
Abstract
Activation of host innate antiviral responses are mediated by retinoic-acid inducible gene I (RIG-I)-like receptors, RIG-I and melanoma differentiation-associated gene 5, and TLRs 3, 7, 8 and 9, recognising different types of viral nucleic acids. The major components of the RIG-I- and TLR pathways have putatively been identified, but previously unrecognised kinases may contribute to virus infection-induced activation of the IFN response. Here, we screened a human kinase cDNA library, termed the kinome, using an IFN-λ1 promoter-driven luciferase reporter assay in HEK293 cells during Sendai virus infection. Of the 568 kinases analysed, nearly 50 enhanced IFN-λ1 gene expression at least twofold in response to Sendai virus infection. The best activators were FYN (FYN oncogene related to SRC, FGR, YES), serine/threonine kinase 24, activin A receptor type 1 and SRPK1 (SFRS protein kinase 1). These kinases enhanced RIG-I-dependent IFN-λ1 promoter activation via IFN-stimulated response and NF-κB elements, as confirmed using mutant IFN-λ1 promoter constructs. FYN and SRPK1 enhanced IFN-λ1 and CXCL10 protein production via the RIG-I pathway, and stimulated RIG-I and MyD88-dependent phosphorylation of IRF3 and IRF7 transcription factors, respectively. We conclude that several previously unrecognised kinases, particularly FYN and SRPK1, positively regulate IFN-λ1 and similarly regulated cytokine and chemokine genes during viral infection.
Keywords
Introduction
First-line defence mechanisms against invading viruses are initiated by evolutionary conserved pathogen recognition receptors (PRRs) that recognise virus-specific genetic material, ssRNA, dsRNA or DNA. Viral nucleic acid-recognising receptors include retinoic acid inducible gene I-like receptors (RLR) retinoic acid inducible gene I (RIG-1) and melanoma differentiation-associated gene 5 (MDA5), TLRs 3, 7, 8 and 9, and intracellular foreign DNA-recognising receptors.1,2 Their ligand-dependent activation leads to stimulation of downstream signalling molecules, adaptor proteins, protein kinases and transcription factors that enhance antiviral cytokine and chemokine gene expression. RIG-I and MDA5 can recognise various types of RNA molecules in the cytoplasm and trigger the intracellular signalling cascades by interacting with the mitochondria-associated adaptor molecule IFN-β promoter stimulator-1 (IPS1), which leads to the activation of inhibitor of κB kinase (IKK) ε/TANK binding kinase 1 (TBK1) complex and interferon regulatory factor (IRF)-3, which enhances cytokine gene expression in the nucleus.3–5 TLRs3, 7 and 8 are transmembrane receptors expressed on the plasma membrane (TLR3) or cytoplasmic vesicles (TLR7 and TLR8), such as endosomes and the endoplasmic reticulum. While TLR3 recognises dsRNA and activates IRF3 via TRIF and IKKε/(TBK1), TLR7 and TLR8 sense foreign ssRNA and interact with the MyD88/IRAK complex to activate IRF7.6,7 Activation of RLR and TLR pathways also leads to activation of NF-κB and MAP kinase-regulated transcription factors. 8
Type I IFNs consist of 14 functional IFN-α subtypes and a single IFN-β. Type II IFN, IFN-γ, has a key role in regulating cell-mediated immunity rather than working as a direct antiviral substance. 9 Recently-discovered type III IFNs, IFN-λ1 and IFN-λ2/310,11 have been found to resemble type I IFNs in their activation and biological functions, even though they are structurally related to IL-10. RLR or TLR3-activated IRF3 enhances the expression of type I IFNs, IFN-β and IFN-α4, as well as type III IFN-λ1. 12 Other type I IFNs, including most of the IFN-α subtypes and type III IFN-λ2/3, are considered to be activated more efficiently by IRF7. 13 IRFs bind to the interferon stimulated response element (ISRE) sites residing on type I and type III IFN gene promoters. IFN-λ genes also contain PRDI-1 (positive regulatory domain 1) elements, which are involved in IRF induced IFN production. NF-κB binding sites are also found on IFN-λ and IFN-β promoters, unlike IFN-α genes, which do not have functional NF-κB binding sites.12,14 In general, the IFN-λ1 gene appears to be regulated in a similar fashion to IFN-β, while IFN-λ2/3 genes are regulated like IFN-α genes.12,15 Type III IFNs are produced by almost all cell types in response to different viral infections.10,11,16–21 IFN-λ genes have also been reported to be induced in certain bacterial infections, similar to type I IFNs.22–24
In the present study we aimed to identify previously unrecognised kinases involved in virus-induced activation of innate immune response using an expression-ready human kinase cDNA collection termed the kinome. 25 We used IFN-λ1 promoter-driven firefly reporter constructs to study whether co-expressed human protein kinases could enhance Sendai virus (SeV)-induced promoter activation; this approach resulted in identification of more than 70 kinases that enhanced virus-induced innate immune responses. Subsequent follow-up studies verified that FYN (FYN oncogene related to SRC, FGR, YES), STK24 (serine/threonine kinase 24), ACVR1 (activin A receptor type 1) and SRPK1 (SFRS protein kinase 1) were the most promising candidates as novel signalling components regulating host antiviral signalling pathways.
Materials and methods
Plasmids
Cytokine promoter-driven firefly luciferase reporter constructs used in the present study have been described previously: pGL3-IFN-λ1-luc, 26 pGL3-RANTES-luc, 27 pGL3-IFN-β-luc and pGL3-IFN-α1-luc. 28 The expression constructs for the various signalling pathway molecules have been described previously: RIG-I and ΔRIG-I, 29 MyD88, 30 IRF1, 31 IRF3, 32 IRF7 33 and FLAG-TBK1. 34 pGL3-FLAG-TBK1 was used as a positive control in kinome screening and transfection experiments. Rous sarcoma virus (RSV)-Renilla expression plasmid and expression plasmids for IKKα, IKKβ, IKKγ/NEMO were kindly provided by Dr John Hiscott (McGill University, Montreal, Canada). The human kinome expression plasmids, including kinases TBK1, FYN, STK24, ACVR1 and SRPK1, have been cloned into pCMV-XL(4-6)-vectors and have been described previously. 25
IFN-λ1 promoter mutants
Plasmid pGL3-IFN-λ1-luc was the template to generate the single ISRE site mutant promoter or the NF-κB1 and NF-κB2 site double-mutant promoter constructs. NF-κB1/2 double-mutant IFN-λ1 promoter construct was further used as a template to generate a triple mutant promoter in which ISRE, NF-κB1 and NF-κB2 sites were mutated. The oligonucleotides used to generate the promoter mutants are described in Supplementary Table 1. Mutations were incorporated using the QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). All plasmids were maintained and propagated in Escherichia coli strain DH5α.
Cell culture, transfections and viral infections
HEK293 cells (ATCC CLR 1573) were grown in Eagle minimal essential medium (MEM) culture medium (Gibco, Carlsbad, CA, USA) supplemented with 90 µg/ml penicillin, 150 µg/ml streptomycin, 2 mM
Human kinome screening and luciferase reporter assays
The automated screening of the human kinome was performed at the High Throughput Center, Biomedicum, Helsinki University. HEK293 cells were seeded on 96-well plates (20,000 cells/well) and grown overnight in Eagle-MEM supplemented with 10% FCS and antibiotics. Plasmids in transfection mixtures were added robotically onto the cells. For each well 20 ng of IFN-λ1-promoter-driven firefly luciferase reporter plasmid, 10 ng of RSV–Renilla-luciferase reporter plasmid and 50 ng of each kinase expression plasmid were added. The expected variation in kinase plasmid concentrations due to robotic extractions and dilutions was from 10 to 100 ng. pGL3-FLAG-TBK1 plasmid was used as a positive control. After 18 h luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer’s instructions with the exception that LARII and Stop&Glow reagents were used at 50 µl/well. In the second screening of the human kinome, cells were infected with Sendai virus at a MOI of 5, 4 h after transfection. RSV–Renilla luciferase activities were used to standardise transfection efficiency. HEK293 cell transfection experiments with individual kinase expression constructs were performed otherwise similarly to the screening experiments with the exception that the TBK1 plasmid from the kinase library was used as a positive control.
SDS-PAGE, Western blot and immunostaining
HEK293 cells were grown in 24-well culture plates at a density of 180,000 cells/well. Transfections were made with 500 ng/well of a given kinase expression plasmid, and the amounts of the expression plasmids of the signalling components transfected simultaneously are indicated in the respective figures. Cells were collected into passive lysis buffer (Dual-Luciferase Reporter Assay System; Promega) containing the protease inhibitors Complete™ (Roche, Basel, Switzerland) at a 1:50 dilution and 1 mM Na3VO4. Proteins from lysed cells were separated on SDS-PAGE gels with the Laemmli buffer system and transferred electrophoretically onto Immobilon-P polyvinylidene fluoride (PVDF) membranes. Membranes were probed with rabbit Abs against IRF3, 36 phosphorylated forms of IRF3 (rabbit-P-IRF3; Cell Signaling, Boston, MA, USA), IRF7 (rabbit-P-IRF7; Cell Signaling), MyD88 (Cell Signaling), c-myc and actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and guinea pig Abs against IRF7 and RIG-I. 37 Secondary HRP-conjugated anti-rabbit or anti-guinea pig IgG Abs were from Dako (Glostrup, Denmark) and immunocomplexes were detected using the enhanced chemiluminescence system and HyperMax films (GE Healthcare, Buckinghamshire, UK).
