
Abstract
TLR2 has a prominent role in host defense against a wide variety of pathogens. Stimulation of TLR2 triggers MyD88-dependent signaling to induce NF-κB translocation, and activates a Rac1-PI 3-kinase dependent pathway that leads to transactivation of NF-κB through phosphorylation of the P65 NF-κB subunit. This transactivation pathway involves tyrosine phosphorylations. The role of the tyrosine kinases in TLR signaling is controversial, with discrepancies between studies using only chemical inhibitors and knockout mice. Here, we show the involvement of the tyrosine-kinase Lyn in TLR2-dependent activation of NF-κB in human cellular models, by using complementary inhibition strategies. Stimulation of TLR2 induces the formation of an activation cluster involving TLR2, CD14, PI 3-kinase and Lyn, and leads to the activation of AKT. Lyn-dependent phosphorylation of the p110 catalytic subunit of PI 3-kinase is essential to the control of PI 3-kinase biological activity upstream of AKT and thereby to the transactivation of NF-κB. Thus, Lyn kinase activity is crucial in TLR2-mediated activation of the innate immune response in human mononuclear cells.
Introduction
TLR2 is an essential innate immune receptor that senses bacterial lipoproteins/lipopeptides inducing host innate immune responses. 1 Such diversity has been attributed to its unique ability to heterodimerize with TLRs 1 and 6 in order to bind triacyl and diacyl lipoproteins, respectively.2,3 TLR2 subsequently initiates the inflammatory signaling cascade that contributes to host defense against infection. 4 Initiation of this cascade requires the constitution of a TLR2-specific activation cluster in membrane microdomains or lipid rafts in order to induce NF-κB transcriptional activity.5,6 Transcriptional activity of NF-κB relies on the engagement of a canonical pathway that induces translocation of the P50 and P65 subunits of NF-κB. 7 In addition, activation of NF-κB also requires phosphorylation of its P65 subunit through a pathway dependent on the Rho-GTPase Rac1 and phosphoinositide-3 kinase (PI 3-kinase).8–11 PI 3-kinase is composed of a p85α regulatory subunit and a p110 catalytic subunit. The p85α regulatory subunit is characterized by a p110- binding domain flanked by two Src-homology 2 (SH2) domains. The p85 SH2 domains control activation of PI 3-kinase via interactions with tyrosine-phosphorylated receptor/adaptor proteins. 12
A significant body of evidence indicates that tyrosine phosphorylation plays an important role in the activation of TLR2-dependent signaling pathways. Stimulation of TLR2 leads to tyrosine phosphorylation of its TIR domain that is essential to the activation of the Rac1-PI 3-kinase pathway. 8 In addition, PI 3-kinase also receives regulatory signals from tyrosine kinases. 12 Sensing of TLR2 agonists also activates Bruton tyrosine kinase, which then contributes to the transactivation of NF-κB through a functional interaction with Mal and PI 3-kinase.13,14 However, the precise sequence of tyrosine phosphorylation events following recruitment of TLR2 in the lipid rafts is still poorly understood and subsequent transactivation of NF-κB may involve additional protein kinases.
Several lines of arguments indicate that Lyn, a microdomain-associated Src kinase, may participate in the regulation of TLR signaling. First, Lyn is required for TLR4-dependent phosphorylation of MD-2 adaptor protein in the human embryonic kidney (HEK)-293 cell line. 15 Second, Lyn plays a pivotal role in TLR2-dependent internalization of Pseudomonas aeruginosa by alveolar epithelial cells. 16 Finally, Lyn also participates in signal transduction downstream TLRs, and either activates NF-κB in human mononuclear cells, 17 or represses its transcriptional activity in murine macrophages. 18 In adaptive immunity, Lyn is noted for its ability to regulate BCR signaling pathways positively or negatively through modulation of PI 3-kinase activity. 19 Lyn also plays negative and positive regulatory roles in dendritic cell generation and maturation.20,21 Whether Lyn functions to promote or inhibit immune cell activation depends on the cell type, the differentiation stage of immune cells, the type of receptor agonists, as well as the activation status of downstream transduction molecules including kinases. Altogether, these data indicate that the consequences of Lyn activity are context dependent.
To further investigate the role of Lyn in TLR2-dependent signaling, we combined pharmacologic and RNA interference approaches to inhibit Lyn activity after stimulation of TLR2 with acylated lipoproteins. We report that Lyn positively contributes to the regulation of NF-κB transactivation through the phosphorylation of the p110 subunit of PI 3-kinase and the control of its biological activity upstream of AKT in human cellular models and monocytes.
