase substrates. Purified GST protein was used as a negative control. The input of GST or GST fusion protein was shown by Coomassie Blue staining. (C) Different amounts of IRF3(5D)-, IRF3(5D)3 A- or IRF3(5D)2D1E-expressing constructs were co-transfected with pLuc-PRD(III-I)3 reporter and GFP expression plasmids into HEK293 cells. At 24 h post-transfection, luciferase activity was measured and normalized with GFP intensity. The readings are means ± SD from duplicated transfections. Data are representative of two independent experiments. * P < 0.05; **P < 0.01; ***P < 0.001 (Student’s t-test). For the Western blotting, ‘T’ indicates transfected IRF3 and ‘E’ indicates endogenous IRF3. F. Luc: firefly luciferase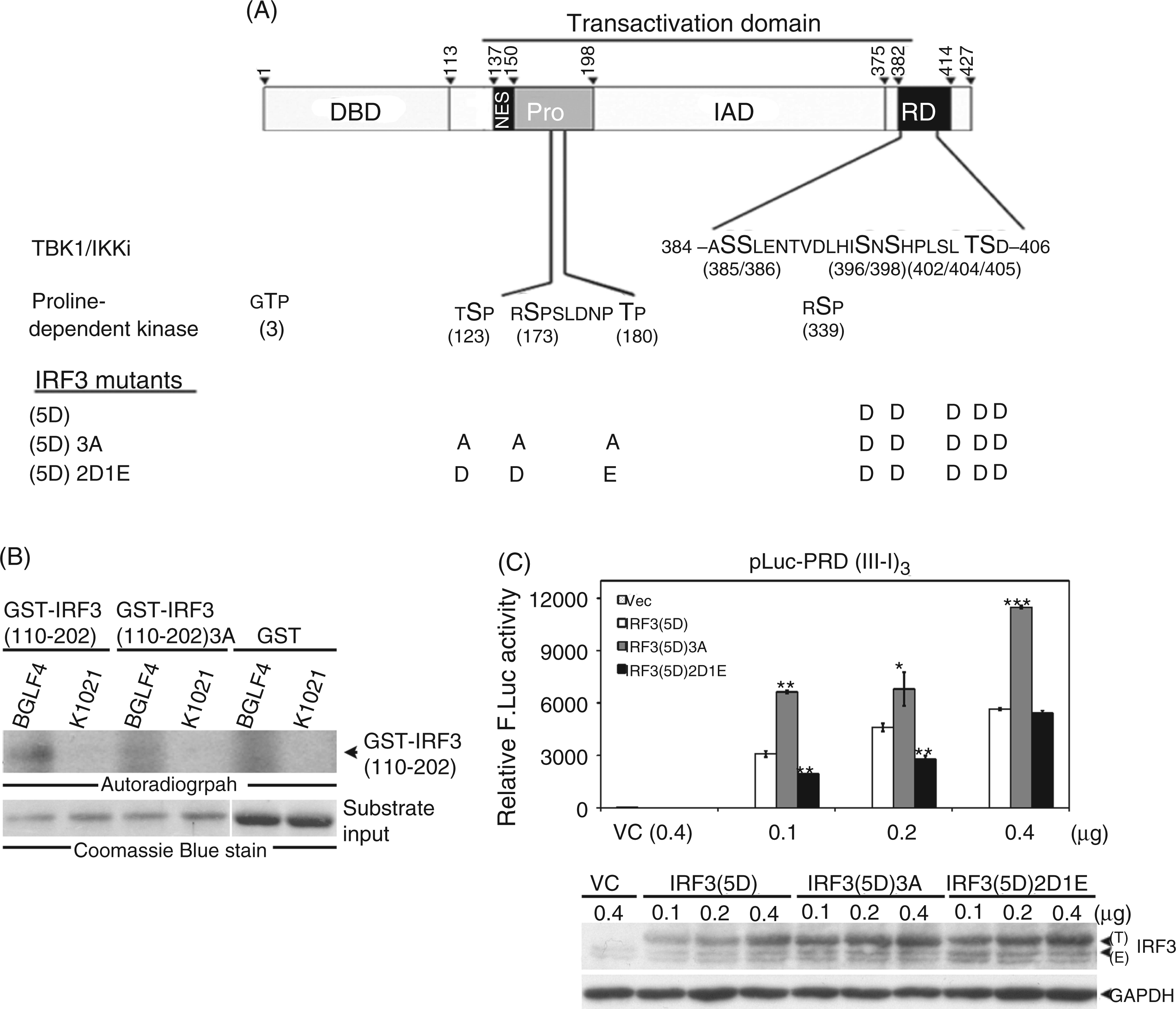
Inhibition of GSK3, but not ERK1 or Cdc2/Cdk5, enhances the transactivation of activity of IRF3(5D)
Based on the observation that transactivation activity of IRF3(5D)3A was stronger than that of IRF3(5D), we wondered whether any cellular kinase may also phosphorylate IRF3 at Ser123, Ser173 or Thr180 to regulate IRF3 function. Candidates of possible cellular kinase were screened by Scansites 2.0 (Figure 2A).