ELISA
For endogenous protein measurements supernatants from transfected HEK293 cells were collected from three replicate wells of 24-well culture plates. IFN-λ and CXCL10 cytokine levels from supernatants were determined with ELISA. IFN-λ ELISA was performed according to manufacturer’s instructions (PBL Interferon Source, Piscataway, NJ, USA). CXCL10 (IP10) ELISA was performed with specific Abs purchased from BD PharMingen (San Diego, CA, USA).
Statistical analyses
Data from promoter-driven luciferase assay and ELISA were analysed with Student’s t-test (two-tailed, unequal variance). The significance of differences was considered significant when P < 0.05 and highly significant when P < 0.005.
Results
Screening of the human kinome cDNA library yielded multiple kinases that enhance SeV infection-stimulated IFN-λ1 promoter activation
The human kinase collection consists of 568 human kinase cDNAs and comprises more than 93% of all human kinases.
25
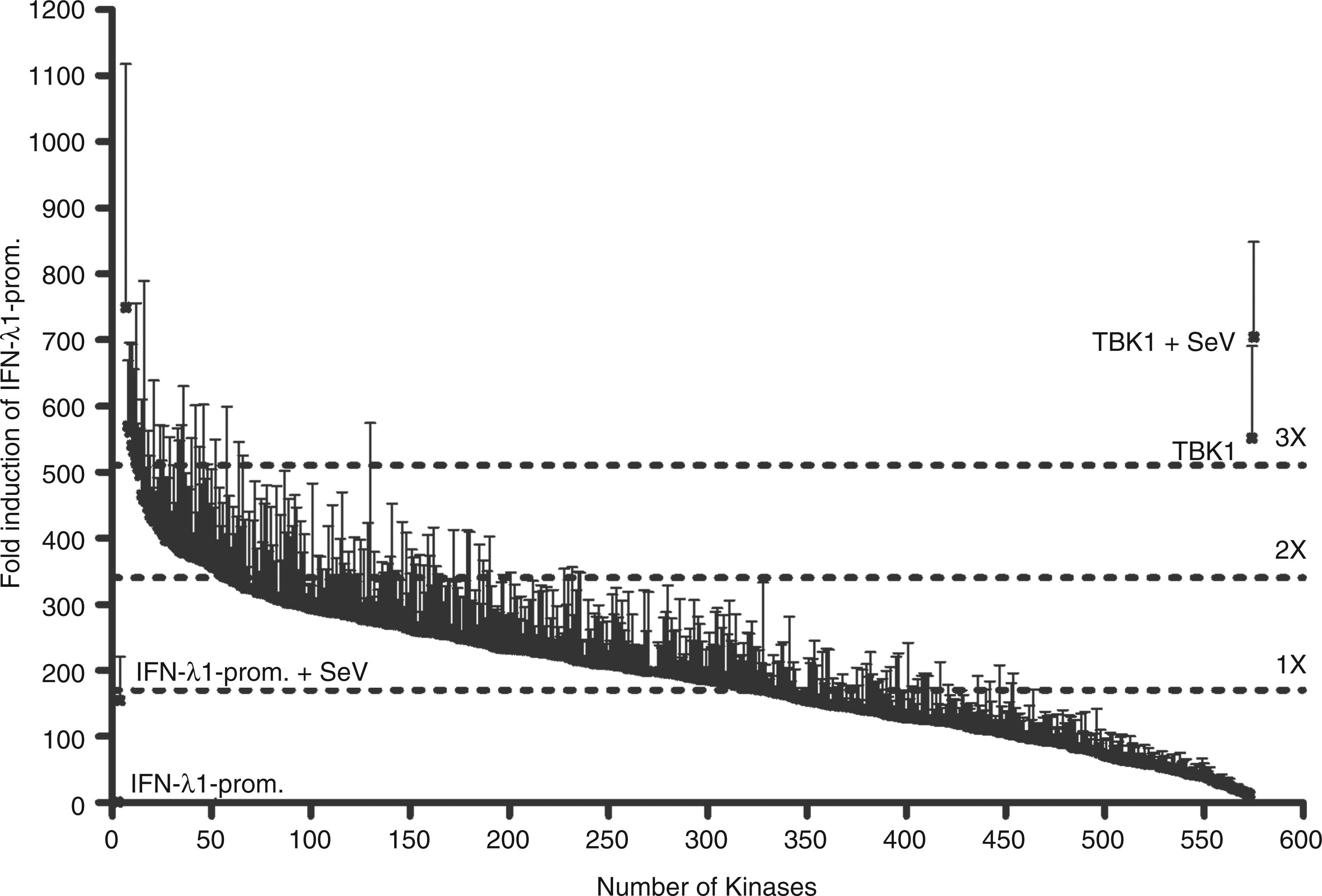
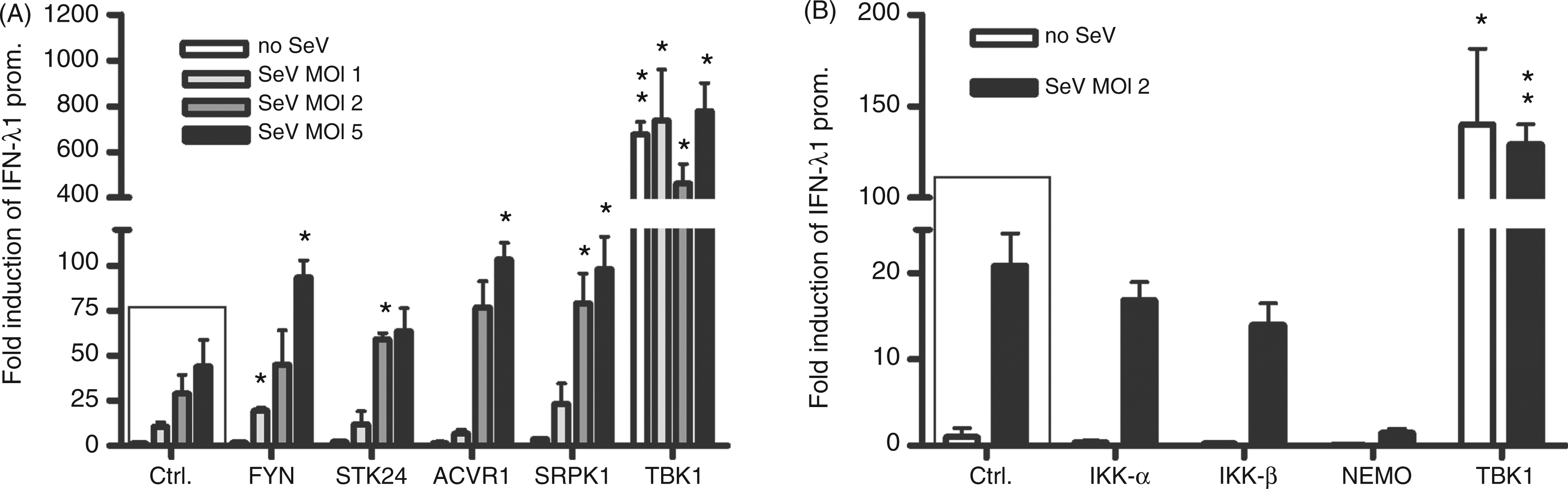
We established a high throughput screening method to find out whether there are novel kinases that can activate type III IFN gene expression with an IFN-λ1 promoter-driven luciferase reporter in HEK293 cells. The experimental model and process is outlined in Figure 1. To prevent screening background and bias we used HEK293 cells in our experiments as they do not express all of the signalling components that need to be ectopically expressed. In uninfected HEK293 cells, only TBK1 and IKKε were able to activate the IFN-λ1 reporter (data not shown). This suggested that in this experimental system only these two kinases, which have been shown previously to be able to auto-activate themselves, activate IRF3 and stimulate IFN gene expression. In order to identify other kinases that would potentiate virus infection-induced IFN-λ1 gene expression, we performed the screening of the human kinome again and infected the transfected HEK293 cells with SeV at 4 h post-transfection at a MOI of 5 for 18 h to induce antiviral signalling pathways. With this approach we identified several novel kinases, which enhanced IFN-λ1 promoter activation at least 2 to 3-fold during SeV infection. The relative IFN-λ1 promoter activation by all kinome kinases are shown in descending order in Figure 2 and the corresponding numeric values are listed in Supplementary Table 2. We selected 29 kinases that were able to enhance virus-induced IFN-λ1 promoter activation by more than 2.5-fold compared with IFN-λ1 promoter activation seen with SeV infection alone for further analyses. In these analyses we used two different kinase expression plasmid concentrations: 25 and 50 ng/well (Supplementary Table 3). Four kinases were observed to activate the IFN-λ1 promoter during SeV infection—FYN, STK24, ACVR1 and SRPK1—and they were selected for more detailed analyses. The ability of these kinases to potentiate SeV-induced IFN-λ1 promoter activation was analysed in relation to virus dose (Figure 3A). At higher MOI values of 2 and 5, STK24, ACVR1 and SRPK1 enhanced IFN-λ1 promoter activity at least twofold. FYN was able to stimulate a twofold increase in reporter activity at a MOI of 1. The positive effects on the IFN-λ1 promoter activation of all four kinases were found to be statistically significant. The positive control kinase, TBK1, efficiently activated the IFN-λ1 promoter with or without SeV infection (Figure 3A and Supplementary Table 3). As an additional control for the screening system we tested whether the components of the classical IKK complex had any effect on IFN-λ1 promoter activation in our experiments. We transfected IKKα, IKKβ and IKKγ/NEMO expression constructs into HEK293 cells, and infected the cells with SeV at a MOI of 2, 4 h after transfection (Figure 3B). The classical IKKs did not activate or enhance SeV-induced IFN-λ1 promoter activation (Figure 3B), similar to the data obtained from kinome screening, further confirming the specificity of our assay.