Materials and methods
Reagents and cell culture
We used the monocytic cell line THP-1 stably transfected with human CD14 (THP1-CD14), and 293 (HEK) cells stably transfected with Flag-TLR2 (HEK 293–TLR2) or parental HEK 293 as described.
8
Of note, HEK 293 cells were previously reported to express endogenous Lyn tyrosine kinase.
22
HEK 293–TLR2 cells were maintained in low Glc DMEM supplemented with 10% heat-inactivated FBS, HEPES (10 mM),
PBMC were isolated from healthy volunteers using whole-blood Ficoll-Plaque PLUS (GE Healthcare, Little Chalfont, UK). Monocytes were sorted by means of an immunomagnetic procedure for CD14+ cell isolation (MACS; Miltenyi Biotec, Bergisch-Gladbach, Germany). Cells labeled with MACS MicroBeads were positively selected using MS, Columns (MACS; Miltenyi Biotec). Purity of the positive fraction was generally >90%, as confirmed by CD14 staining analyzed by flow cytometry (see Figure 4D). CD14+ human cells were seeded into six-well culture dishes for stimulation studies with synthetic lipopeptides.
Pam3-Cys-KKKK (Pam3; EMC Microcollections, Tübingen, Germany) mimics bacterial triacyl component after ligation to the heterodimer TLR1/2. Pam2-Cys-FEPPPATTT (Pam2; EMC Microcollections) mimics bacterial diacyl lipoprotein after ligation to the heterodimer TLR2/6. PP2, a src kinase inhibitor, was from Calbiochem (La Jolla, CA, USA). Protein G sepharose was from Sigma (St. Louis, MO, USA). Polyclonal anti-p85, anti-Rac and anti-Lyn were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Polyclonal Abs to phospho-AKT (Ser473), AKT, phospho-P65 (Ser536), P65, phospho-P38, phospho-ERK, phospho-SAP-JNK, ERK, SAP-JNK, IκB and mAb P38 were from Cell Signaling (Danvers, MA, USA). mAb to Flag was from Sigma. mAbs against CD14 and aminoacid 800-1139 of human p110 isoforms were from Santa Cruz Biotechnology. Anti-phosphotyrosine 4G10 and anti-Rac were obtained from Upstate (Charlottesville, VA, USA).
Transfection
NF-κB responsive luciferase reporter (5 × NF-κB-Luc, 40 ng) and β-galactosidase (40 ng) plasmids (Promega, Madison, WI, USA) were used for measuring luciferase activity. Luciferase assays were done using the Luciferase Assay System (Promega) according to the manufacturer’s instructions. Luciferase activity was normalized to β-galactosidase to standardize transfection efficiency. Lyn pcDNA constructs [wild type (WT), DN LynK275D] and GFP-Akt-PH31 were used as previously described. 23 cDNAs for Rac1 WT was subcloned into pRK5 containing an NH-terminal Myc tag. These constructs were transiently transfected using LipofectAMINE PLUS (Invitrogen, Carlsbad, CA, USA). Lyn siRNA SMARTpools and negative control siRNA non-targeting pool and GAPDH siRNA were obtained from Dharmacon (Logan, UT, USA). Transient transfection of siRNA constructs was done using LipofectAMINE 2000 (Invitrogen) according to the manufacturer's protocol.
Western blotting assay
Cells (1 × 106) stimulated or not with Pam2 or Pam3 were washed three times in PBS and then lysed in a buffer containing 25 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 5 mM MgCl2, 10% glycerol, 1 mM sodium vanadate, 1 mM dithiotreitol (DTT), protease inhibitors from Roche (Meylan, France), and 1% Nonidet P-40 (NP40). Protein extracts were incubated for 5 min at 95℃ in Laemmli buffer [65 mM Tris (pH 6.8), 20% glycerol, 5% β-mercaptoethanol, 0.01% bromophenol blue and 2% SDS]. Proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane (BioRad, Hercules, CA, USA). Membranes were incubated in TBS-Tween (10 mM Tris pH 7.5, 150 mM NaCl, 0.1% Tween 20) and 5% low-fat milk for 2 h to saturate non-specific sites, and then in specific Ab overnight at 4℃. The membranes were then washed with TBS-tween and incubated with secondary Ab for 1 h at room temperature (∼ 21℃). In some experiments, mouse TrueBlot (1:1000; Cliniscience Laboratories, Montrouge, France) was used as the second Ab for enhanced chemiluminescence to avoid immunodetection of immunoglobulins. Proteins were revealed with a chemoluminescent kit according to the manufacturer’s instructions (Amersham Pharmacia Biotech, Little Chalfont, UK).
