
Abstract
The mammalian target of rapamycin (mTOR) plays critical roles in immunity. We previously showed that infection of human embryonic kidney (HEK) cells with Sindbis virus (SIN), an enveloped RNA alphavirus, profoundly suppresses Akt/mTOR signaling, and host translation late during infection. To understand how SIN mediated suppression of mTOR affects innate response, we analyzed phosphorylation of interferon regulatory factor 3 (IRF3) and expression of two antiviral genes. Here we show strong phosphorylation of IRF3, and an increase in mRNA levels for antiviral genes interferon stimulated gene (ISG)56 and interferon gamma inducible protein (IP)-10 when intracellular viral RNA levels are high during late infection. The mTOR inhibitors rapamycin and torin1 do not block, but mildly upregulate these responses. Even after prolonged treatment with Ly294002, the PI3K inhibitor only partially blocks SIN induced phosphorylation of IRF3. While Ly294002 treatment downregulated the SIN induced expression of ISG56 mRNA levels, it had no effect on SIN induced upregulation of IP-10 expression. These results point to SIN replication-mediated activation of IRF3, independent of mTOR function, when host protein synthesis is severely suppressed by virus infection.
Introduction
An early event during innate response to RNA virus infections is the activation of a set of constitutively expressed transcription factors, including interferon regulatory factor 3 (IRF3) and NF-κB, that induce the expression of antiviral proteins. 1 Recognition of different viral RNAs by cellular TLR 3,7,8, and cytoplasmic helicases, retinoic acid inducible gene (RIG-I), and melanoma differentiation associated gene 5 (MDA5), leads to activation of IRF3 by phosphorylation, dimerization, and nuclear translocation.2,3 In association with other regulatory proteins, IRF3 binds to the interferon stimulated response elements (ISRE) located within the promoter regions of interferon stimulated genes (ISGs) such as ISG56 and interferon gamma inducible protein 10 (IP-10). 4 The phosphorylation of IRF3 in virus infected cells is controlled by the phosphatidylinositol-3 kinase, PI3K/Akt pathway.5,6 Recent work indicates that the mammalian target of rapamycin (mTOR) is a PI3K regulated sensor of cellular signals.7,8 However, the involvement of mTOR in the phosphorylation of IRF3 in RNA virus infected cells is unknown.
The PI3K/Akt/mTOR axis of signaling regulates translation, cell proliferation and metabolism.7,8 Recent studies indicate that mTOR signaling impacts innate immune response to bacterial and viral agents, and their components, in a cell type-dependent manner. 9 The mTOR is a serine/threonine kinase and forms two multiprotein complexes, mTORC1 and mTORC2, 7 of which mTORC1 promotes translation by phosphorylating ribosomal protein S6 via S6Kinase, and eukaryotic initiation factor 4E binding protein, 4E-BP1. mTORC2 activates Akt1 which can further activate mTORC1. 7 The widely used immunosuppressant, rapamycin blocks mTORC1 function, but not mTORC2; whereas, torin1 inhibits both mTORC1 and mTORC2. 10 While activation of mTOR by PI3K through Akt is well established, Akt-independent activation of mTOR pathway is also known. 8 Many DNA and RNA viruses upregulate the PI3/Akt/mTOR pathway to support virus replication and persistence. 11 The role of the mTOR pathway in innate response to different viruses is just beginning to be understood.9,12
Sindbis virus (SIN) is an enveloped positive-sense RNA virus that belongs to the alphavirus genus of the Togaviridae family.13,14 Sindbis virus and other alphaviruses are excellent model systems to study virus-host interactions.13,14 Genetically engineered SIN is a powerful expression system and a proven oncolytic agent in preclinical models.15,16 Systemically delivered SIN targets tumor tissues with minimal adverse effects. 16 Oncolytic virotherapy in combination with rapamycin is a promising approach in oncology.17,18 Therefore, we initiated a study to uncover the molecular events in rapamycin-treated SIN infected cells. We have recently shown that SIN replication in HEK293T cells leads to suppression of the mTOR pathway late during infection 19 . As mTOR is implicated in innate response, we chose to determine the extent of phosphorylation of IRF3 and expression of IRF3 inducible candidate genes, namely ISG56 and IP-10. We show that IRF3 phosphorylation is associated with high intracellular viral RNA and is insensitive to mTOR inhibition.
Materials and Methods
Cells
Human embryonic kidney cells (HEK 293T) were obtained from ATCC (Manassas, VA, USA). The HEK293T cells were cultured in humidified 5% CO2 incubator in Dulbecco’s modified eagle medium (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum. Cell viability checked by trypan blue dye exclusion was more than 90%. Minimal essential medium (Invitrogen) containing 10% fetal bovine serum was used for maintaining baby hamster kidney (BHK) and African green monkey kidney (Vero) cells.
Virus preparations
Sindbis virus, Toto1101, was produced by in vitro transcription of plasmid Toto1101, followed by transfection into BHK cells. Propagation of the virus and titering was done in vero cells as previously described. 20 Virus in the culture supernatants was purified by ultracentrifugation at 160,092 g, using 20% sucrose cushion. The virus particles were UV-inactivated by two cycles of program C4 in a Bio-RAD UV-chamber (Bio-Rad, Richmond, CA, USA). Absence of any infectious virus particle in the UV-inactivated virus (UV-SIN) preparations was confirmed by plaque assay.