25
To dissect which kinase has a regulatory function on these sites, transfected HEK293 cells expressing IRF3(5D), IRF3(5D)3A or IRF3(5D)2D1E were treated with inhibitors against each of the candidate kinases, including ERK1, Cdc2/Cdk5 and GSK3, for 24 h. A luciferase assay was performed to determine IRF3 transactivation activity. Presumably, the inhibitor for the putative kinase involved in the phosphorylation of Ser123, Ser173 and Thr180 on IRF3 should enhance transactivation activity of IRF3, but not that of phosphorylation-defective IRF3(5D)3 A or phosphorylation-mimicking IRF3(5D)2D1E in IRF3-dependent reporter assays. The results showed that GSK3 inhibitor LiCl (Figure 2D), but not Cdc2/Cdk5 inhibitor roscovitine (Figure 2B) or ERK1 inhibitor U0126 (Figure 2C), met these criteria. LiCl treatment enhanced the transactivation activity of IRF3(5D) about twofold, but had no effect on IRF3(5D)3 A or IRF3(5D)2D1E (Figure 2D). It is known that GSK3 phosphorylates β-catenin, leading to the ubiquitination of β-catenin and followed by proteasome-dependent degradation. Inhibition of GSK3 thus restores the protein level of β-catenin.26,27 Here, the increased level of β-catenin was detected as a positive control of LiCl treatment. Thus, we speculated that GSK3 may repress IRF3 function through phosphorylating Ser123, Ser173 and Thr180 residues of IRF3.
Inhibition of GSK3, but not Cdc2/Cdk5 or ERK1, enhances the transactivation activity of IRF3(5D). (A) According to the phosphorylation prediction program Scansite 2.0, Ser123, Ser173 and Thr180 of IRF3 could be phosphorylated by several cellular kinases as listed. (B–D) IRF3(5D), IRF3(5D)3 A or IRF3(5D)2D1E was co-transfected with pLuc-PRD(III-I)3 reporter plasmid and GFP control plasmid into HEK293 cells. At 20 h post-transfection, cells were treated with (B) Cdc2/Cdk5 inhibitor roscovitine (10 μM) or solvent control DMSO, (C) ERK1 inhibitor U0126 (20 μM) or DMSO, or (D) GSK3 inhibitor LiCl (20 mM) or