Immunoprecipitation and PBD pulldown assays
Cells were lysed in 25 mM Tris-HCl (pH 7.5), 1 mM EDTA, 5 mM MgCl2, 0.1 mM DTT, 0.1 mM EGTA, 100 mM NaCl, 10% glycerol, 1% NP-40, 1 mM orthovanadate, phosphatase inhibitor cocktails I and II (1/100) and protease inhibitor cocktail 1/100 (Sigma). Protein extracts were quantified with BCA colorimetric assay (Pierce, New-York, NY, USA) using SMARTpec3000 spectrometer (Biorad). Cell lysates (600 µg) obtained from HEK 293–TLR2 before and after stimulation were incubated at 4℃ either with primary Ab for immunoprecipitation, or with 10 µg of recombinant GST-PBD beads for PBD assay as described. 24
Fluorescence analysis and video imaging
HEK 293–TLR2 cells (12.5 × 103/ml) were plated in their culture medium on glass coverslips mounted on 25-mm Petri dishes and were incubated at 37℃ for 2 d. Cells were starved for 20 h before the experiment in basal culture medium. For Lyn inhibition experiments, HEK 293–TLR2 cells were either transiently transfected with dominant negative LynK275D (LynDK) plasmid construct, or preincubated at 37℃ for 60 min with PP2 (25 µM) before TLR2 agonist Pam3 was added. Cell video imaging was obtained with Zeiss Axiovert 100 M inverted microscope equipped with the Metafluor imaging system (Universal Imaging, Bedford Hills, NY, USA). Transmitted light images were obtained at 37℃, every 15 s in turn with images of GFP. Acquisition started 2 min before the addition of Pam3 and lasted 30 additional min. Superimposed movies were made with QuickTime software.
Confocal microscopy
THP1-CD14 cells (1 × 106) were seeded on six-well dishes and starved for 20 h before stimulation with Pam3. Cells were then washed by PBS, fixed with 4% paraformaldehyde and then washed with glycin 0.1 M. Cells were then permeabilized with PBS-BSA 1% containing 0.05% saponin 20 min and labeled with anti-CD14 and anti-Lyn Abs for 1 h at room temperature. Cells were subsequently incubated with anti-mouse Alexa Fluor 488 and TexasRed anti rabbit secondary Abs, for 30 min. Cells were washed with PBS and mounted on slides. All images were obtained using a confocal Leica DMIRE2 microscope and analyzed using ImageJ 1.40 software.
Measurement of cytokine production
THP1 and HEK 293–TLR2 (1 × 106/ml) were stimulated by Pam3 in the absence or presence of PP2. Concentrations of TNF-α and IL-6 in culture supernatants were determined by ELISA according to the manufacturer’s protocol (R&D Systems, Minneapolis, MN, USA).
Results
Lyn contributes to TLR2-dependent NF-κB activation
Stimulation of TLR2 initiates canonical MyD88-dependent signaling and activation of a Rac1–PI 3-kinase pathway that are both required to activate NF-κB.8,9 To test whether Lyn contributes to this activation process, we monitored the activity of NF-κB in HEK 293–TLR2 cells exposed to TLR2 agonists Pam2 or Pam3. Whereas stimulation with both agonists for 6 h induced a significant increase in luciferase activity (Figure 1A), treatment with the Src-kinase inhibitor PP2 resulted in a dose-dependent decrease in NF-κB activity. In addition, expression of LynDK led to a strong inhibition of Pam2- and Pam3-induced NF-κB activity (Figure 1B). To confirm the importance of Lyn in NF-κB activation, HEK 293–TLR2 cells were transfected with siRNA targeting Lyn expression. As Lyn is part of the Src-kinase family, we first tested the specificity of the siRNA used by Western blot with Src Abs as negative control. As shown in Figure 1C, only Lyn was silenced. Silencing Lyn significantly decreased luciferase activity after stimulation with Pam3 (Figure 1D) or Pam2 (data not shown).