Reagents
Gene-specific primers for IRF3, IP-10, ISG56 and β-actin were obtained from Integrated DNA technologies (Coralville, Iowa, USA) and RealTimePrimers.com. Rapamycin, Ly294002 and cycloheximide were purchased from Sigma (St Louis, MO, USA). Torin1 was a kind gift from N. Gray and D.M. Sabatini, Boston. Primary Abs for p-IRF3 (Ser 396), total IRF3 and β-actin were from Cell Signaling Technology (Danvers, MA, USA) and the anti-mouse and anti-rabbit secondary Abs were from Invitrogen.
Infection and plaque assays
The HEK cell line was cultured to subconfluence in a six-well plate with 10% FBS-containing DMEM. Cells were pretreated with 100 n
Isolation of RNA and quantitative real time PCR (qPCR)
Total RNA from cell cultures was isolated using Trizol reagent (Invitrogen). The RNA was treated with DNAse, (RQ1 RNAse free DNAse; Promega, Madison, WI, USA) and cDNA synthesis was done with random hexamers using M-MuLV reverse transcriptase (Invitrogen). SYBR green was used as the reporter flourophore for the quantitative real time PCR (qPCR) with β-actin as internal control. The gene-specific primers used were: IRF3 (forward, ACC AGC CGT GGA CCA AGA G; reverse, 5’ TAC CAA GGC CCT GAG GCA C); ISG56 (forward, 5’GCT GAA GTG TGG AGG AAA GA; reverse, 5’ AGC AAA GAA AAT GGC TTG TG); IP-10 (forward, AAC CTC CAG TCT CAG CAC CAT GAA; reverse, 5’ CTG ATG CAG GTA CAG CGT ACA GTT); and, β-actin (forward, 5’GGA CTT CGA GCA AGA GAT GG; reverse, 5’ AGC ACT GTG TTG GCG TAC AG).
Western blotting
The cells were washed twice with ice-cold PBS and directly lysed with Laemmli SDS-loading buffer. Additionally, an aliquot of the cells was lysed with mammalian protein extraction reagent (Thermo Scientific, Rockford, IL, USA) for quantification of total protein levels by the Bradford method (Sigma). The Laemmli lysates were incubated for 5 min at 95°C and then subjected to a short impulse of ultrasonication to minimize the viscosity before loading onto 4-15% and 10% SDS gels. Proteins were separated and electro-transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with fat-free milk (Cell Signaling) and incubated overnight with the primary Abs for p-IRF3 (Ser396), total IRF3 and β-actin, followed by incubation with HRP-conjugated anti-rabbit/anti-mouse secondary antibodies. After treating with the substrate (Super signal west Pico, Thermo Scientific) for 1 min, the membranes were exposed to X-ray films (Kodak, Rochester, New York, USA).
Results
Sindbis virus (SIN) replication induces phosphorylation of IRF3 and expression of ISG56 and IP10
Sindbis virus replicates in a number of cell types including muscle, connective tissue and endothelium, and elicits a robust inflammatory response in vivo.
14
As fibroblast is an important target for SIN replication, and much information is available for signaling events in the human fibroblast, we initiated studies in HEK293T cells. To determine innate response, HEK cells were infected with SIN at a multiplicity of infection (MOI) of 10, along with a UV-inactivated SIN as a control. Culture supernatants were recovered at 4 h and 24 h p.i and analyzed for IFN-α using ELISA. No significant IFN-α was demonstrable for SIN or UV-SIN infected cultures (data not shown). This result suggested that one or more steps in signaling leading to IFN induction was defective. As activation of IRF3 is a critical step in the induction of antiviral response, we examined the level of total and phosphorylated IRF3 (p-IRF3) by western blot using a specific antibody to Ser 396 of IRF3, which is an early marker for its activation.3,6 As shown in Figure 1, little, or no, p-IRF3 was detectable at 4 h in either infected or uninfected cells. However, at 24 h, there was a drastic upregulation of p-IRF3 in SIN infected cells, but not in other cultures. These results suggested that robust activation of IRF3 occurs late in SIN infection, when intracellular viral RNA levels are high.
Sindbis virus replication induces IRF3 phosphorylation at 24 h: HEK293T cells were infected with SIN or ultraviolet (UV)-inactivated SIN (UV-SIN) at a MOI of ten and the whole cell lysates obtained at indicated times were analyzed by western blot using Abs specific for phospho-IRF3 (Ser396) and total IRF3, with β-actin as loading control. Data is representative of three different experiments. * Band intensity normalized to β-actin by densitometry.
Upon phosphorylation, IRF3 is translocated to the nucleus and activates the transcription of virus stress inducible genes, including ISG56 and IP-10.
4
To determine the level of these specific mRNAs, cellular RNA isolated at 4 and 24 h cultures were subjected to qPCR using gene specific primers. As shown in Figure 2A, the level of ISG56 expression was not significantly altered at either 4 h or 24 h. However, the level of IP-10 (Figure 2B) expression was upregulated eight-fold upon SIN infection at 24 h which may be as a result of phosphorylation of IRF3. The level of IRF3 mRNA was 1.6-fold higher in SIN infected cells at 24 h (Figure 2C). The precise events that contribute to differential expression of these mRNAs may be related to virus-induced phosphorylation of IRF3 or other signaling events related to virus replication (see below).