NaCl (20 mM) for 24 h. Luciferase activity was determined and normalized with GFP intensity. The statistical significance of differences between LiCl and NaCl is indicated at the top of the bars. ** P < 0.01 (Student’s t-test). ‘T’ indicates transfected IRF3 and ‘E’ indicates endogenous IRF3. Immunoblotting of the accumulated protein levels of β-catenin, which can be phosphorylated by GSK3 and then degraded by proteasome, indicates the inhibition of GSK3 activity. F. Luc: firefly luciferase.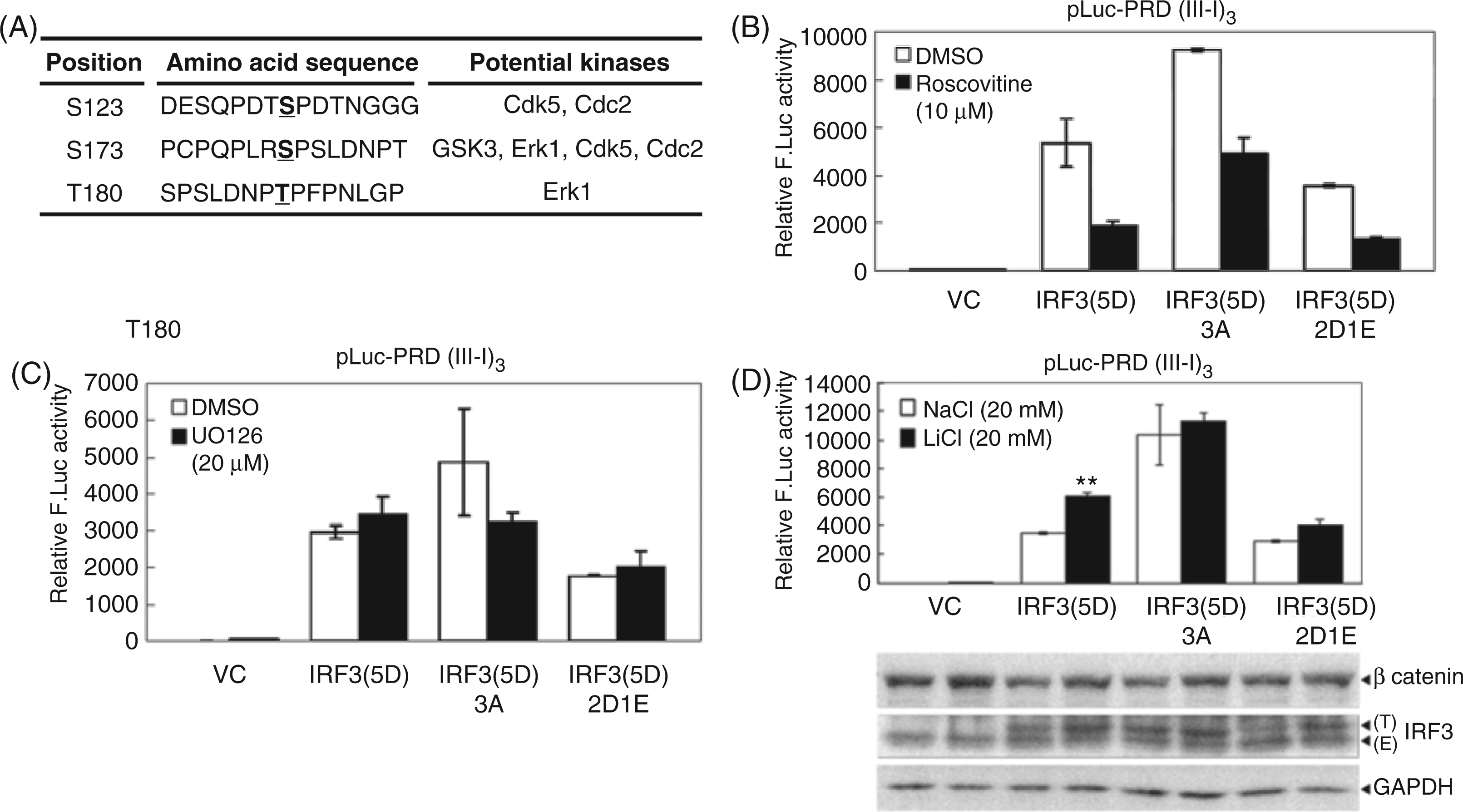
GSK3 suppresses IRF3 transactivation activity
In addition to the LiCl-mediated enhancement on the active form IRF3(5D), the effect of LiCl on the transactivation activity of endogenous IRF3 or exogenous expressed wild type IRF3 was also examined. In the presence of LiCl, the transactivation activities of both endogenous and transfected IRF3 in HEK293 cells were enhanced about threefold compared with the NaCl control (Figure 3A), suggesting that GSK3 functions as a negative repressor for IRF3 in a situation without activation signals. To further confirm the ability of GSK3 to repress IRF3 transactivation, constitutive active form GSK3β(S9A) was transfected into HEK293 cells before LiCl treatment to see if it could reverse the LiCl-mediated enhancement of IRF3 transactivation activity. The data showed that GSK3β(S9A) reversed LiCl-mediated enhancement of IRF3 transactivation activity in a dose-dependent manner. We also examined whether overexpression of dominant negative GSK3β(KI) can enhance IRF3(5D) transactivation activity further and found the dominant negative GSK3β(KI) only slightly enhanced IRF3(5D) transactivation activity in the reporter assay (Figure 3C). It is possible that residual GSK3 activity in the cell still plays a suppressor function for IRF3. Nevertheless, these results provide evidence that GSK3 functions as a cellular kinase, repressing both wild type IRF3 and the constitutively active form IRF3(5D).