Schematic illustration of the innate immunity signalling pathways and IFN-λ1 promoter reporter system used in the present work to study the potential involvement of new kinases. The figure outlines the most important events after viral recognition by PAMP receptors in human cells resulting in antiviral cytokine and chemokine promoter activation. The activation of signalling pathways are known to involve kinases TBK1 and IKKe. The kinases tested for positive effects have been shown as one universal kinase, and the possible sites where the respective kinase would function are shown as dashed arrows followed by question marks. The following activation of transcription factors and consequent attachment to the promoter regions leads to IFN-λ1 promoter activation, as well as other cytokine and chemokine genes regulated in a similar fashion to type I/III IFNs. The activation of IFN-λ1 promoter by 568 human kinases during SeV (MOI 5) infection. HEK293 cells were co-transfected in 96-well culture plates with 20 ng of reporter plasmids carrying firefly luciferase gene under IFN-λ1 promoter, 10 ng of Renilla luciferase under a constitutive RSV promoter and 50 ng of kinase expression plasmids. The activation of IFN-λ1 promoter is shown for each kinase in a descending order. IFN-λ1 promoter–reporter luciferase expression was normalized with Renilla luciferase reporter. The sample transfected with the two reporter constructs was assigned as 1 and relative inductions by a given kinase expression construct were calculated in relation to this sample. SeV-infected IFN-λ1 promoter activation gave a relative induction of 160 (on the left). Two positive controls, cells transfected with TBK1 in the absence or presence of SeV infection are indicated in the furthest right figure.The numerical values for IFN-λ1 promoter activity induced by each kinase expression plasmid can be found in Supplementary Table 2. Two hundred and sixty-three kinases activated the IFN-λ1 promoter more than onefold, 66 kinases activated the promoter more than twofold, and 8 activated it more than threefold compared with IFN-λ1 promoter activation by SeV infection. These limits are shown as dotted lines. FYN, STK24, ACVR1, SRPK1 and TBK1 kinases enhance SeV-induced IFN-λ1 promoter activation. HEK293 cells grown on 96-well culture plates were transfected with FYN, STK24, ACVR1 or SRPK1 (A) or classical IKK (B) expression plasmids (50 ng/well) together with IFN-λ1 promoter–reporter (20 ng/well) and Renilla control plasmids (10 ng/well) followed by SeV infection at MOIs of 1, 2 or 5 (A) or a MOI of 2 (B), or left untreated. Control cells (ctrl) were transfected with IFN-λ1 promoter–reporter and RSV–Renilla promoter, and the promoter activation in these cells is designated as 1. The results are the means (±1 SD) IFN-λ1 promoter activities normalised with Renilla from three technical replicates. Results are representative of three independent experiments. Results were considered statistically significant when P < 0.05 (*) and highly significant when P < 0.005 (**) compared with the boxed bars.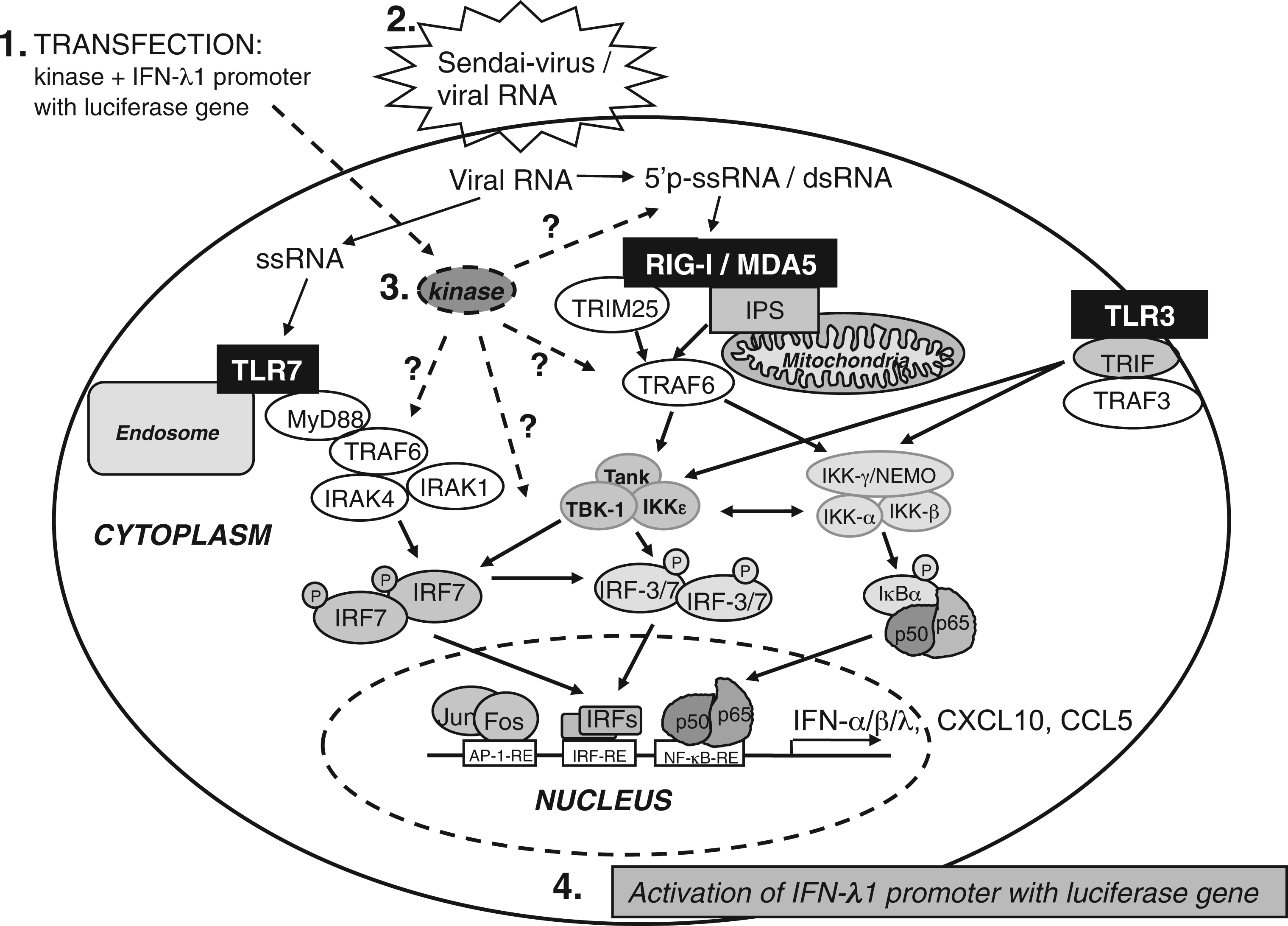
FYN, STK24, ACVR1 and SRPK1 kinases enhance IFN-λ1 promoter activation elicited by the RNA helicase RIG-I
The RNA helicase RIG-I and its downstream transcription factor IRF3 are known to activate type I and III IFN gene transcription after sensing viral RNA in the cytoplasm.