Increased expression of interferon stimulated genes upon SIN replication: HEK293T cells were infected with SIN or UV-SIN [MOI, five] and the mRNA levels of ISG 56 (A), interferon gamma inducible protein (IP)-10 (B) and IRF3 (C) were determined by SYBR green based quantitative PCR, with β-actin as internal control. Data represent mean ± SE from three different experiments. P-value calculated using student's t-test (P < 0.05 considered statistically significant). UI: uninfected.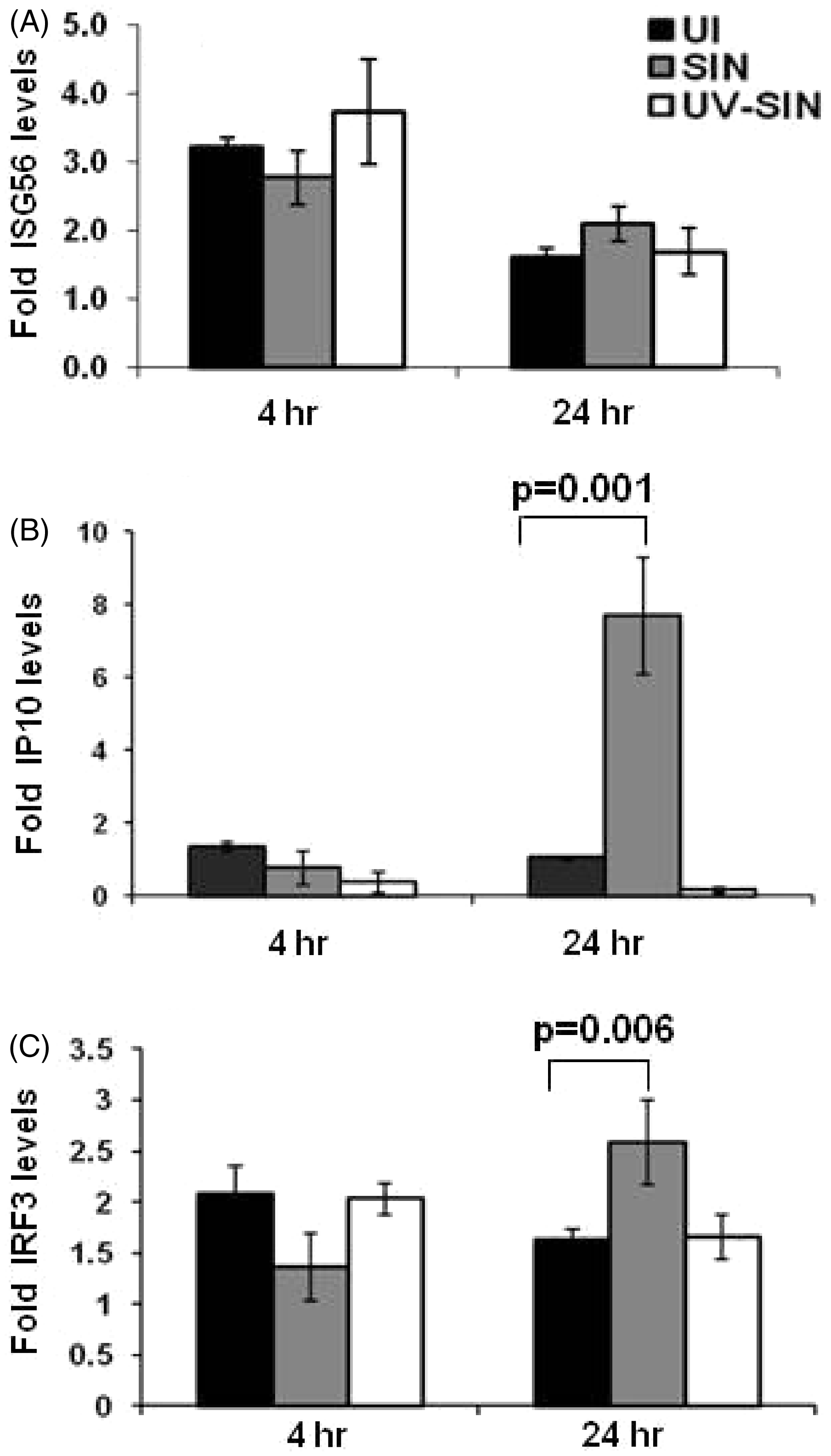
mTOR function is not required for IRF3 phosphorylation
A flurry of recent reports indicate opposing roles for mTOR in immunity and innate response.
9
To understand the role of mTOR in SIN induced activation of IRF3, we used mTOR-specific inhibitors, rapamycin and torin1. Cells pretreated with these drugs were infected with SIN or UV-SIN, harvested at 4 h and 24 h, and analyzed for IRF3 phosphorylation. As shown in Figure 3A, at 4 h, no p-IRF3 was detectable in either drug- treated or untreated cultures. At 24 h, cells infected with SIN showed very high levels of p-IRF3, which was unaltered by rapamycin and torin1. The prominent appearance of p-IRF3 in SIN infected cells was accompanied by lowered total IRF3. Similarly, at 24 h, torin1 treated cells also had low levels of total IRF3 (Figure 3A). As expected, rapamycin and torin1 blocked phosphorylation of S6 (Figure 3B), a key substrate for mTOR regulated S6K1. As shown in Figure 3C, SIN-induced, rapamycin-resistant IRF3 phosphorylation was demonstrable at 16 h and 24 h, but not at 8 h post-infection. These results suggest that SIN-induced IRF3 phosphorylation is insensitive to mTOR specific inhibitors and, therefore, mTOR function may not be required for IRF3 phosphorylation.