Lyn modulates the activity of NF-κB after TLR2 stimulation. HEK 293–TLR2 cells were co-transfected with NF-κB responsive luciferase reporter (40 ng) and β-galactosidase (40 ng) plasmids and then stimulated for 6 h with Pam2 or Pam3 (100 ng/ml). In vitro NF-κB-driven luciferase activity was measured (A) after inhibition of Lyn using various concentrations of PP2, or (B) after co-transfection with various concentrations of LynDK. Control conditions (−) correspond to DMSO or empty pcDNA vector (500 ng). (C) Endogenous or exogenous expression of Lyn was controlled by Western blot. The specificity of the Lyn siRNA was controlled by Western blot with Src Abs as negative control. Control condition (−) corresponds to GAPDH siRNA (32 ng/ml). β-Actin was used as loading control. (D) HEK 293–TLR2 cells were co-transfected with reporter, β-galactosidase plasmids and various concentrations of Lyn siRNA before stimulation with Pam3 (100 ng/ml; 30 min). Control condition (−) corresponds to GAPDH siRNA (32 ng/ml). In all experiments, NF-κB activity was expressed as arbitrary units corresponding to ‘fold activation’. NF-κB luciferase activity was rationalized upon β-galactosidase expression. Expression of Lyn (endogenous and overexpressed) was tested in HEK 293–TLR2 cells by Western blot for each condition. Data are representative of four independent experiments and all data are mean ± SD. *P < 0.05 compared with control stimulated by Pam2 or Pam3, unpaired t-test or ANOVA.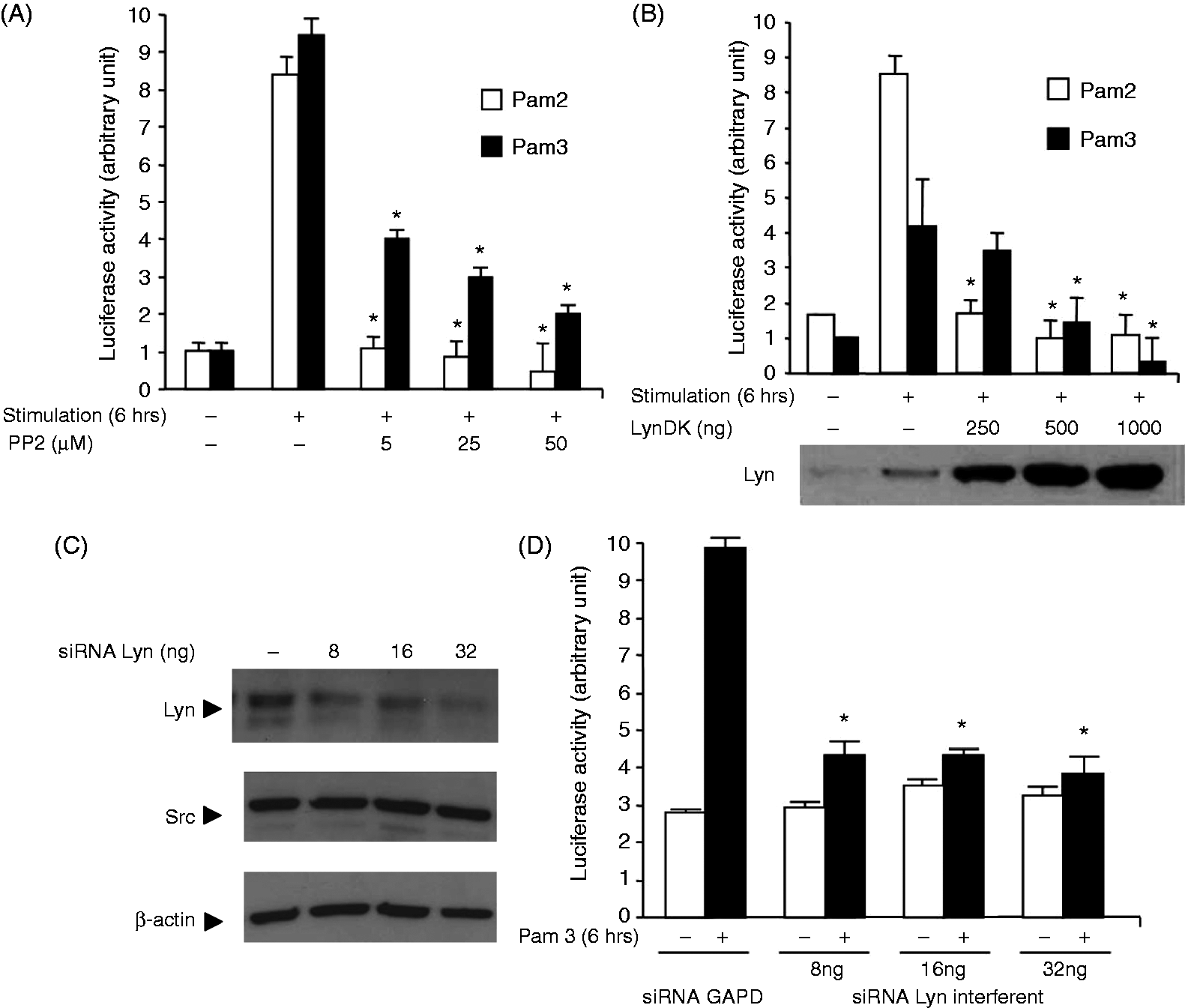