38
We studied the possible role of FYN, STK24, ACVR1 and SRPK1 on the RIG-I-mediated signalling pathway. RIG-I, IRF3 or both were transfected into HEK293 cells with each kinase. Four h after transfection the cells were infected with SeV at a MOI of 2 for 18 h and IFN-λ1-promoter-driven luciferase reporter activities were measured. RIG-I overexpression alone did not induce the IFN-λ1 promoter significantly and required SeV infection for activity (Figure 4A). Interestingly, overexpressed RIG-I, together with FYN, STK24 or SRPK1, led to a clear and statistically significant increase in IFN-λ1 promoter activation (Figure 4A, white bars). Under SeV infection expression of FYN, STK24, ACVR1 or SRPK1 kinases induced a 2–3-fold increase in RIG-I-mediated IFN-λ1 promoter activity (Figure 4A, black bars). We further analysed whether FYN, STK24, ACVR1 or SRPK1 would also enhance IFN-λ1 promoter activation in response to IRF3 overexpression during viral infection, but no other enhancing kinases apart from TBK1 were identified (Figure 4B). Co-expression of RIG-I and IRF3 in HEK293 cells modestly activated the IFN-λ1 promoter and, in the absence of SeV infection, FYN, STK24, and SRPK1, along with TBK1, further enhanced this activation statistically significantly (Figure 4C, white bars). Under SeV infection IFN-λ1 promoter activation was further enhanced by FYN, STK24 and TBK1 (Figure 4C, black bars). TRIM25, which is an essential component of the RIG-I signalling pathway,
39
had no effect in these experiments (data not shown). The requirement of SeV-induced activation of the RIG-I pathway can be circumvented by transfecting cells with a constitutively activated form of RIG-I, ΔRIG-I. We transfected HEK293 cells with three different amounts of ΔRIG-I (5, 20 and 50 ng/well) and the kinase expression plasmids. There was a clear dose-dependent activation of the IFN-λ1 promoter in ΔRIG-I-expressing cells, and FYN, STK24, ACVR1 and SRPK1 increased the promoter activation between 1.5 and 3-fold, depending on the dose of ΔRIG-I. The kinases had highly significant positive effects on IFN-λ1 promoter activation (Figure 4D). The addition of IRF3 (5 ng/well) with 50 ng of ΔRIG-I stimulated IFN-λ1 promoter activity above the dynamic range of the reporting system (Figure 4C) and it was difficult to observe any additional stimulatory effects by the kinases. Statistically significant effects were only seen with FYN or SRPK1 with the lowest amounts of ΔRIG-I (Figure 4E, white and grey bars). The natural ligand of the RIG-I receptor is viral 5’ppp-dsRNA. We transfected HEK293 cells with the IFN-λ1 promoter, RIG-I and respective kinase expression plasmids, and, 4 h after transfection, an 88-base pair 5’ppp-dsRNA was added to the cells as indicated (Figure 4F). The 5’ppp-dsRNA activated the IFN-λ1 promoter on its own, and FYN, STK24, ACVR1 and SRPK1 enhanced this activation. All the kinases except STK24 enhanced the IFN-λ1 promoter activation 3–4 fold, and this was highly significant (Figure 4F, black bars). Figure 4 shows that FYN and SRPK1 kinases enhance IFN-λ1 promoter activation via RIG-I- and IRF3-mediated signalling pathways, and when ΔRIG-I -mediated activation signal is stronger (with higher plasmid amounts) STK24 and ACVR1 kinases also show promoter-enhancing activity.
FYN, STK24, ACVR1, SRPK1 and TBK1 kinases enhance the IFN-λ1 promoter activation induced by the RIG-I pathway in HEK293 cells. Kinase expression plasmids (50 ng/well) together with RIG-I expression constructs (20 ng/plasmid/well) or IRF3 (5 ng/well) or their combinations were transfected into HEK293 cells in 96-well culture plates. At 4 h after transfection cells were infected with SeV at a MOI of 2 or left untreated (A, B, C). A ΔRIG-I expression construct was transfected at concentrations 5, 20 or 50 ng/well (D, E) with or without IRF3 expression plasmids (20 ng/well) (E). Samples were similarly transfected as indicated and, 4 h after initial transfection, 88 bp 5’ppp-dsRNA was transfected to the samples indicated at 20 ng/well (F) Control cells (ctrl) were transfected with IFN-λ1 promoter–reporter and RSV–Renilla control plasmids, and reporter activity in these cells was designated as 1. The mean luciferase activities with SDs of the means are shown from three replicates. Results are representative of three independent experiments. Results were considered statistically significant when P < 0.05 (*) and highly significant when P < 0.005 (**) compared with the boxed bars.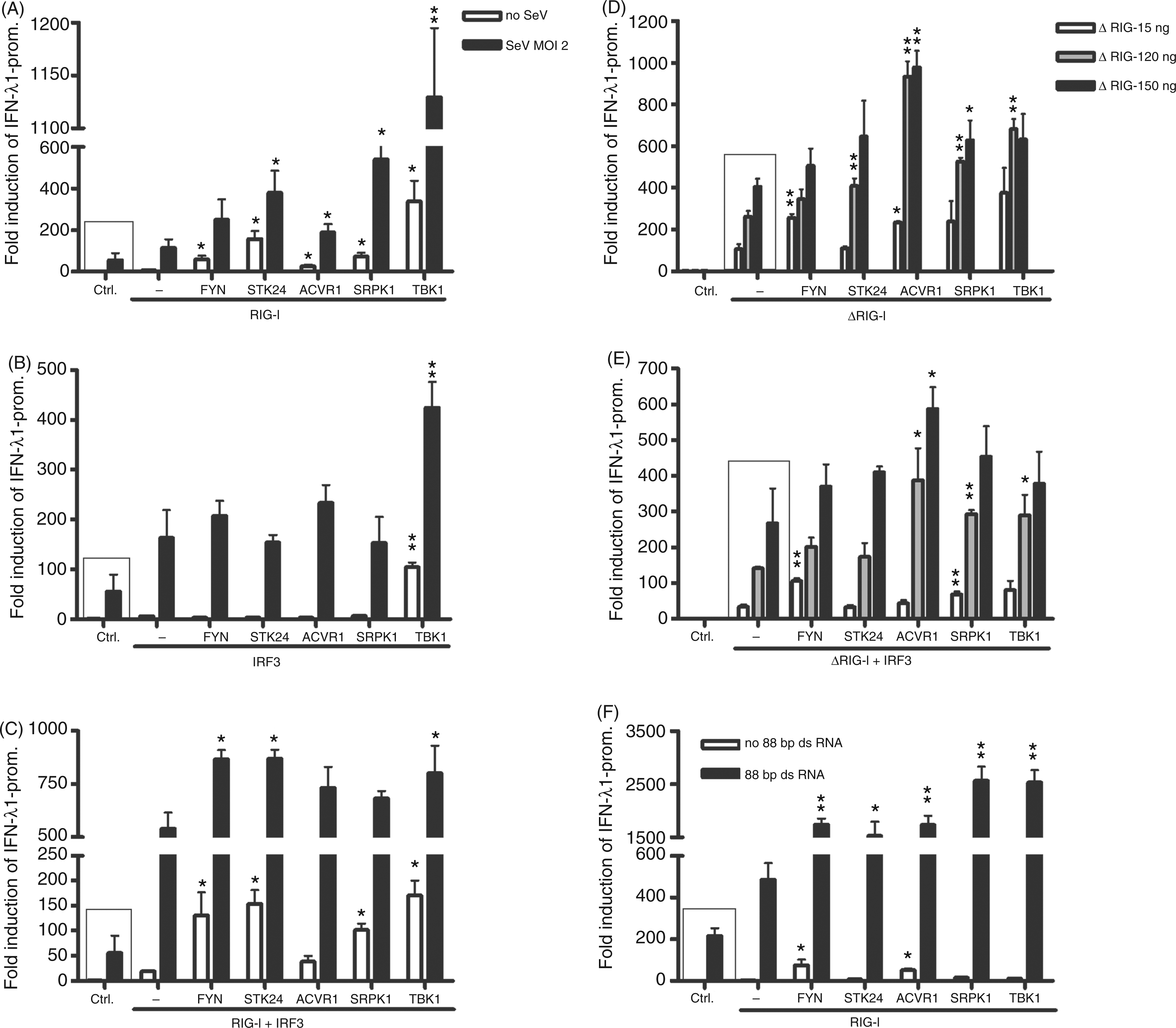