Sindbis virus-induced IRF3 phosphorylation is insensitive to mTOR inhibition. (A) HEK293T cells were pretreated with 100 n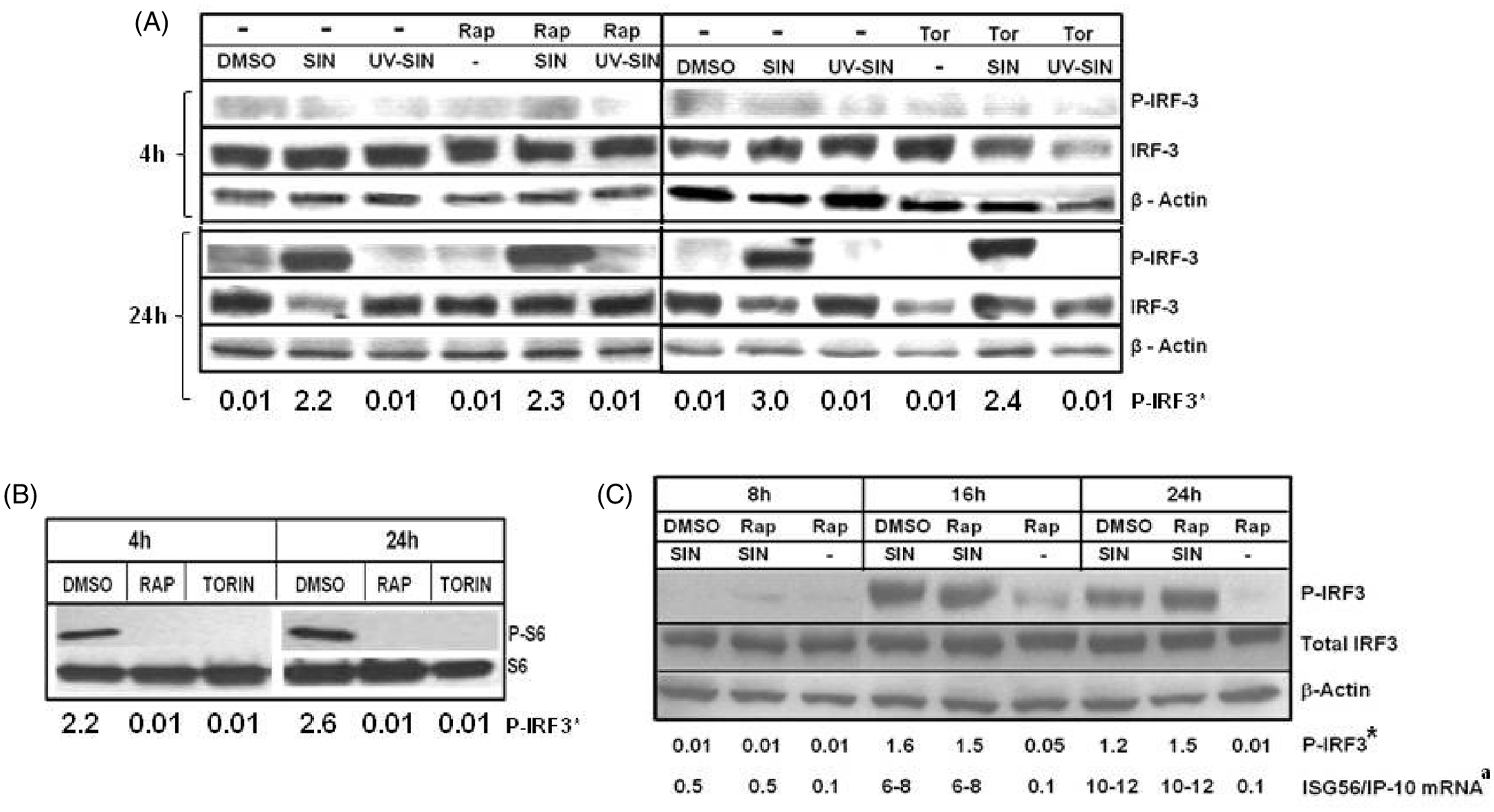
mTOR inhibitors moderately upregulate mRNA levels of ISG56, IP-10 and IRF3
The mRNA level of ISG56 was very mildly altered by rapamycin at 4 h (Figure 4A). However, at 24 h, rapamycin upregulated ISG56 mRNA 1.6-fold and 5.4-fold inthe absence and presence of SIN, respectively. Rapamycin also upregulated the mRNA levels of IP-10 3.3-fold and 13.4-fold in the absence and presence of SIN infection (Figure 4B). Similarly, the mRNA level of IRF3 was also upregulated 2.2-fold by SIN in the presence ofrapamycin. The effect of torin1 on the level of ISG56 mRNA was less pronounced at 4 h (Figure 4D). However at 24 h, torin1 upregulated ISG56 mRNA 2.5-fold and IP-10 mRNA level 38-fold, both in the absence and presence of the virus (Figure 4E). The IRF3 mRNA level was also mildly increased by torin1 (Figure 4F). These results suggest that mTOR function is not required for the expression of ISG56, IP-10 and IRF3 mRNAs during SIN infection, and the mTOR inhibitors may variably augment ISG56 and IP-10 levels.