NF-κB activation following stimulation of TLR2 results in the secretion of inflammatory cytokines, such as TNF-α or IL-6.25,26 To test whether inhibition of Lyn modified the production of TNF-α, HEK 293–TLR2 cells were stimulated with Pam2 or Pam3 in the presence of PP2 or vehicle (DMSO) and concentrations of cytokines in the culture media were quantified. As expected, HEK 293–TLR2 cells released significant amounts of TNF-α after incubation for 6 h with either Pam3 (Figure 2A) or Pam2 (data not shown). Pretreatment of HEK 293–TLR2 cells with various concentrations of PP2 resulted in a dose-dependent decrease of TNF-α concentrations in the culture media. Similarly, inhibition of Lyn activity after transfection of HEK 293–TLR2 with LynDK decreased the amounts of TNF-α released in the media compared with control cells (Figure 2B). We also tested the ability of PP2 to modulate the release of TNF-α and IL-6 after stimulation of THP1 cells. Whereas stimulation with Pam3 or Pam2 (data not shown) increased both TNF-α (Figure 2C) and IL-6 (Figure 2D) release, treatment with PP2 reduced concentrations of both pro-inflammatory cytokines in a dose-dependent manner.
Lyn modulates TLR2-dependent release of pro-inflammatory cytokines. Concentrations of pro-inflammatory cytokines were measured by ELISA in supernatants of cell cultures following a 6-h stimulation with Pam3 (100 ng/ml). HEK 293–TLR2 cells were incubated with (A) various concentrations of PP2 or transfected with (B) LynDK, and were stimulated with Pam3 (100 ng/ml) for 6 h. Control conditions (−) correspond to DMSO or pcDNA empty vector (500 ng/ml). THP1–CD14 monocytic cell line was incubated with various concentrations of PP2 or DMSO and then stimulated 24 h with Pam3 (100 ng/ml) and (C) TNF-α or (D) IL-6 (D) were measured in the supernatant. Results are expressed as quantities of IL-6 and TNF-α (pg/ml) detected in the culture media and derived from three independent experiments; all data are mean ± SD. *P < 0.05 compared with control stimulated by Pam2 or Pam3, unpaired t-test or ANOVA.