As type I and III IFN genes are regulated by the RIG-I/IRF3 pathway we analysed the role of these kinases on other IFN promoters, including IFN-β and IFN-α4 promoters, as well as on a chemokine promoter, CCL5/RANTES, which can be activated via the same pathways as the classical IFNs.27,40,41 We transfected cells with 10 or 20 ng of ΔRIG-I, thus bypassing other stimulatory signals post-transfection, and FYN, STK24, ACVR1, SRPK1 or TBK1 along with IFN-λ1, IFN-β, IFN-α4 or CCL5 promoter reporter constructs. The data show that IFN-λ1 and IFN-β promoters were very similarly activated by ΔRIG-I, as expected. FYN and SRPK1 kinases expressed with ΔRIG-I-activated IFN-λ1 and IFN-β promoters were highly statistically significant, whereas STK24 and ACVR1 only enhance promoter activation with the higher amount of ΔRIG-I (Figure 5A, B). IFN-α4 promoter was only weakly induced by ΔRIG-I, as expected, with considerable variation, so the possible stimulatory effect of the kinases can not be verified reliably (Figure 5C). The trend for CCL5 promoter enhancement by the kinases was similar to that seen with IFN-λ1 and IFN-β promoters The effect of FYN, STK24, ACVR1, SRPK1 and TBK1 kinases on RIG-I mediated IFN and chemokine promoter activation. FYN, STK24, ACVR1 or SRPK1 kinase (50 ng/well) and ΔRIG-I (10 or 20 ng/well) expression plasmids, and IFN-λ1 (A), IFN-β (B), IFN-α4 (C) or CCL5 (D) promoter–reporter, together with RSV–Renilla plasmids, were transfected into HEK293 cells grown on 96-well culture plates. Control cells (ctrl) were transfected with respective promoter–reporter and RSV–Renilla promoter, and the promoter activation in these cells is designated as 1. The mean luciferase activities with SDs of the means are shown from three replicates. Results are representative of three independent experiments. Results were considered statistically significant when P < 0.05 (*) and highly significant when P < 0.005 (**) compared with the boxed bars.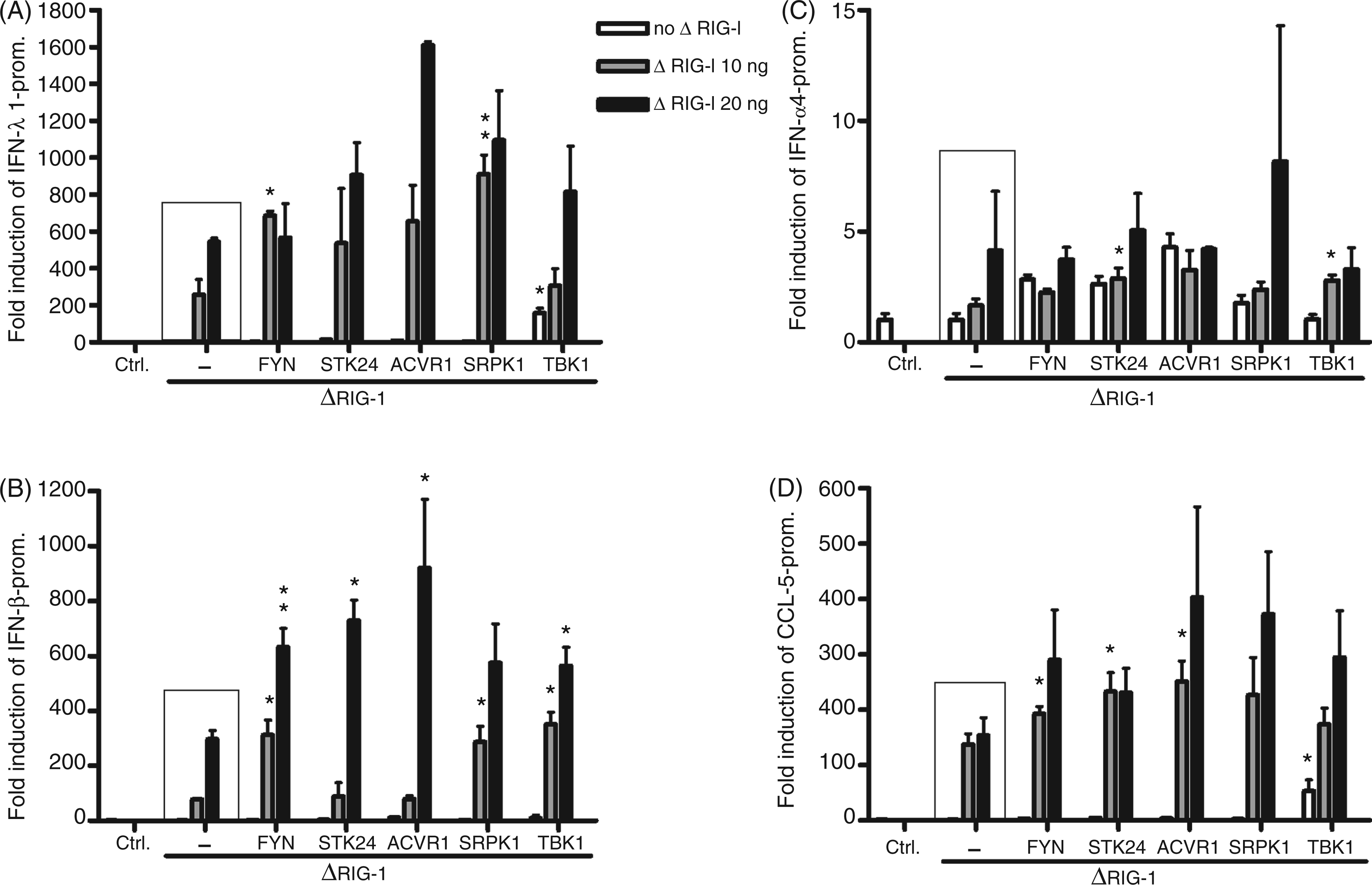
The role of kinases in the MyD88-IRF1/7 pathway
Viral RNA sensing by TLR7/8 in endosomal membranes is signalled via MyD88. It is known that, in transfected HEK293 cells, MyD88 co-operates with IRF1 and IRF7 to induce IFN promoter activation, including IFN-λ1.
12
Similarly to the RIG-I experiments, we studied the effects of overexpression of MyD88 and IRF1/7 alone or in combination with the kinases on IFN-λ1 promoter activation in the presence and absence of SeV infection (Figure 6A–E). The IFN-λ1 promoter was poorly induced with MyD88 and only TBK1 could efficiently activate the promoter (Figure 6A, white bars). IRF1 overexpression with STK24, as well as with TBK1, led to significant enhancement of promoter activation (Figure 6B, white bars), and, under SeV infection, all kinases established statistically significant positive effects (Figure 6B, black bars). MyD88 and IRF1 overexpression with kinases led to similar results (Figure 6C).
The effect of FYN, STK24, ACVR1, SRPK1 and TBK1 kinases on MyD88-, IRF1- and IRF7-regulated IFN promoter activation. HEK293 cells grown on 96-well culture plates were transfected with FYN, STK24, ACVR1 and SRPK1 expression plasmids (50 ng/well), together with MyD88 or IRF1 (20 ng/well) or IRF7 (10 ng/well) expression constructs, or their combinations with IFN-λ1 (A-D) or IFN-α4 (E, F) promoter–reporter and RSV–Renilla plasmids, as indicated in the figure. Transfected cells were left uninfected or were infected with SeV (MOI 2) at 4 h after transfection followed by measurements at 18 h after infection. Promoter activation in control cells (ctrl) transfected with IFN-λ1 or IFN-α4 promoter–reporter and RSV–Renilla promoter were designated as 1. The mean luciferase activities with SDs are shown from three replicates. Results are representative of three individual experiments. Results were considered statistically significant when P < 0.05 (*) and highly significant when P < 0.005 (**) compared with the boxed bars.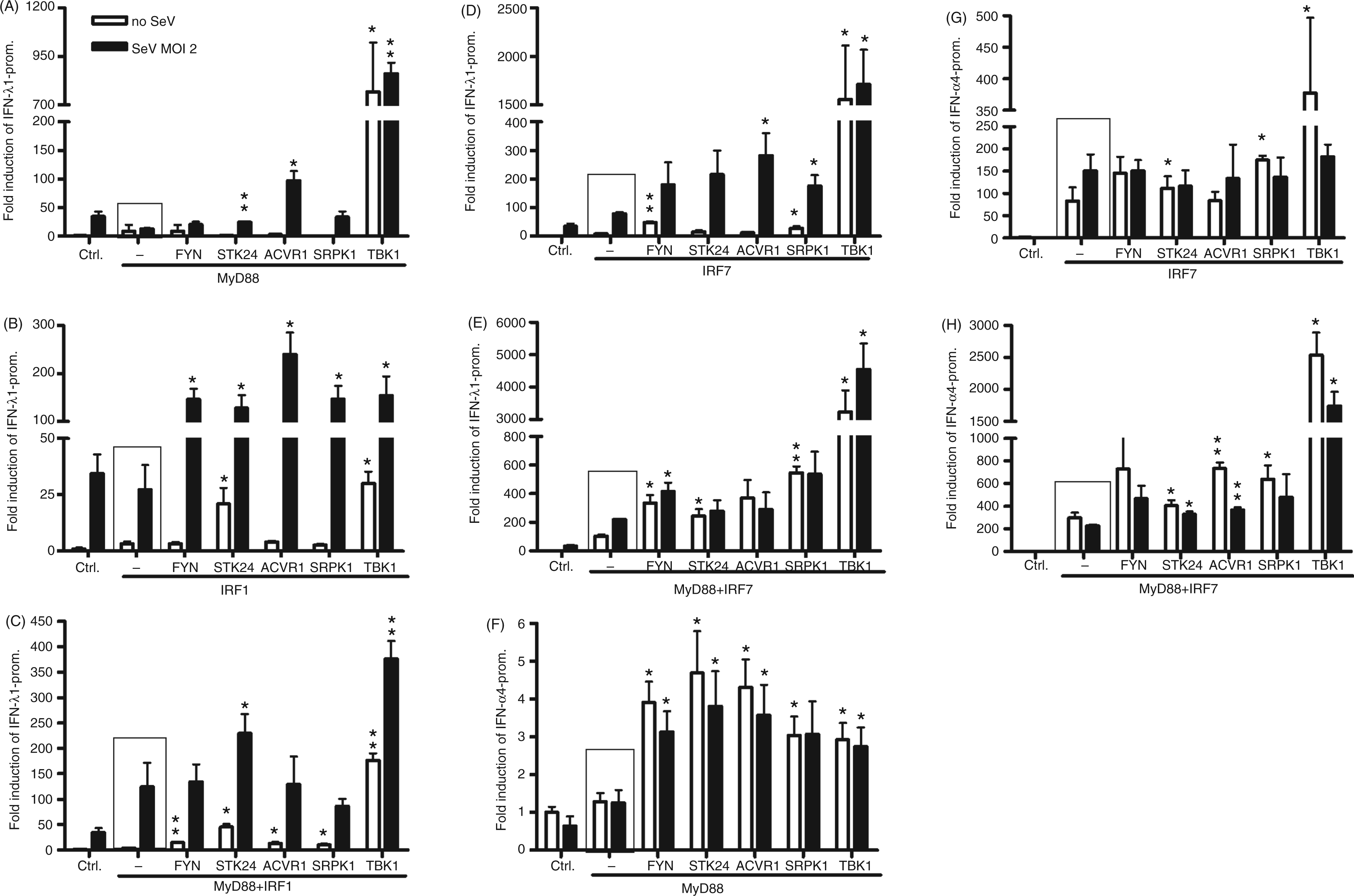
We continued to study the role of the kinases with IRF7-mediated IFN-λ1 promoter activation. IRF7 alone only moderately activated the IFN-λ1 promoter—FYN or SRPK1 can enhance this activation to some extent; however, TBK1 was the most effective kinase (Figure 6D, white bars). Under SeV infection all four kinases can increase IFN-λ1 promoter activation (Figure 6D, black bars). Positive control TBK1 can activate the promoter so strongly with IRF7 that it does not require SeV infection. Co-expression of MyD88 and IRF7 is a powerful activator of type I and III promoters; therefore, the additional effect of SeV infection on promoter activation was low (Figure 6E, white and black bars). Nevertheless, we observed a 2–3-fold stimulation of the promoter activity in FYN and SRPK1 transfected cells, and a strong effect with TBK1 (Figure 6E). As IFN-α gene expression is known to be activated primarily by the TLR-MyD88-IRF7 pathway, we wanted to analyse the effect of kinases on IFN-α4 promoter. The IFN-α4 promoter was only weakly induced by MyD88 and the observed effects on IFN-α4 promoter activation remain controversial (Figure 6F). IRF7 expression in the absence or presence of MyD88 activated the IFN-α4 promoter to the maximum level. There was a drop in IFN-α4 promoter activation with SeV infection compared with uninfected cells, resulting from over-stimulation of the system (Figure 6G, H). Still, FYN and SRPK1 contributed to the IFN-α4 promoter activation to some extent, and the relative effects were statistically significant or highly significant (Figure 6G, H). The data presented in Figures 4–6 suggest that FYN, STK24, ACVR1 and SRPK1 have a stronger role in enhancing RIG-I than MyD88-mediated pathways, and that FYN and SRPK1 have the best positive effects on the RIG-I-mediated promoter activation. Therefore, we decided to characterise, in more detail, the role of these four kinases on the RIG-I pathway.