Mammalian TOR signaling may not be involved in SIN-induced expression of IFN inducible genes: Induction of SG56, interferon gamma inducible protein (IP)-10 and IRF3 mRNA expression by SIN was determined in the presence of mTOR inhibitors. The HEK293T cells were pretreated with 100 n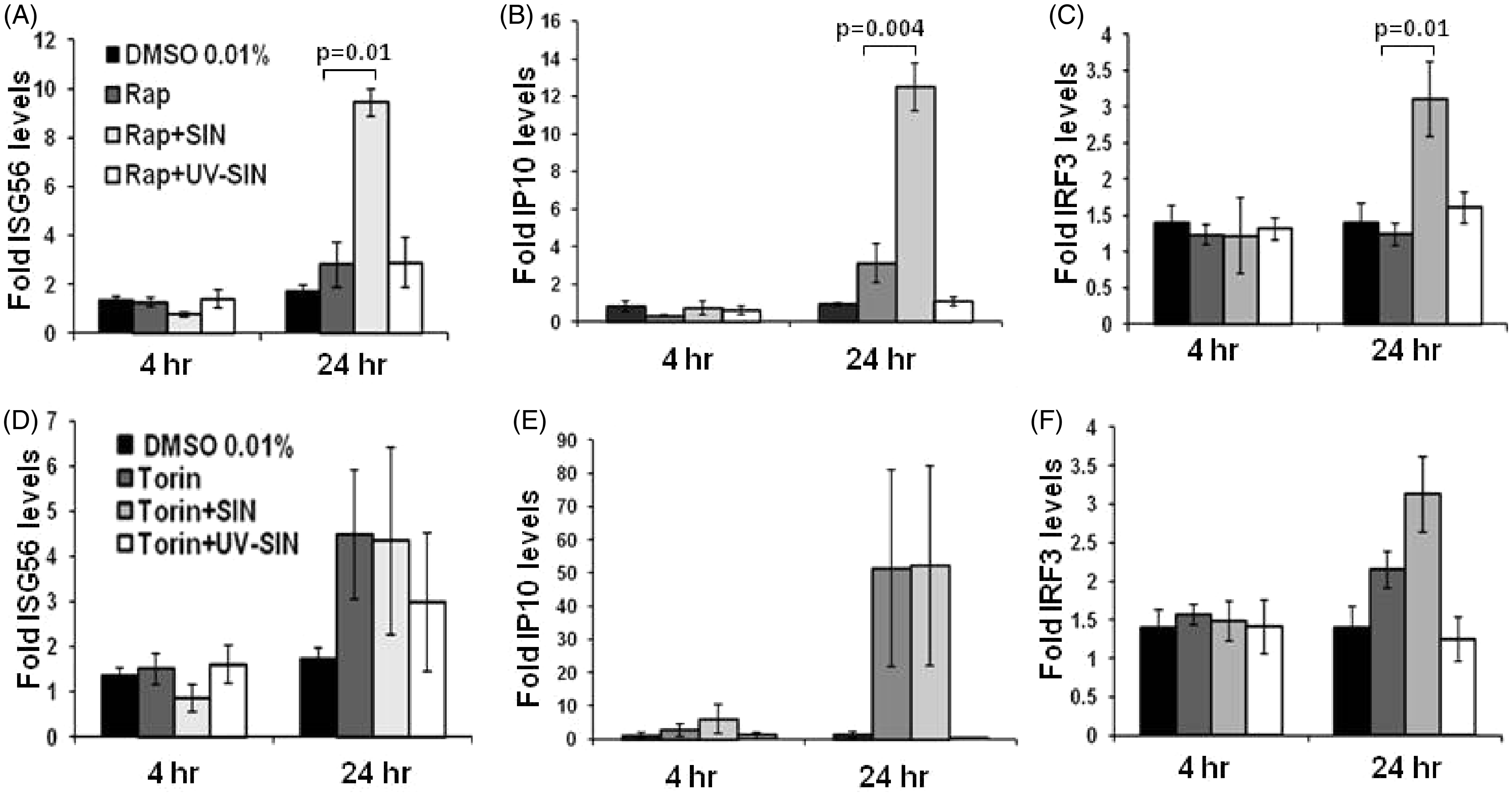
Partial requirement of PI3K in SIN-mediated IRF3 phosphorylation
Phosphorylation of IRF3 is a multistep process, involving the TBK and PI3K pathways.5,6,21 Treatment of fibroblasts with Ly294002 was shown to block a critical PI3K-mediated phosphorylation of IRF3 that results in incomplete association of IRF3 with ISRE in the nucleus.
5
To determine if PI3K plays a role in SIN- induced phosphorylation of IRF3, cells were treated with Ly and further infected with SIN or UV-SIN. The cells were harvested at 4 h and 24 h, and IRF3 phosphorylation was determined. At 4 h, p-IRF3 was undetectable in all conditions (Figure 5A); therefore, the effect of Ly could not be ascertained. However, at 24 h, there was a partial reduction in IRF3 and p-IRF3 in SIN + Ly treated cells, compared to cells infected with SIN alone (Figure 5A). Of note, Ly blocked the phosphorylation of Akt1 indicating its effectiveness (Figure 5B). These results suggest mild degradation and inhibition of phosphorylation of IRF3 in Ly treated SIN infected cells. It is to be noted that even prolonged treatment of Ly does not eliminate IRF3 phosphorylation in SIN infected cells.