GSK3 inhibits IRF3 transactivation activity. (A) IRF3 expression plasmid was co-transfected with pLuc-PRD(III-I)3 and GFP expression plasmid into HEK293 cells. At 20 h post-transfection, cells were treated with LiCl (20 mM) or NaCl (20 mM) for 24 h. Luciferase activity was determined and normalized with GFP intensity. Enhanced protein levels of β-catenin were detected in immunoblotting as an indicator of LiCl-mediated GSK3 inhibition. (B) HEK293 cells were transfected with pLuc-PRD(III-I)3, GFP and different amounts of constitutively active HA-GSK3β(S9A) expression plasmids. At 20 h post-transfection, cells were treated with LiCl (20 mM) or NaCl (20 mM) for 24 h. Luciferase activity was measured and normalized with GFP intensity. (C) Constitutively active IRF3(5D) and increasing amounts of dominant negative GSK3β(KI) were co-transfected with pLuc-PRD(III-I)3 and GFP expression plasmid into HEK293 cells. At 24 h post-transfection, luciferase activity was measured and normalized with GFP intensity. F. Luc: firefly luciferase.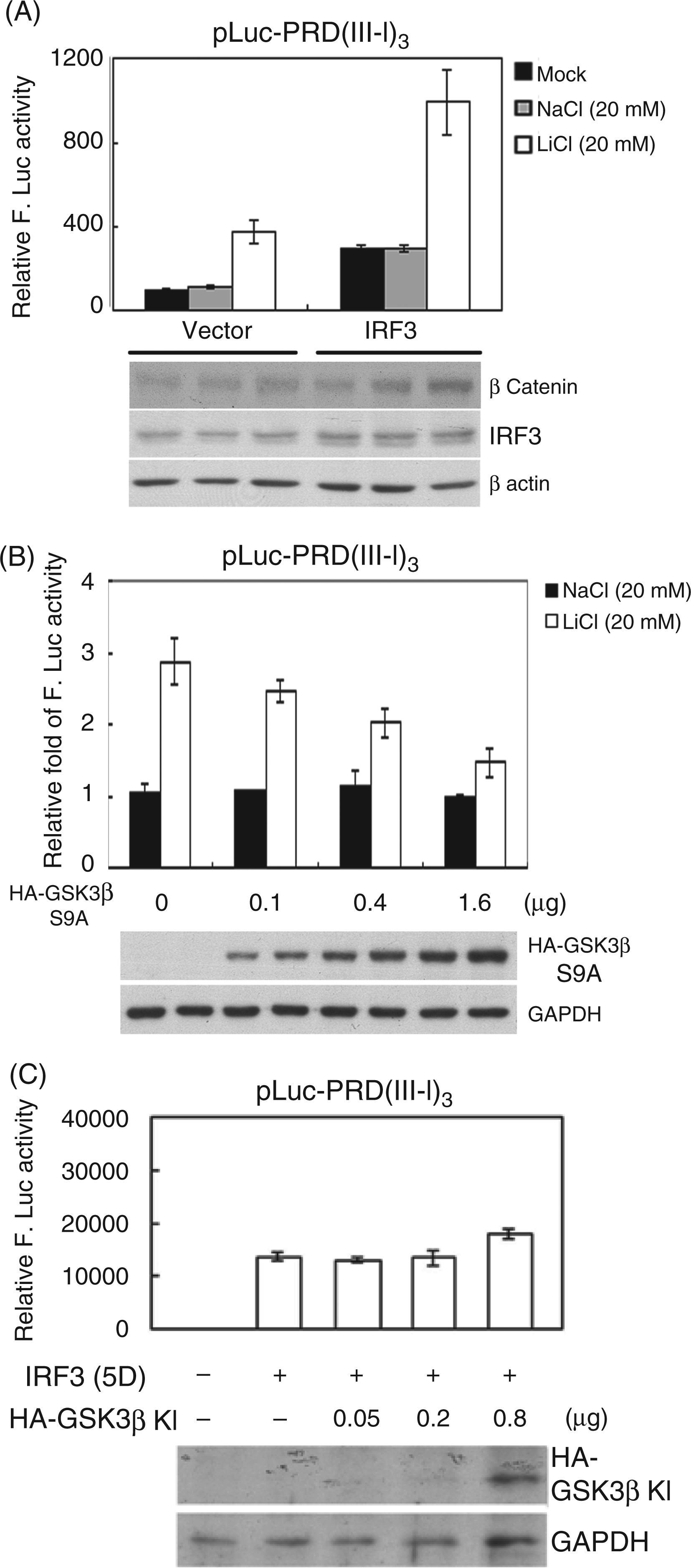
GSK3-mediated phosphorylation of IRF3 contributes to the repression of IRF3 transactivation
To confirm the hypothesis that GSK3 modulates IRF3 function through phosphorylation, an in vitro kinase assay was performed with recombinant GSK3α or GSK3β, and purified GST–IRF3(110–202) or GST–IRF3(110–202)3 A as the substrate. The result showed that both GSK3α and GSK3β phosphorylate GST–IRF3(110–202) but not the GST–IRF3(110–202)3 A (Figure 4A), suggesting that IRF3 is a substrate of GSK3, and Ser123, Ser173 or Thr180 could be the target site of GSK3. To further investigate the effect of GSK3-mediated phosphorylation site(s) on IRF3, HEK293 cells were transfected with various mutants of IRF3 containing alanine substations at Ser123, Ser173 or Thr180, and pLuc-PRD(III-I)3 reporter plasmid. After treatment with LiCl, a luciferase assay was performed to determine IRF3 activity (Figure 4B). Without LiCl treatment the basal activity of IRF3(5D)3 A was higher than that of IRF3(5D), as observed previously, whereas the basal activity of IRF3(5D)2D1E was lower than that of IRF3(5D). With