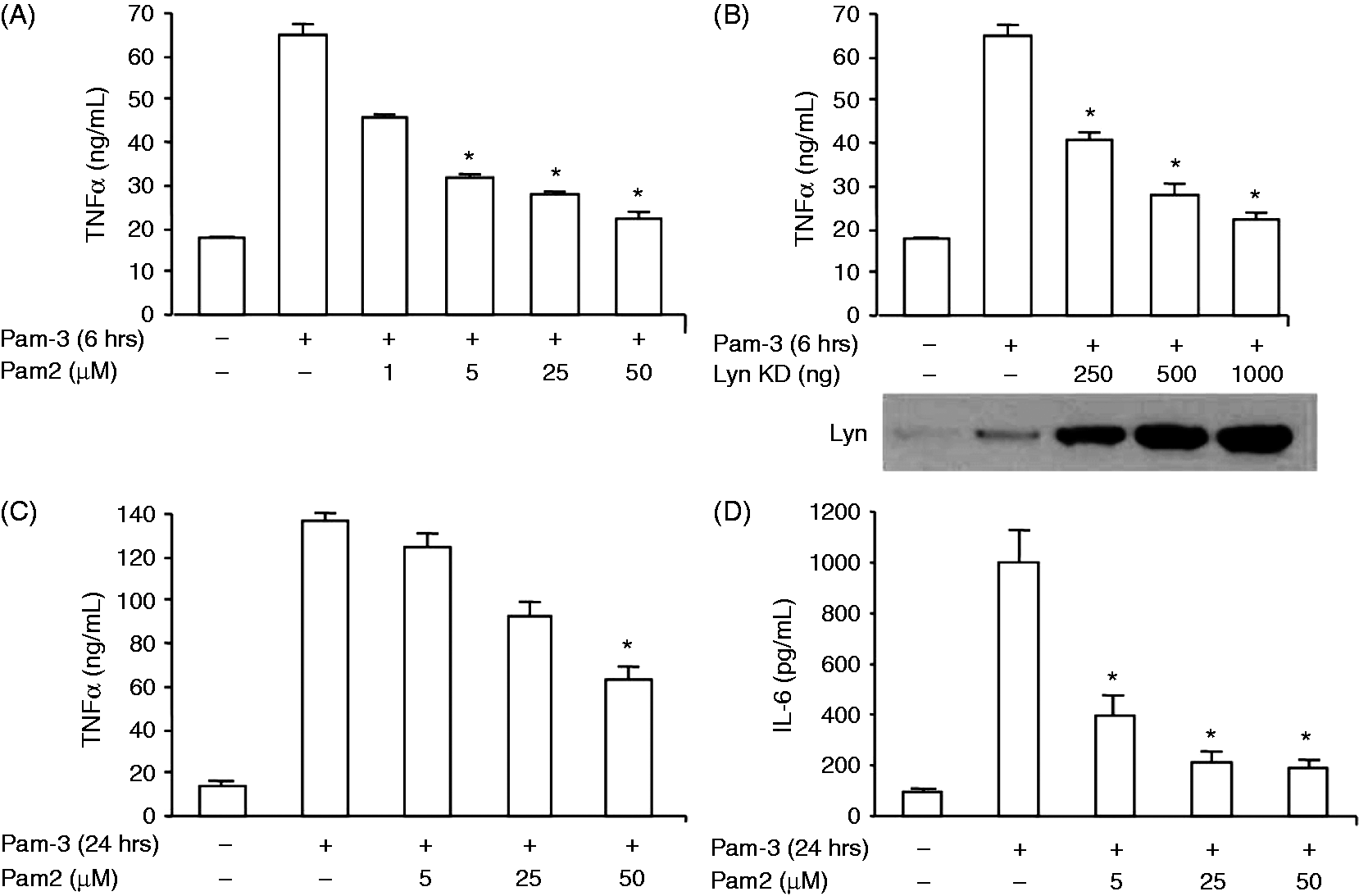
These results suggest that Lyn contributes to TLR2-dependent activation of NF-κB and subsequent release of pro-inflammatory cytokines, irrespective of the TLR2 heterodimers engaged.
Lyn is required for transactivation of NF-κB
MyD88-dependent canonical pathway downstream TLR2 leads to nuclear translocation of NF-κB subunits, after IκB degradation.
7
To test whether Lyn participates in this pathway, we measured the degradation of IκB in HEK 293–TLR2 (Figure 3A, upper panel) or THP1 cells (Figure 3A lower panel) stimulated by Pam3 by immunoblot. In both cell types, IκB decreased following Pam3 stimulation. Neither exposure to PP2 nor transfection of Lyn DK prevented the decrease in IκB levels detected by immunoblot, suggesting that inhibition of Lyn does not impair nuclear translocation of NF-κB subunits. Moreover, stimulation of TLR2 also initiates a signaling pathway leading to activation of MAP kinases. To test whether Lyn activity is necessary to TLR2-dependent activation of MAP kinases, we stimulated HEK 293–TLR2 cells with Pam3 and assessed expression of activated forms of ERK1/2, JNK and P38 by immunoblotting. Stimulation with Pam3-induced phosphorylation of ERK1/2, SAP-JNK and P38 (Supplementary Figure 1). Inhibition of Lyn by PP2 or LynDK did not modify MAP kinases activation, suggesting that Lyn does not contribute to the activation of MAP kinases in TLR2-stimulated HEK 293–TLR2 cells.