(A) Sindbis virus-induced phosphorylation of interferon regulatory factor 3 (IRF3) is partially dependent on PI3K/Akt. HEK293T cells were treated with the PI3 kinase inhibitor, LY294002 (40 µM), 1 h before infection with SIN or UV-SIN. The extracts obtained at indicated times were analyzed by western blotting using Abs specific for phosphorylated, and total forms, of IRF3, with β-actin as loading control. (B) LY294002 efficiently inhibits Akt phosphorylation. Pretreatment of HEK cells with Ly, inhibited phosphorylation of Akt at both 4 h and 24 h. A representative blot of three experiments is given. * p-IRF3 band intensity for 24 h normalized to corresponding β-actin.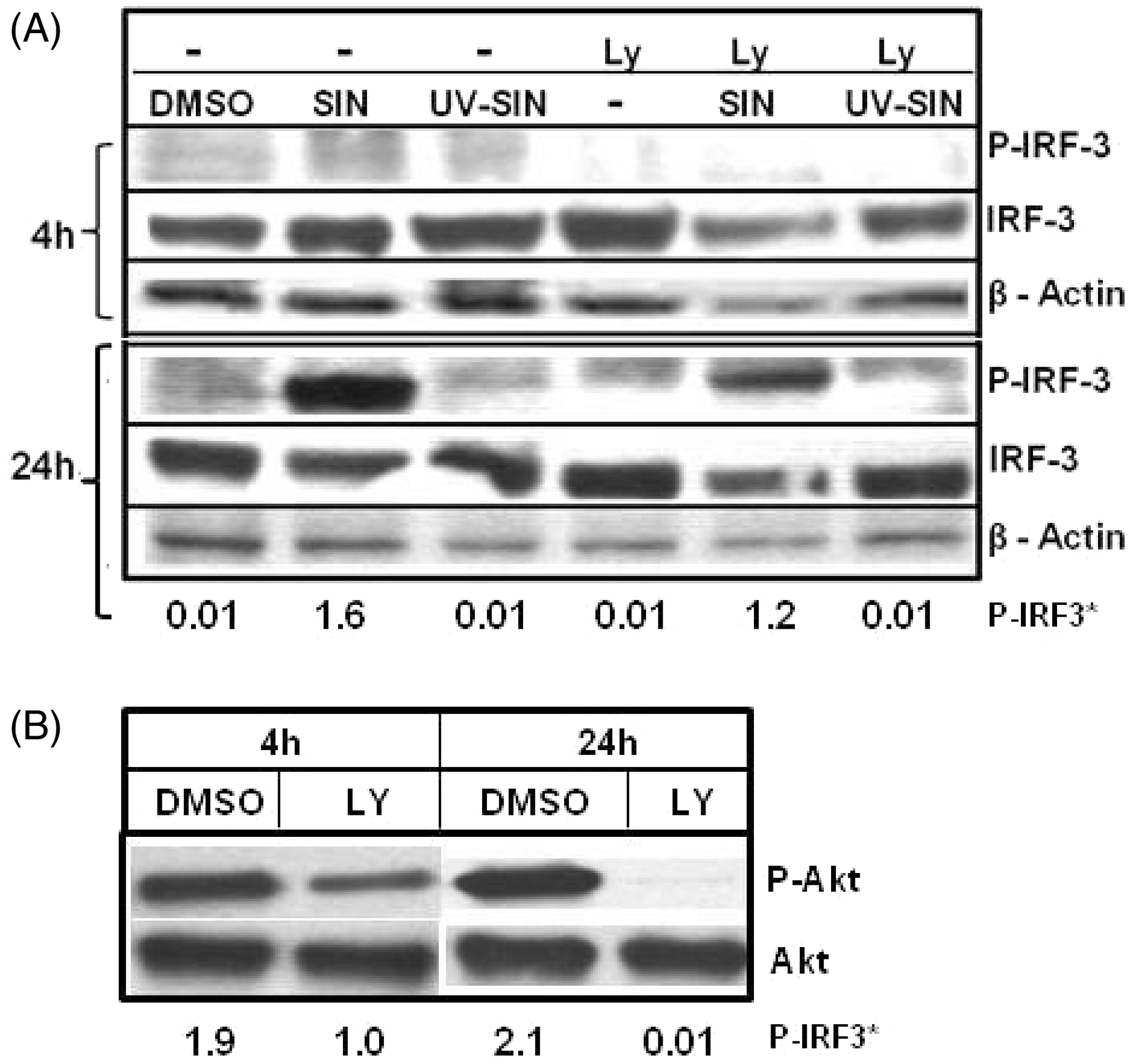
Differential requirement of PI3 kinase for ISG56 and IP-10 expression in SIN infected cells
Ly treatment partially decreased the level of p-IRF3, which could impact the expression of IRF3 regulated genes. Total RNA isolated from the above experiment was used to determine the level of ISG56 and IP-10 mRNAs by qPCR. At 4 h, Ly treated cultures showed five-fold and six-fold lower levels of ISG56 in the absence and presence of SIN, compared to cells treated with vehicle alone. At 24 h, while Ly decreased the ISG56 mRNA five-fold, Ly + SIN treated cells showed only two-fold lower mRNA (Figure 6A). These results suggest that inhibition of PI3K by Ly effectively decreases ISG56 mRNA expression, during both early and late infection. However, Ly increased the IP-10 mRNA 1.7- fold at 4 h, but Ly + SIN decreased the mRNA 1.4-fold (Figure 6B). At 24 h, Ly upregulated the IP-10 expression 1.7-fold, and Ly + SIN treatment increased the expression 4.9-fold. These results point to the differential role of the PI3K/Akt pathway in regulating ISG56 and IP-10 expression by alternate mechanisms. Interestingly, Ly treatment decreased mRNA levels of IRF3 two-fold at 4 h, but at 24 h this effect was not seen.