Role of protein kinases in RIG-I-induced artificial ISRE and NF-κB promoters, and wild type and mutated IFN-λ1 gene promoter
IRF and NF-κB transcription factors are known to regulate the activation of the IFN-λ1 promoter by binding to ISRE and NF-ĸB binding sites in respective IFN promoters (Figures 4–6 and Osterlund et al.
12
). We studied the role of FYN, STK24, ACVR1, SRPK1 and TBK1 kinases on the activation of an artificial 5xISRE-luc or 3xNF-κB-luc promoter by SeV infection at a MOI of 2, 4 h after transfection, or by ΔRIG-I overexpression. In the presence of SeV infection all kinases had significant positive effects on 5xISRE-promoter (Figure 7A), and FYN and SRPK1 enhanced the 3xNF-κB promoter activation almost as well as TBK1—SRPK1 having highly significant effects (Figure 7B). When the cells were stimulated with ΔRIG-I, FYN and SRPK1 were able to activate the ISRE and NF-κB promoters as efficiently as the positive control TBK1, though the effects of TBK1 or the other kinases were not statistically significant (Figure 7C, D). To further characterise the role of the kinases on ISRE- and NF-κB-mediated gene activation we used three different IFN-λ1 mutant promoter reporter constructs (see Supplementary Table 1), an ISRE site mutant, an NF-κB1 + NF-κB2 site double mutant and a third construct with all three sites mutated (Figure 7E). The effects of FYN, STK24, ACVR1 and SRPK1 were analysed on wild type and mutant IFN-λ1 promoter constructs stimulated by 5 ng of ΔRIG-I expression plasmid, which efficiently activates the IFN-λ1 promoter. The wild type IFN-λ1 promoter activation was further enhanced by FYN and SRPK1, as well as by TBK1, as expected from previous experiments (Figure 7E, white bars). The ISRE site mutant and the double NF-κB mutant promoters were both activated weakly by ΔRIG-I compared with the wild type promoter, and some positive effects of the kinases were observed (Figure 7E, light and dark grey bars). The triple mutant IFN-λ1 promoter did not respond to ΔRIG-I stimulation, and no activation was elicited by any of the kinases (Figure 7E). In this experiment, a lower amount of ΔRIG-I expression plasmid (5 ng) was used. These data are consistent with the results in Figures 4 and 5, and may indicate that the relative ability of STK24 and ACVR1 to stimulate RIG-I mediated signalling is weaker compared with FYN, SRPK1 and TBK1.
The effect of FYN, STK24, ACVR1, SRPK1 and TBK1 kinases on ISRE- and NF-κB promoters, and IFN-λ1 mutated or natural promoter activation. HEK293 cells grown on 96-well plates were transfected with protein kinase expression plasmids and artificial 5xISRE promoter–reporter (A and C), 3xNF-κB promoter–reporter (B and D) or IFN-λ1 natural promoter, IFN-λ1-ISRE-mutant, IFN-λ1-INF-κB1/NF-κB2-mutant and IFN-λ1-triple-mutant (E), and RSV–Renilla promoter plasmids. The cells were left uninfected or infected with SeV (A, B) at 4 h after transfection and cells were collected 18 h after infection. Alternatively, promoter activation was stimulated with ΔRIG-I expression plasmid (20 ng/well), which was transfected together with the kinase expression and promoter–reporter constructs (C–E) followed by collection of cells at 22 h after transfection. Promoter activation in control cells (ctrl) transfected with different promoter–reporter and RSV–Renilla promoter constructs were designated as 1. The mean luciferase activities with SDs are shown from three replicates. Results are representative of three individual experiments. Results were considered statistically significant when P < 0.05 (*) and highly significant when P < 0.005 (**) compared with the boxed bars.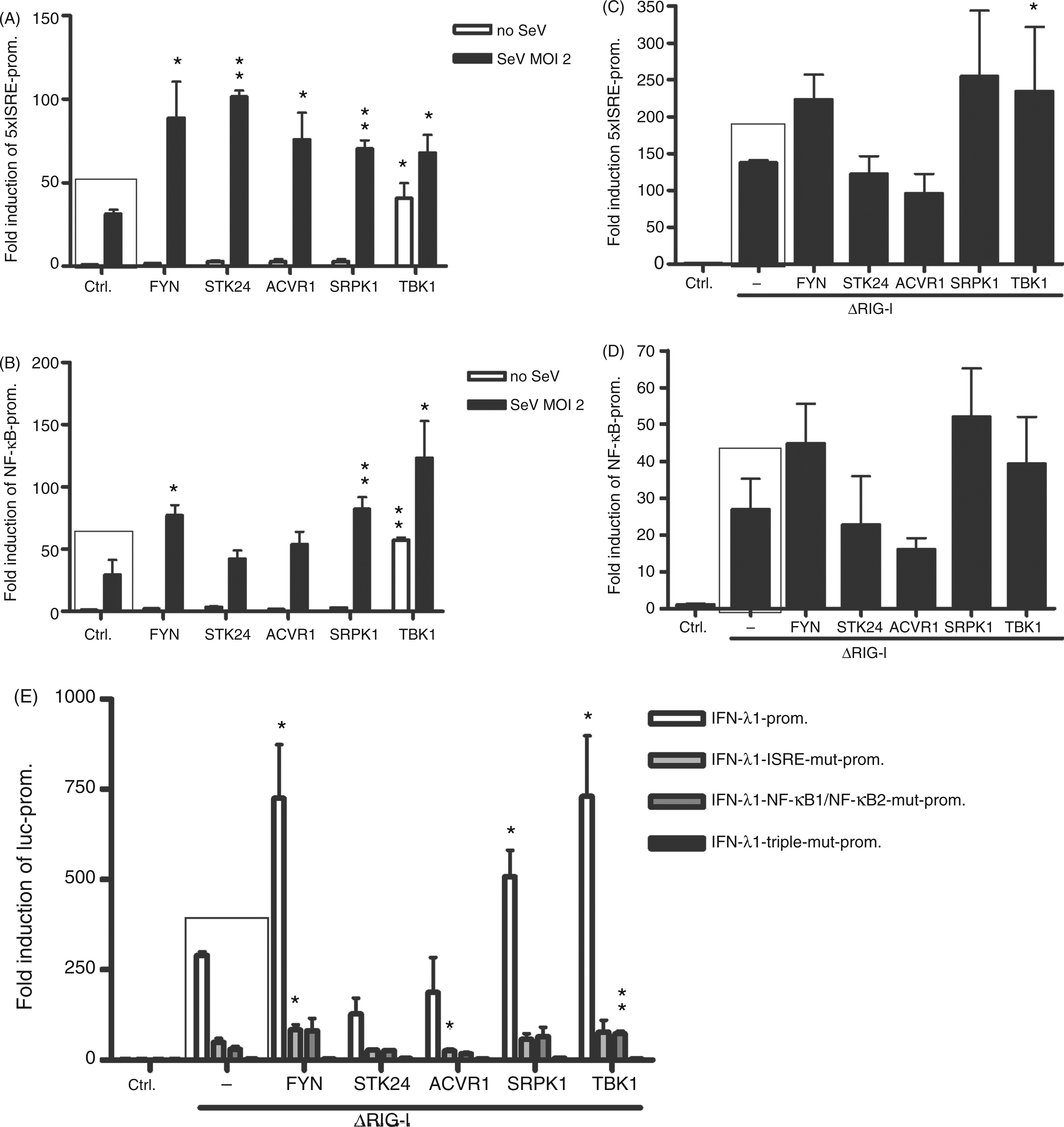
The role of kinases in enhancing endogenous IFN-λ1 and CXCL10 protein production
Of the 568 kinases we screened initially, we have been able to show that FYN and SRPK1 kinases stimulate IFN-λ1 promoter via the RIG-I pathway in an ISRE- and NF-κB-dependent fashion. Next, we wanted to find out whether the positive regulatory effects by FYN and SRPK1 could be detected on endogenous IFN-λ1 and CXCL10 protein expression. HEK293 cells were transfected with ΔRIG-I, IRF3 and the kinase expression plasmids in the amounts indicated in Figure 8. Cells were collected at 16 and 24 h after transfection. Endogenous IFN-λ protein expression was enhanced with all kinases compared with protein measured from the control sample at the 24 h. FYN- and TBK1-transfected samples showed the highest rate of increase in IFN-λ protein detected. The positive effects of the kinases were not visible at 16 h (Figure 8A). CXCL10 protein production was slower, and detectable levels of the cytokine were only seen in the 24-h supernatants. Similar to IFN-λ1 protein data, CXCL10 protein production was enhanced 1.5–2.5-fold by FYN, STK24, ACVR1, SRPK1 and TBK1 (Figure 8B). Only samples expressing FYN were considered statistically significant (Figure 8A, B). This data shows that ectopic expression of the tested kinases not only activates cytokine promoter–reporter constructs, but also stimulates authentic cytokine genes leading to enhanced production of the respective proteins.