Lyn is essential to the transactivation of the p65 subunit of NF-κB. HEK 293–TLR2 cells were incubated with PP2 (25 µM) or transfected with LynDK expression plasmid (500 ng/ml). (A, upper panel) Cells were then stimulated with Pam3 (100 ng/ml), lysed and subjected to SDS-PAGE. Degradation of I-κB after stimulation with Pam3 was quantified by Western blot. (A, lower panel) β-Actin was used as loading control. THP1–CD14 cells were stimulated with Pam3 (100 ng/ml) in the presence of PP2 (25 µM), lysed and IκB degradation observed by Western blot. (B, upper panel) β-Actin was used as loading control. Phosphorylation of p65 (pP65) was quantified by Western blot on cells treated with a range of PP2 or various concentrations of LynDK and stimulated with Pam3 (100 ng/ml, 30 min). (B, bottom panel) Control conditions correspond to DMSO and empty pcDNA vector (500 ng/ml). THP1–CD14 cells were incubated with PP2 (25 µM) for 1 h, stimulated with Pam3 (100 ng/ml, lysed, subjected to SDS-PAGE and pP65 was quantified by Western blot. Control conditions correspond to DMSO. (C) HEK 293–TLR2 cells were stimulated with Pam3 (100 ng/ml, 30 min) following transfection with various concentrations of Lyn-targeting siRNA or non-targeting scrambled siRNA. Expression of IκB and pP65 was quantified by Western blot analysis; total P65 was used for loading control, as indicated. Results are representative of three independent experiments.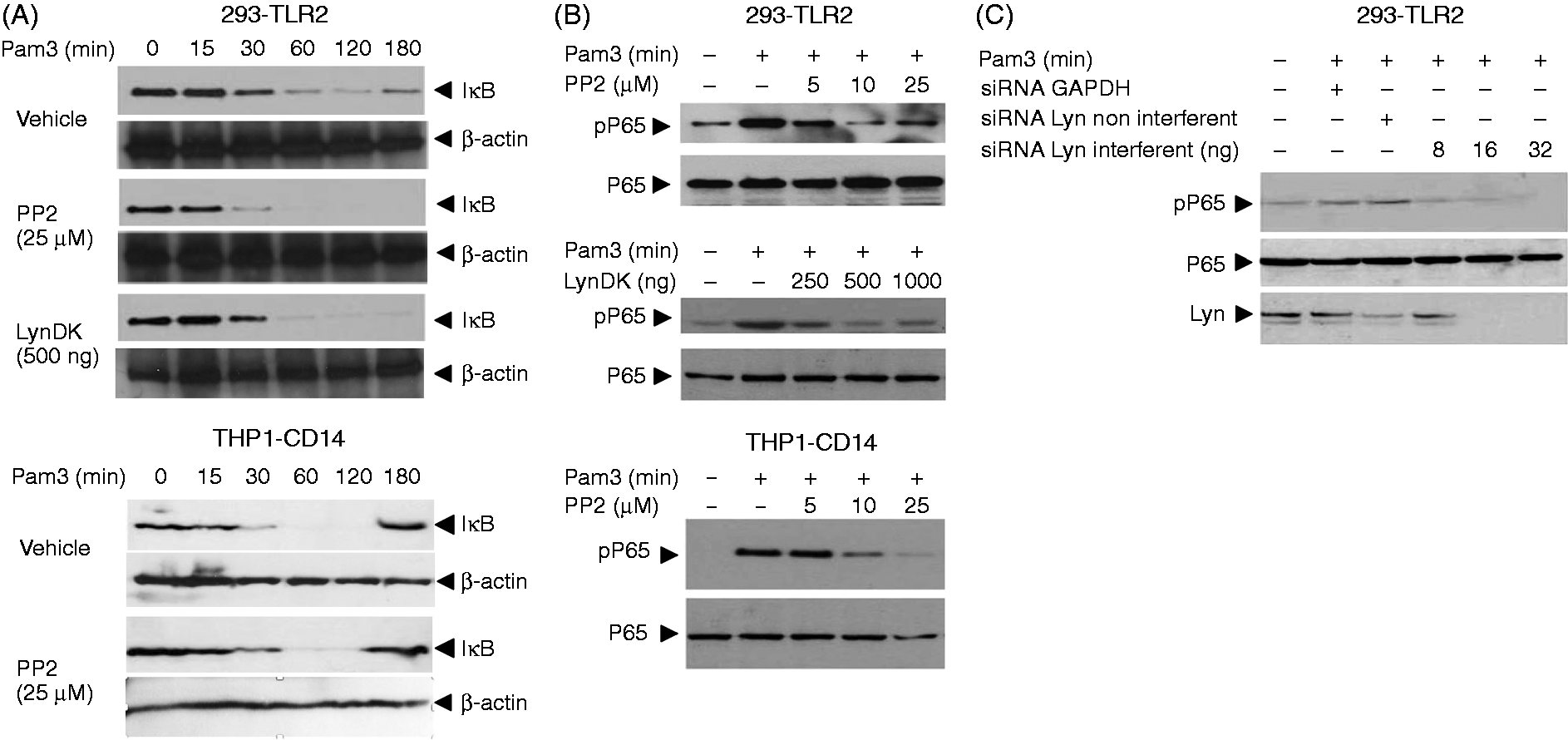
Activation of NF-κB via TLR2 also requires a Rac-PI 3-kinase pathway. To test whether Lyn contributes to the transactivation of NF-κB, we assessed phosphorylation of the P65 subunit in HEK 293–TLR2 cells. Engagement of TLR2 with Pam3 induced the phosphorylation of P65 30 min after cell stimulation (Figure 3B, upper panel). In contrast, exposure of HEK 293–TLR2 cells to PP2 prevented phosphorylation of P65 in a dose-dependent manner. Similarly, LynDK expression inhibited P65 phosphorylation (Figure 3B, lower panel). Similar results were found in THP1 cells incubated with PP2 (Figure 3B, lower panel). Finally, nearly undetectable levels of phospho-P65 (pP65) were observed after Pam3 exposure in HEK 293–TLR2 transfected with Lyn-specific siRNA (Figure 3C).
Altogether, these results suggest that Lyn is necessary for NF-κB transactivation through phosphorylation of P65 but is dispensable for NF-κB nuclear translocation.
Lyn controls PI 3-kinase activity upstream of AKT