PI3K/Akt differentially regulates transcription of IFN stimulated genes. Pretreatment of HEK cells with PI3 kinase inhibitor Ly, decreases ISG56 (A) and IRF3 (C), but not IP-10 (B), mRNA levels in uninfected and infected cells. HEK293T cells were treated with 40 µM Ly, 1 h before infection with SIN or UV-SIN and the mRNA levels of IRF3, ISG56 and IP10 were determined by qPCR. Data represent mean ± SE from four different experiments. P-value indicates statistically significant differences between groups by Student's t-test.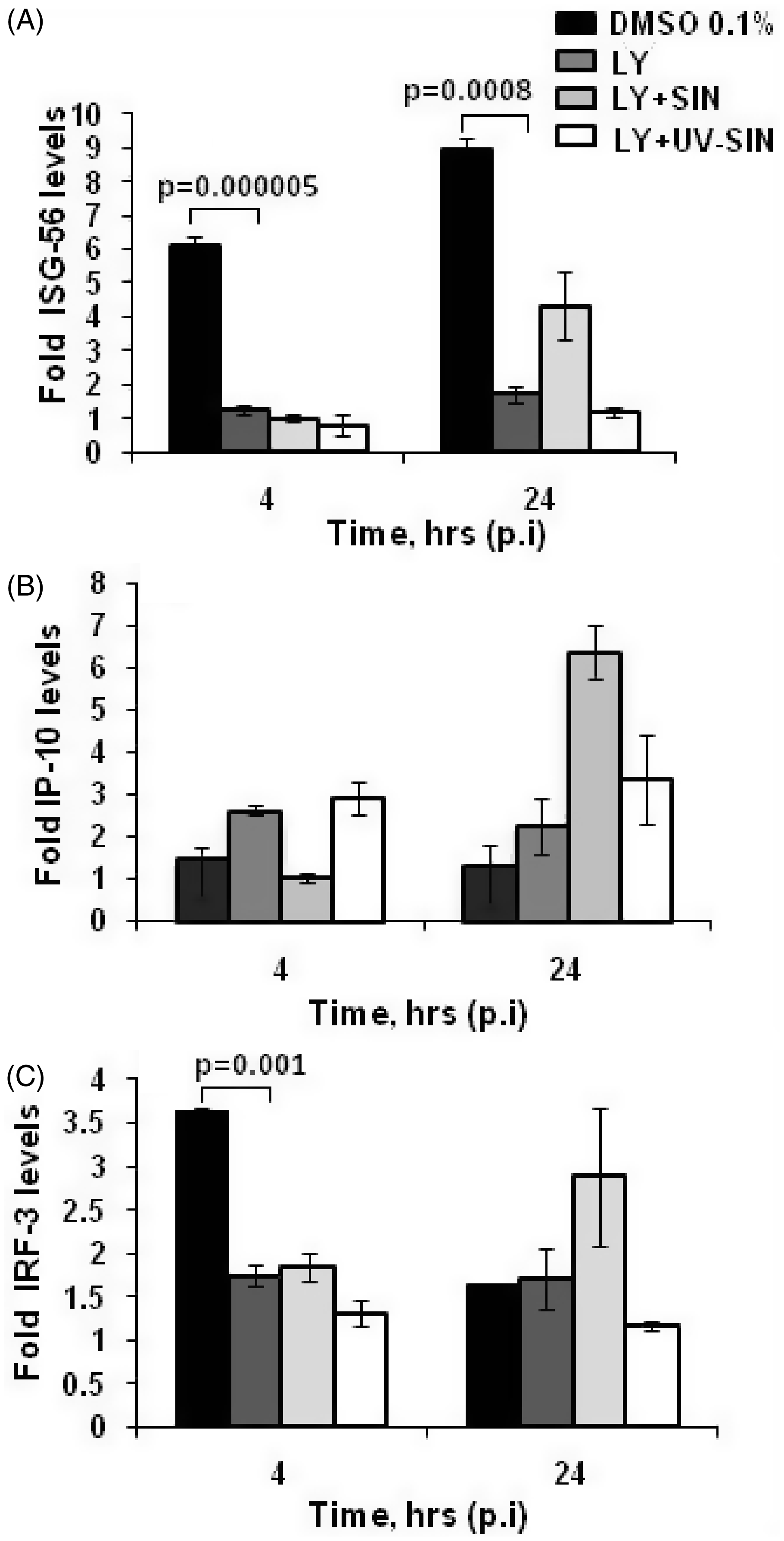
Relative levels of viral RNA in SIN, and UV-SIN infected HEK cells
The abundant phosphorylation of IRF3 in SIN infected HEK cells at 24 h may be due to accumulation of viral RNA that activates the cytoplasmic RNA sensing system.
2
To determine the level of viral RNA accumulation, HEK cells were infected with SIN and UV-SIN in the presence and absence of cycloheximide. Cycloheximide was used to block synthesis of new viral RNA so that the amount of viral RNA in the virus inoculum could be determined accurately. As shown in Figure 7A, the level of viral RNA at 4 h was 390-fold lower compared to 24 h. However the input level of UV-SIN was 160-fold lower compared to SIN (Figure 7). Although the MOI equivalent of both SIN and UV-SIN are same, the UV inactivation could have cross-linked the RNA, leading to poor quantitation by qPCR. These results suggest that the drastic accumulation of viral RNA at 24 h after SIN infection could have served as the trigger for the observed innate response.