Regulation of endogenous IFN-λ1 and CXCL10 protein production by protein kinases. In order to study the expression of endogenous IFN-λ1 and CXCL10 genes HEK293 cells grown on 24-well plates were transfected with FYN, STK24, ACVR1, SRPK1 and TBK1 expression plasmids (500 ng/well) together with IRF3 and ΔRIG-I expression constructs (100 ng/well), as indicated in the figure. Supernatants were collected at 16 or 24 h after transfection. Cytokine production into cell culture supernatants was analysed by IFN-λ1 (A) and CXCL10 (B) ELISA assays. ELISA values are the means and SDs of triplicate cell culture supernatants. Results are representative of at least twice-repeated experiments. Results were considered statistically significant when P < 0.05 (*) and highly significant when P < 0.005 (**) compared with the boxed bars.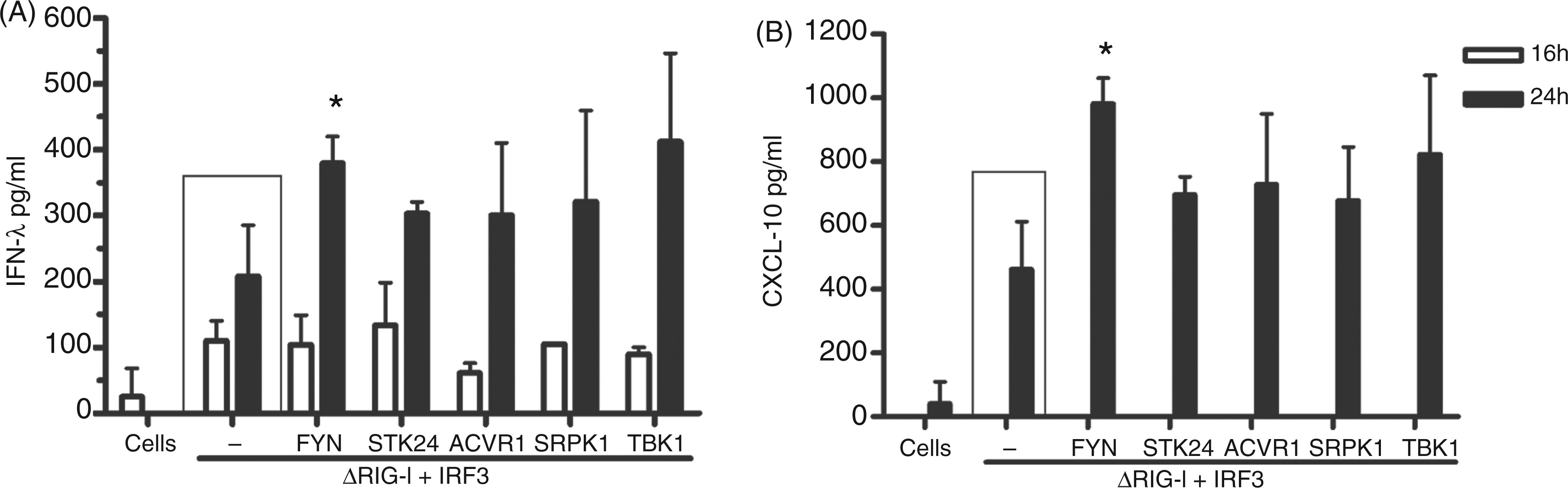
Role of kinases in enhancing the phosphorylation of IRF3 and IRF7
The data presented in Figures 3–8 confirm that kinases FYN, STK24, ACVR1 and SRPK1 all have similar affects on the activation of IFN-λ1 and other cytokine genes, and that some of the effects seen are at the same level as observed with the positive control kinase, TBK1. We continued to study the possible mechanisms of action of these kinases by exploring the possible positive effects of FYN, STK24, ACVR1 or SRPK1 on the phosphorylation of IRF3 and IRF7. HEK293 cells were transfected with IRF3 and the individual kinase constructs, then either infected with SeV at a MOI of 5, 4 h after transfection, or left uninfected. Sixteen or 24 h after infection cells were collected and phosphorylated IRF3 and other indicated proteins were detected. Here, we confirmed that TBK1 does, indeed, enhance IRF3 phosphorylation 16 h after transfection, also without SeV infection (Figure 9A). At 24 h after SeV infection FYN and SRPK1 kinases phosphorylated IRF3 (Figure 9A). STK24 may also have a minor IRF3 phosphorylation-enhancing effect. Next, HEK293 cells were transfected with ΔRIG-I, IRF3 and the kinases, and, 24 h later, cells were analysed for IRF3 phosphorylation. ΔRIG-I induced the phosphorylation of transfected IRF3 and this activation was further enhanced by FYN and SRPK1 (Figure 9B). In this experiment other kinases had no detectable activating effect on IRF3 phosphorylation at 24 h post transfection (Figure 9A). Analogously, we tested whether the kinases have any positive effects on the activation of IRF7. MyD88 and IRF7 were overexpressed, together with the kinases, and, 24 h later, the cells were collected. The results showed that the phosphorylation of IRF7 was enhanced the most by FYN, but also that SRPK1 and TBK1 could enhance it (Figure 9C).