LiCl treatment, the transactivation activity of IRF3(5D)3 A or IRF3(5D)2D1E was not significantly enhanced, suggesting that phosphorylation of IRF3 at Ser123, Ser173 and Thr180 down-regulates IRF3 transactivation activity. The activity of single- or double-site mutated IRF3 mutant was also examined. Compared with NaCl treatment, LiCl treatment inhibited GSK3 function and enhanced the transactivation activity of IRF3(5D) with a 3.2-fold increase, IRF3(5D)S123A with a 2-fold increase, IRF3(5D)S173A with a 1.6-fold increase and IRF3(5D)T180A with a 1.9-fold increase. The enhancement of double-site mutated IRF3 mutants was lower, with only a 1.1- to 1.3-fold increase. Because Ser173 was predicted as the GSK3 target site, we suggest that Ser173 is the primary site responsible for the GSK3-mediated repression of IRF3 function. However, IRF3(5D)S123/180 A was also defective for LiCl-mediated enhancement of IRF3 transactivation activity, which was probably owing to the fact that neighboring phosphorylation may affect GSK3 phosphorylation ability.
Both GSK3α and GSK3β phosphorylate IRF3 in vitro, and GSK3-mediated phosphorylation of IRF3 contributes to the repression of IRF3 transactivation. (A) Recombinant active GSK3α or GSK3β was used as the kinase source. GST–IRF3(110–202) or GST–IRF3(110–202)3A, which Ser123, Ser173 and Thr180 residues were mutated to alanine residues, were used as kinase substrates. The amount of input GST fusion proteins was shown by Coomassie Blue staining. (B) pLuc-PRD(III-I)3 reporter plasmid and GFP control plasmid were co-transfected with wild type IRF3 or IRF3 mutants into HEK293 cells. At 20 h post-transfection, cells were treated with LiCl or NaCl for 24 h. Luciferase activity was measured and normalized with GFP intensity. F. Luc: firefly luciferase.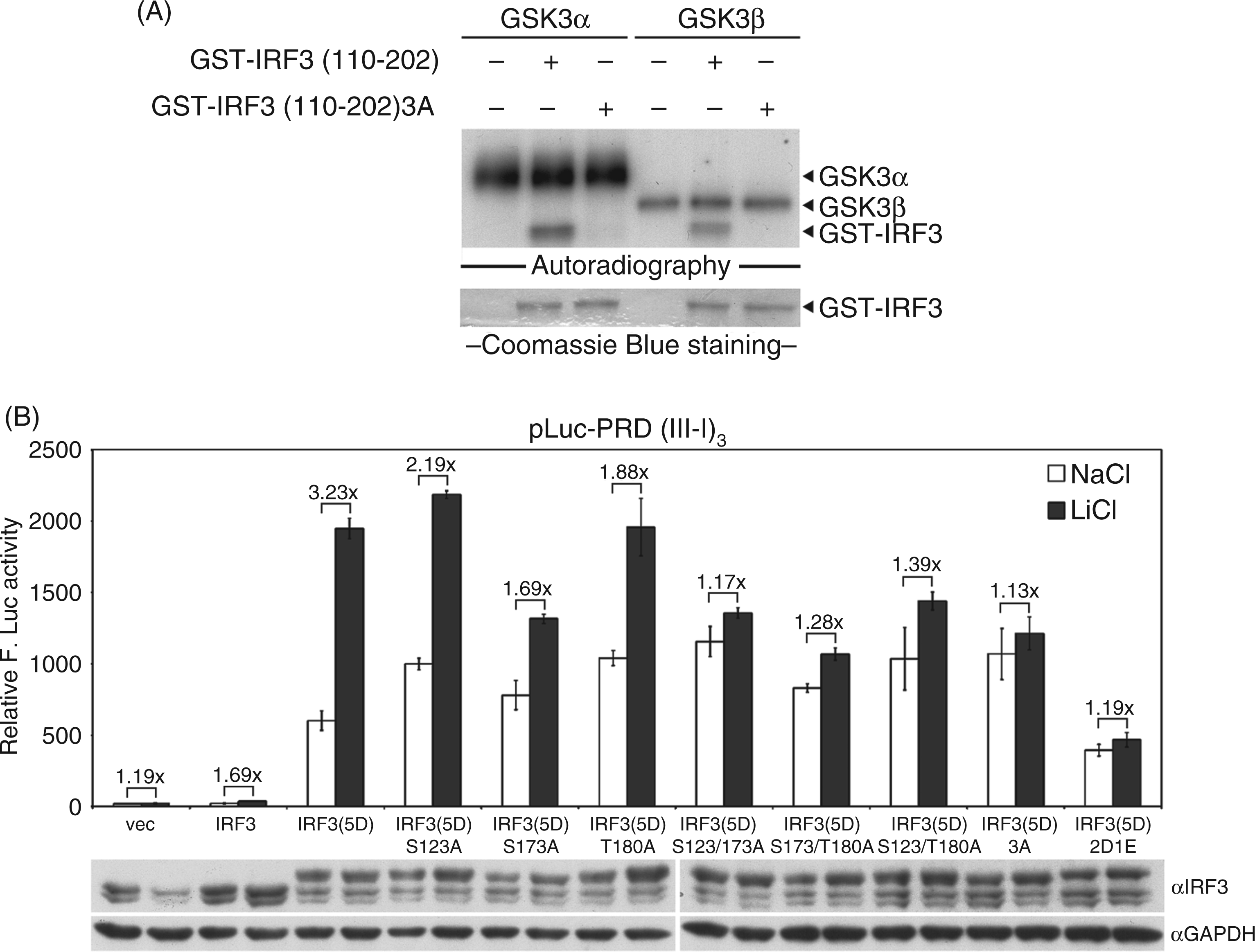
Discussion