Viral RNA levels in SIN and UV-SIN infected cells. Human embryonic kidney cells were infected with SIN (A) or UV-SIN (B) at a MOI of 10 or its equivalent, respectively, with and without 2 h of cycloheximide pretreatment. Cells were recovered at indicated times, RNA isolated, and virus-specific RNA determined by qPCR. CHX: cyclohexamide.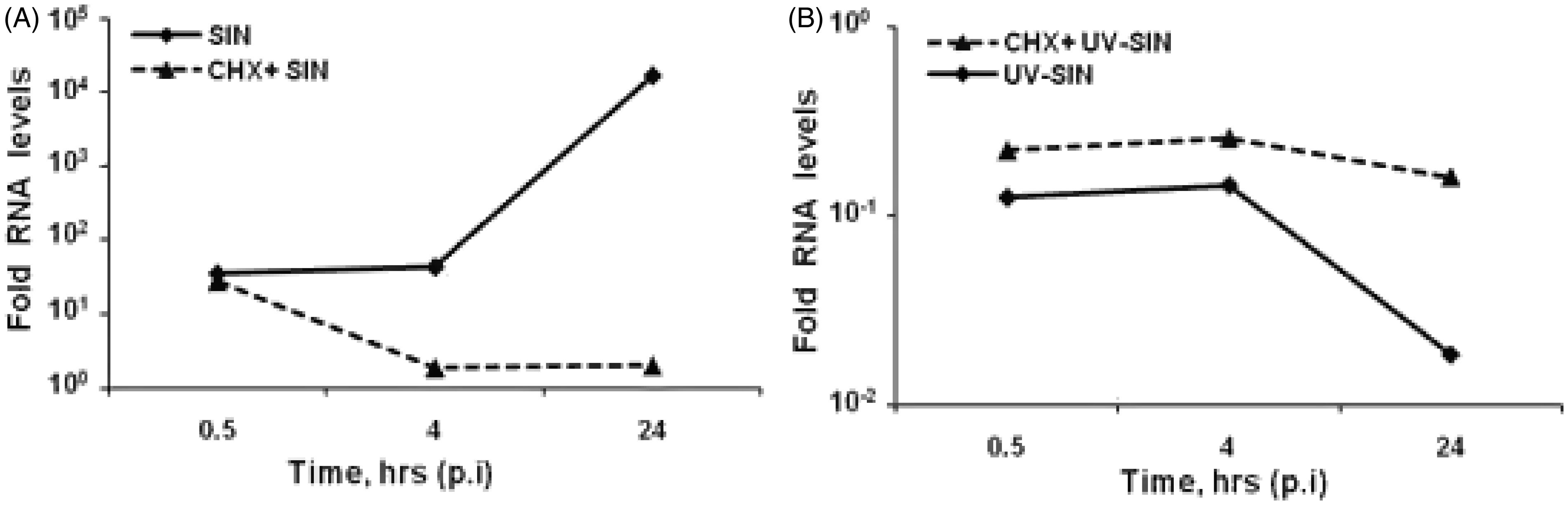
Discussion
The use of oncolytic viruses in combination with rapamycin in preclinical therapeutic models of cancer warrants in depth studies on virus-host drug-induced alterations in innate immunity and signaling.17,18 Here we have shown that the early phosphoactivation of IRF3, a key regulator of antiviral signaling, is refractory to mTOR inhibitors in HEK cells infected with SIN. Rapamycin upregulates expression of inflammatory cytokines from macrophages in response to bacterial ligands, but downregulates IFN synthesis in plasmacytoid dendritic cells and attenuates immune response to an enveloped virus. 9 In human keratinocytes, rapamycin downregulates cytokine expression in response to Poly I:C. 22 Our findings that SIN induces IRF3 phosphorylation, in the absence of IFN production, is reminiscent to the work of Burke et al. who reported high IRF3 activation in SIN infected mouse embryo fibroblasts, but poor IFN production, indicating viral antagonism. 23
The mechanism of phosphorylation and activation of IRF3 may depend on whether the signals originate from membrane bound TLRs or cytosolic sensors such as RIG-I and MDA5.3,24 The HEK cells express little or no TLRs, including TLR3. As phosphorylation of IRF3 occurred only late during SIN infection, when the viral macromolecules including RNA are abundant, it appears that recognition of cytosolic viral RNA by the helicases may have activated IRF3. Although expression of ISG56 is mediated by IRF3, IP-10 expression is also mediated by NFκB. 25 Therefore, we speculate that cytosolic recognition of SIN viral RNA, and activation events involving IRF3 and NFκB, may be mTOR independent. A TLR, IFN, and RIG-I independent virus particle entry that mediated IRF3 activation in mouse fibroblasts was reported for enveloped UV-inactivated viruses. 26 Similarly, a particle entry mediated, IRF3-dependent IFN α/β induction was reported for Semliki forest virus in myeloid dendritic cells. 27 However, in our experiments, UV-SIN failed to activate IRF3 or alter antiviral gene expression in HEK cells. These observations may be explained by different pathways of IRF3 activation in different cell types.3,24
We have previously shown that, HEK cells infected with SIN show drastic suppression of Akt/mTOR signaling, increased apoptosis and strong inhibition of protein synthesis. 19 It is remarkable that early during infection, the magnitudes of these events are minimal; however, late during infection they become pronounced. It is clear that SIN is able to effectively antagonize host antiviral strategies during early hours of infection. The strong appearance of p-IRF3 and apoptosis of HEK cells late in SIN infection is in agreement with the observation of others.28,29 It is also striking that SIN induced suppression of the Akt/mTOR pathway coincides with high levels of p-IRF3. Although mTOR controls host translation, as virus-induced alterations in signaling can occur independent of host translation 30 , the ramifications of high levels of p-IRF3 late in SIN infection needs further scrutiny.
Footnotes
Acknowledgements
This work was supported by NIH grant SC1AI081655. We thank N. Gray and D. Sabatini for torin1. The use of CHDR/HIV core for flow cytometry, MMC Morphology Core and Molecular Biology Core (U54NS041071, G12RR03032 and U54CA91408) RCMI supported Molecular Biology Core, VICTR and MeTRC resources are gratefully acknowledged.