The role of FYN, STK24, ACVR1, SRPK1 and TBK1 kinases on SeV infection or RIG-I- or MyD88-stimulated IRF activation. HEK293 cells grown on 24-well culture plates were transfected with different kinase expression plasmids (500 ng/well) (A–C) together with IRF3 (100 ng/well) (A), ΔRIG-I (50 ng/well), IRF3 (100 ng/well) (B), MyD88 (100 ng/well) and IRF7 (50 ng/well) (C). At 4 h after transfection cells were left uninfected or were infected with SeV (MOI 5) for 16 or 24 h (A). Cells were collected, proteins were separated on 12% SDS-PAGE (30 µg/lane) and electrophoretically transferred onto Immobilon P membranes followed by Western blot analysis with respective Abs against the indicated signalling components. Anti-actin staining functions as a loading control. For panels B and C cells were collected for Western blot analysis at 24 h after transfection and processed as described in panel A. Anti-actin staining functions as a loading control for both panels. Results are representative of five independent experiments. Control cells (cells) represent uninfected/untransfected (in panel A) and untransfected (panels A–C) cells.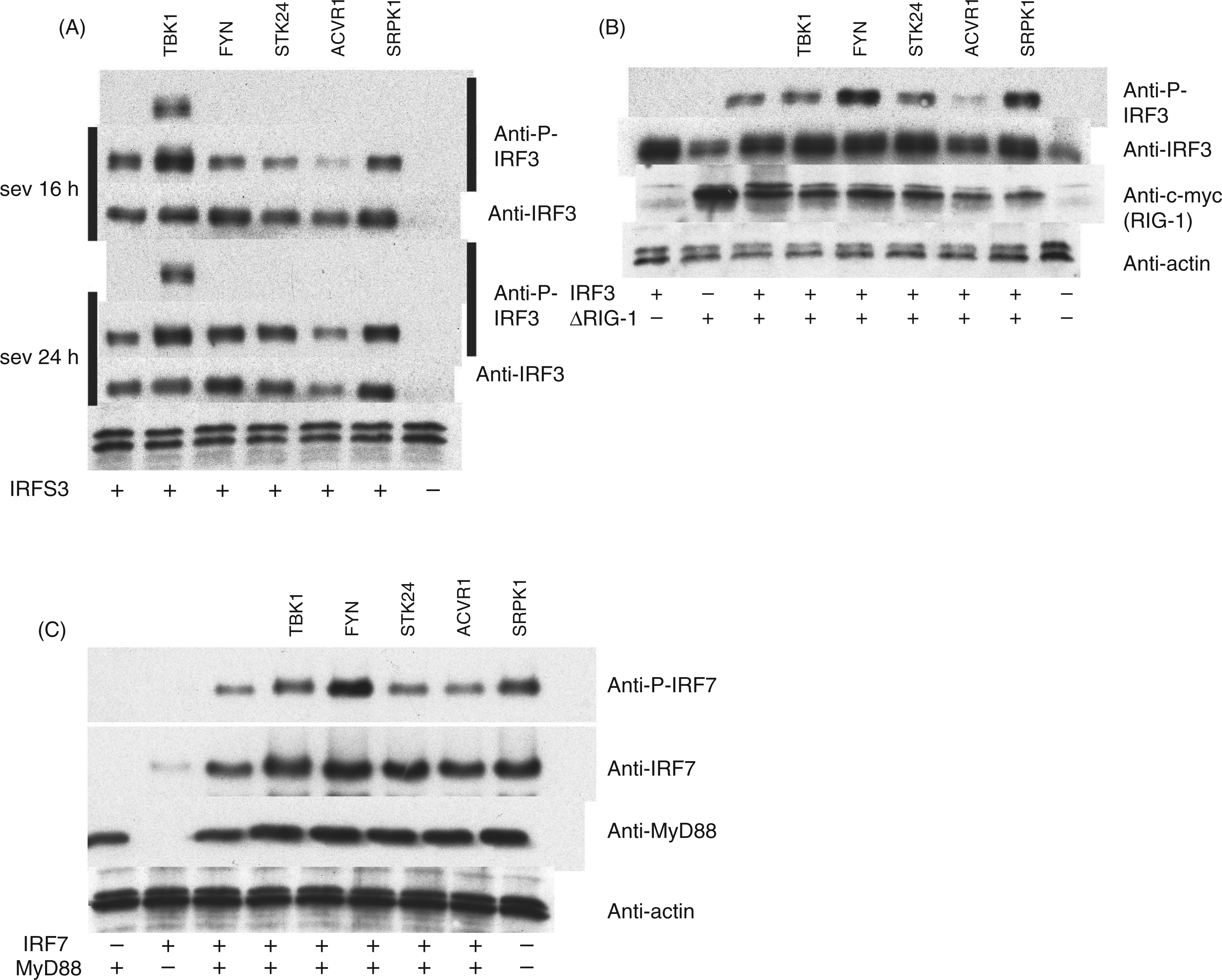
Discussion
In the present study we have identified novel kinases that contribute to enhanced expression of antiviral cytokine genes during virus infection by screening a human kinase collection, termed the kinome. The kinome collection comprises more than 93% of all human kinases in expression-ready cDNA form. 25 We found nearly 50 kinases not previously associated with activation of innate immunity that enhanced IFN-λ1 promoter activation at least twofold during SeV infection. There is a possibility that some kinases may have remained undiscovered, as high throughput methods often suffer from significant variation. However, we consider our screening method reliable as TBK1 and IKKε also gave a positive signal in the absence of virus infection. These kinases are known to auto-activate themselves and stimulate target gene expression without virus infection or upstream activating factors, such as TRIF or ΔRIG-I. 42 As another level of control, in the second kinome screen under SeV infection, classical IKK complex kinases IKKα/β/γ, continued to remain neutral, as well as in our later confirmatory assay. It was of interest that a number of kinases that seemed to repress IFN-λ1 gene expression were also discovered. The mechanism of action of at least some of these molecules awaits further analyses.
After several intermediary analyses, followed by the screening, we ended up with four kinases that were the most potent in activating the IFN-λ1 promoter under SeV infection. FYN, STK24, ACVR1 and SRPK1 were found to enhance IFN-λ1 and IFN-β promoter activation via the RIG-I pathway, and the seen effects of these kinases were roughly at the same level as with the positive control kinase, TBK1. With SeV infection, or cells overexpressing MyD88 and IRF7, TBK1 showed a stronger stimulatory effect than the other four kinases. It also appeared that STK24 and ACVR1 kinases were less active than FYN and SRPK1 as they enhanced IFN-λ1/β promoter activation when their expression was combined with more strongly activating co-stimuli, such as SeV infection or high ΔRIG-I plasmid amounts. The same observation was made with the 5xISRE promoter–reporter construct. Activation of IFN-λ1 promoter via the RIG-I pathway by FYN, SRPK1 and TBK1 was dependent on the promoter ISRE- and NF-κB sites, as evidenced by the IFN-λ1 promoter mutant experiments. This activation also led to endogenous IFN-λ1 and CXCL10 protein production, with FYN and TBK1 being the best stimulators of cytokine production. Indeed, we were also able to show that IRF3 and IRF7 phosphorylation is enhanced by these kinases, stimulating IFN and cytokine production via the RIG-I or MyD88 pathway, thus providing a mechanistic explanation for the functions of these kinases in antiviral RLR and TLR signalling pathways.
There are emerging data on the functions of FYN, STK24, ACVR1 and SRPK1 kinases. STK24 (also Mst3) has been suggested to inhibit cell migration 43 and oxidative stress-induced trophoblast apoptosis in the placenta. 44 ACVR1 mutations have been linked to fibrodysplasia ossificans, ossification of soft tissues45,46 and polycystic ovary syndrome. 47 SRPK1 belongs to SR-protein kinases, which have important functions in mRNA splicing,48,49 and has thus been suggested to be one target for inhibition in epithelial cancers as its knockdown disrupts multiple mRNA modification pathways. 50 This function is also utilised by human papilloma viruses. 51 Among the four kinases analysed, FYN is the only one that has so far been linked with the immune system. FYN belongs to the non-receptor SFK tyrosine kinase family and it has important functions in T cell biology. 52 These functions are important, for example in HIV infection. Patients with asymptomatic HIV infection have elevated cellular levels of FYN, and patients rapidly developing AIDS have impaired function of FYN. 53 FYN is also known to be important in B cell biology, with its family member kinase Lyn. 54 FYN has also been shown to interact with hepatitis C virus (HCV) NS5A protein 55 and somehow enhance HCV replication. 56 Recently, two SFK family kinases, SRC and HCK (hematopoietic cell kinase) were linked to RIG-I and TLR signalling. SRC has been shown to interact with TRAF3, a critical downstream signalling component of the RIG-I pathway, and facilitate IFN-β production. 57 SRC kinase is also involved in TLR3-regulated cell migration and interferes with cellular functions upon viral dsRNA recognition. 58 HCK was reported to be part of LPS-triggered TLR4-mediated TNF-α and IL-6 cytokine production by activating AP-1 transcription factors. 59
In the present study we have provided important information on how the expression of type III IFN genes are regulated. Very recent data have revealed that IFN-λs are the major IFNs produced during several virus infections in epithelial tissues and they seem to have been evolved to interfere with pathogens invading the epithelial surfaces. 60 In nasal epithelia IFN-λ1 is the dominant IFN subtype produced during respiratory syncytial, measles or mumps virus infections. 61 Several studies have highlighted the importance of type III IFNs against respiratory tract infections caused by severe acute respiratory syndrome (SARS)-coronavirus,62,63 influenza A or B,62,64,65 RSV or human metapneumovirus. 62 There are also reports emphasising the importance of IFN-λs against intestinal SARS 62 or rotavirus infections. 66 Recently, skin has been shown to be the source for IFN-λ, as in keratinocytes and melanocytes the main IFN types produced are type III IFNs.67,68
Conclusions
We have identified previously unrecognised kinases regulating innate immunity, including IFN gene expression. We found that FYN, STK24, ACVR1 and SRPK1 enhance IFN-λ1 promoter activation and protein production. In particular, FYN and SRPK1 were found to have strong positive effects, and they were found to phosphorylate IRF3 and IRF7 transcription factors, offering a mechanistic explanation to the phenomena seen. These kinases act somewhere in between the adaptor molecules MyD88 or IPS-1, and the respective transcription factors, yet the precise interacting counterparts in innate immune pathways are to be determined. We conclude that, from the 568 human kinases screened, the Src kinase FYN and the SR-kinase SRPK1, in particular, are possible new mediators of the innate antiviral signalling from pathogen-associated molecular pattern (PAMP) receptors. The present study gives us more detailed understanding of the fine tuning of the host antiviral pathways, which may provide us with better means to fight against viral infections.
Footnotes
Funding
This work was supported by the Medical Research Council of the Academy of Finland (project contracts 252252 and 256159) and the Sigrid Juselius Foundation.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.