The N-terminal DNA-binding and the C-terminal dimerization domains of IRF3 are connected by a 75-residue proline-rich linker, which contains the nuclear export signal and a region of high helical propensity. Recent structural analysis indicated that the linker represents a folded structural domain.28,29 Strong binding of the dimeric IRF3 to the PRDII–PRDI tandem recognition sites needs to bend DNA by about 100o (Figure 5). Our current study shows that the phosphorylation status at Ser123, Ser173 and Thr180 residues of IRF3(5D) affects its transactivation activity. Treatment of the cells with LiCl enhanced IRF3(5D) transactivation activity, but not the transactivation activity of phosphorylation-defective or phosphorylation- mimicking mutant at Ser123, Ser173 and Thr180 (Figure 1C and 2D). Furthermore, we proved that GSK3 was involved in the LiCl-mediated enhancement of IRF3 transactivation activity (Figure 3B).
A summary of GSK3- and EBV BGLF4-mediated suppression of IRF3 transactivation. (I) Under unstimulated situations, IRF3 shuttles dynamically between cytoplasm and nucleus, and IRF3 monomer may bind to IRF3 responsive promoters with a very low affinity. (II) Upon infection, the activity of VAKs, such as TBK1 and IKKɛ, can be induced, and IRF3 is then phosphorylated by VAK to permit its homodimerization and nuclear translocation, activating the transcription of IRF3 regulating genes. The stable IRF3 dimer DNA complex formation relies on the twist of DNA for about 100˚, which can subsequently activate transcription of IRF3 responsive promoter. To terminate the signaling, the activated IRF3 is then phosphorylated at Ser339 by unidentified kinase(s), and, subsequently, propyl isomerase Pin1-induced conformational change of IRF3 then leads to the degradation of IRF3. (III) Here we proposed that constitutive active GSK3 may function as a housekeeping negative regulator for the unexpected activation of IRF3. GSK3-mediated phosphorylation of IRF3 at proline-rich linker region, which bridges the DNA binding domain and transactivation domain of IRF3, may interfere with stable DNA binding of IRF3 to inhibit IRF3 transactivation. Upon virus infection, PI3K/Akt is usually activated to inhibit GSK3 activity, resulting in the activation of IRF3 to induce interferon expression. The inactivated GSK3 also plays a role in promoting TBK1 activity for IRF3 activation. (IV) During EBV replication, BGLF4 phosphorylates IRF3 through a GSK3-like mechanism to suppress the activation of innate immune response.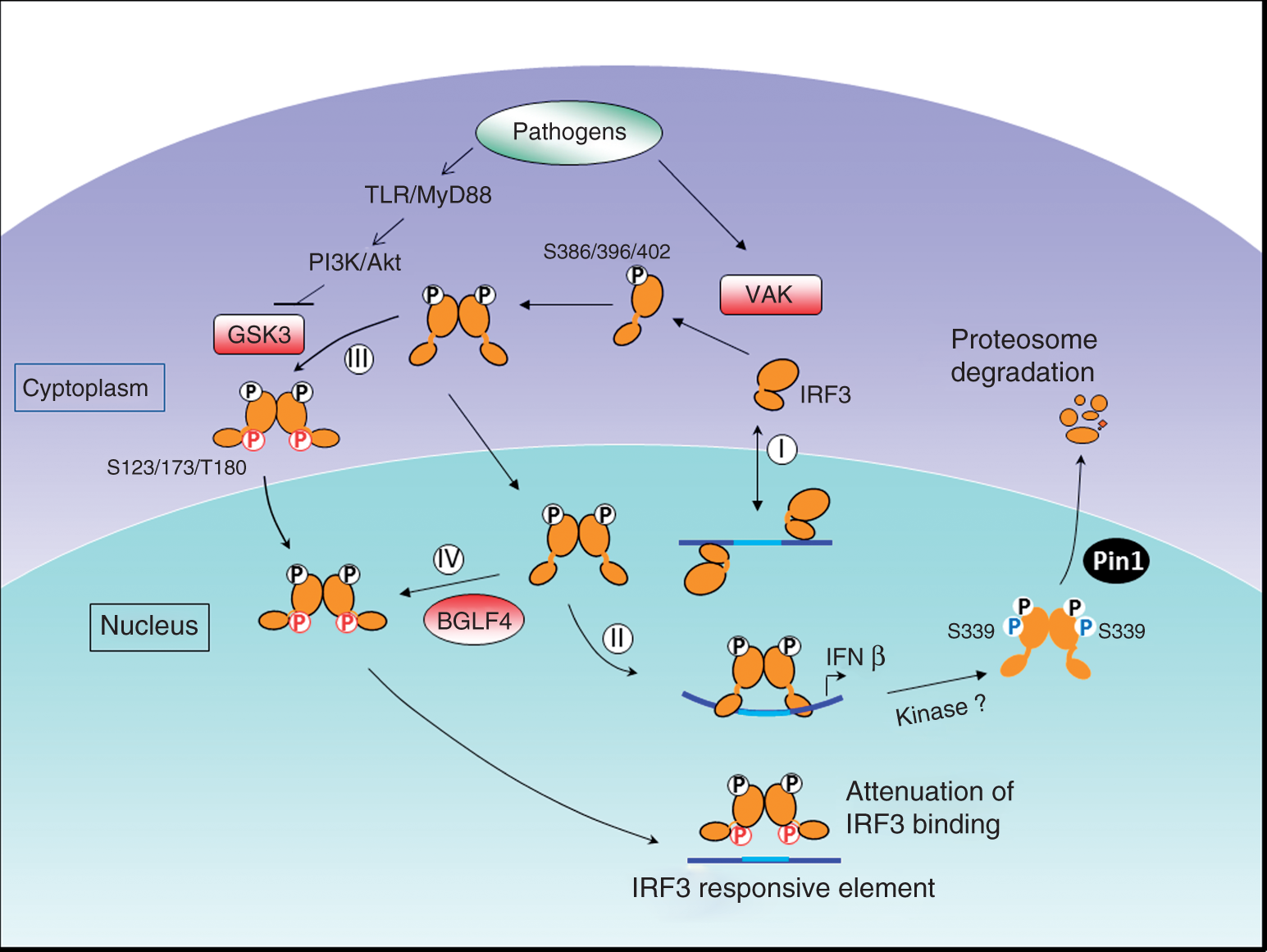
GSK3 is known as an important example for its function to limit and dampen the immune response along with the destructive signals against pathogens. 30 Upon pathogen infection, the microbial molecules bind to TLRs and result in the recruitment of MyD88, which can subsequently lead to the activation of phosphoinositide 3 kinase (PI3K) and Akt, leading to GSK3 inactivation through phosphorylation. GSK3 differentially regulates TLR-mediated production of pro- and anti-inflammatory cytokines through NF-κB and cAMP response element-binding protein (CREB). 14 It was also demonstrated that GSK3β can down-regulate IFN-β production by LPS-stimulated macrophages. 15 In the cell culture system, the levels of the immunoprecipitated nuclear ATF-2 and its associated phospho-c-Jun were dramatically increased in the presence of the ATP competitive GSK3 inhibitor SB216763, whereas the nuclear levels of NF-κB and IRF3 did not change. It suggested that the levels and phosphorylation of c-Jun is the key mediator for the GSK3 regulated suppression of IFN expression. The administration of GSK3 inhibitor before challenge of a sublethal dose of LPS augments the systemic levels of IFN-β in mice, 15 suggesting that GSK3 modulates LPS-mediated IFN production. Interestingly, GSK3β was found to physically associate with TBK1 upon Sendai virus infection for activating IRF3 signaling in a GSK3β kinase activity independent manner in a separate study. 16 To avoid possible complications, we did not test the regulatory effect of GSK3 on double-stranded RNA or virus-mediated activation of IRF3 in this study. However, we found the addition of SB216763 did not stimulate the IRF3 reporter activity significantly, possibly because it was less effective in our system, as revealed by the unclear increase of β-catenin levels (data not shown). Thus, small amounts of the constitutive active GSK3 may be enough to block IRF3 activity. Alternatively, we do not discount that other cellular kinase(s) may have a GSK3-like function to regulate IRF3 through phosphorylation at the linker region.
Phosphorylation of linker regions has been reported in several molecules to modulate their function, including the well-documented Smads. 31 Smad proteins are a family of transcription regulators that consist of three major domains and mediate TGF-β signaling. The N-terminal phosphorylation domain associates with nuclear translocation and binds to DNA. The central linker region contains multiple S/TP sites and interacts with prolyl isomerase or ubiquitin ligases, whereas the C-terminus of Smad binds to type I receptors, and mediates dimer formation and association with various transcriptional regulators. Upon TGF-β stimulation, Smads are phosphorylated at the carboxyl termini and translocate into nucleus, form dimers and bind to specific target promoters through interaction with various transcription factors. Simultaneously, TGF-β also turns on MAPK to phosphorylate the linker region of Smads. 32 Linker region phosphorylation of the Smad heretocomplex was found to down-regulate TGF-β signaling by either inhibiting nucleus translocation or the transcriptional activity of the complex if it translocates into nucleus. It was found that GSK3 phosphorylates Smad3 at the linker region Ser204 and down-regulates its interaction with CREB3. 33 Linker regions of Smads can also be phosphorylated by Cdks in the nucleus.32,34 Notably, constitutive linker region phosphorylations of Smad2 and Smad3 were identified in melanoma cells, contributing to the resistance of melanoma cells to TGF-β-mediated growth inhibition. 35 Thus, Smad3 linker phosphorylation is considered as a negative feedback control of TGF-β.
The domain structure and activation mechanism of IRF3 are very similar to those of Smads. The structural analysis indicated dimeric IRF3 bound to the PRDII–PRDI tandem recognition sites cause a bending of DNA,28,29 so it is possible that the phosphorylation of IRF3 linker region may reduce the flexibility required for the strong binding to the target DNA (Figure 5). Consequently, it may then affect the proper assembly of the transactivation complexes. Because GSK3 is usually constitutively active in the cells, we propose that the GSK3-mediated IRF3 linker region phosphorylation may protect cells from unexpected IRF3 reactivation in the absence of pathogen invasion.
In this study, we found that GSK3 can directly phosphorylate IRF3 at its linker region, possibly leading to attenuation of IRF3 binding to its target DNA sequence. A hypothetic model is proposed to summarize cellular and viral protein kinase-mediated regulation of IRF3 (Figure 5). Upon virus infection, the initial TLR/MyD88 signaling can activate Akt to down-regulate GSK3 activity and therefore the GSK3-mediated IRF3 repression is removed. Furthermore, while virus activated kinase (VAK, e.g. TBK1) phosphorylates IRF3 at its carboxyl terminus and leads to the activation of IRF3, inactivated GSK3β can still associate with and promote the activation of TBK1 to amplify IRF3 signaling. 16 Thus, GSK3 can play both roles in suppressing unexpected activation of IRF3 and in amplifying IRF3 signaling upon virus infection. Overall, the negative regulatory effect of GSK3-mediated IRF3 phosphorylation can function either in the absence of, or at the terminal stage of, pathogen infection to modulate IRF3 transactivation activities. GSK3 predominantly distributes in the cytoplasm, whereas BGLF4 is detected mainly in the nucleus. It is possible that IRF3 can either be phosphorylated in the cytoplasm or in the nucleus by GSK3 or BGLF4. Taken together, GSK3 appears to modulate the immune response at different stages in the absence or presence of various infections. EBV thus evolved to hijack the cellular mechanism through BGLF4 kinase-mediated phosphorylation to evade host antiviral response.
Footnotes
Funding
This study was supported by National Health Research Institute, Taiwan (NHRI-EX101-9928BI and NHRI-EX102-10201-BI).
Acknowledgements
We are grateful for Dr Yi-Ling Lin (Institute of Biomedical Sciences, Academia Sinica, ROC) for providing plasmids pCR3.1-huIRF3 and pCR3.1-huIRF3(5D), Dr Katherine A. Fitzgerald (Division of Infectious Diseases and Immunology, University of Massachusetts Medical School, Worcester, MA, USA) for pLuc-PRD(III-I)3 and Dr Junichi Sadoshima (University of Medicine and Dentistry of New Jersey) for plasmids of HA-GSK-3β (S9A), and kinase dead HA-GSK-3β (K85A).