Phosphorylation of the NF-κB P65 subunit after TLR2 stimulation depends upon AKT activation.8,9,27 To test whether Lyn plays a role in the TLR2-mediated activation of AKT, protein extracts from HEK 293–TLR2 or THP1 cells stimulated with Pam3 were analyzed by immunoblotting using anti-phospho-AKT Abs. Whereas Pam3 efficiently induced phosphorylation of AKT, treatment with PP2 significantly decreased phosphorylation of AKT on Ser473 in THP1 (Figure 4A) or HEK 293–TLR2 cells (data not shown). Similar results were observed when LynDK was expressed in Pam3-stimulated HEK 293–TLR2 cells (Figure 4B). To strengthen the hypothesis that Lyn activity contributes to phosphorylation of AKT, we silenced Lyn expression using Lyn-specific siRNA and assessed phosphorylation of AKT in HEK 293–TLR2 cells stimulated with Pam3. Lyn inhibition with specific siRNA resulted in a dose-dependent decrease of phosphorylated AKT (Figure 4C). In human peripheral blood monocytes, inhibition of Lyn activity by PP2 totally prevented AKT from serine-phosphorylation, confirming our previous findings (Figure 4D).
Lyn is required for the phosphorylation of AKT. (A) THP1–CD14 cells were submitted to various concentrations of PP2 for 1 h before stimulation with Pam3 (100 ng/ml, 30 min). Cells were lysed and subjected to SDS-PAGE. Phosphorylation of AKT (pAKT) was detected by Western blot. Control condition corresponds to cells incubated with DMSO. (B) HEK 293–TLR2 cells transfected with various concentration of LynDK were stimulated with Pam3 (100 ng/ml, 30 min) and then subjected to Western blot analysis. Control condition corresponds to cells incubated transfected with pcDNA empty vector (1 µg/ml). (C) HEK 293–TLR2 cells were transfected with various concentrations of Lyn-specific siRNA, control GAPDH siRNA (32 ng/ml) or non-targeting scrambled siRNA (32 ng/ml), and were stimulated with Pam3 (100 ng/ml, 30 min). Phosphorylation of AKT was quantified by Western blot, total AKT was used for loading control and the silencing of Lyn expression was confirmed by Western blot. (D) Human blood cell monocyte isolation was confirmed by flow cytometry following CD14 staining. Monocytes were incubated with MPA or DMSO for 2 h and then stimulated with Pam3 (100 ng/ml, 30 min), lysed and subjected to SDS-PAGE. The phosphorylated form of AKT and total AKT were visualized by Western blot. These results are representative of three independent experiments.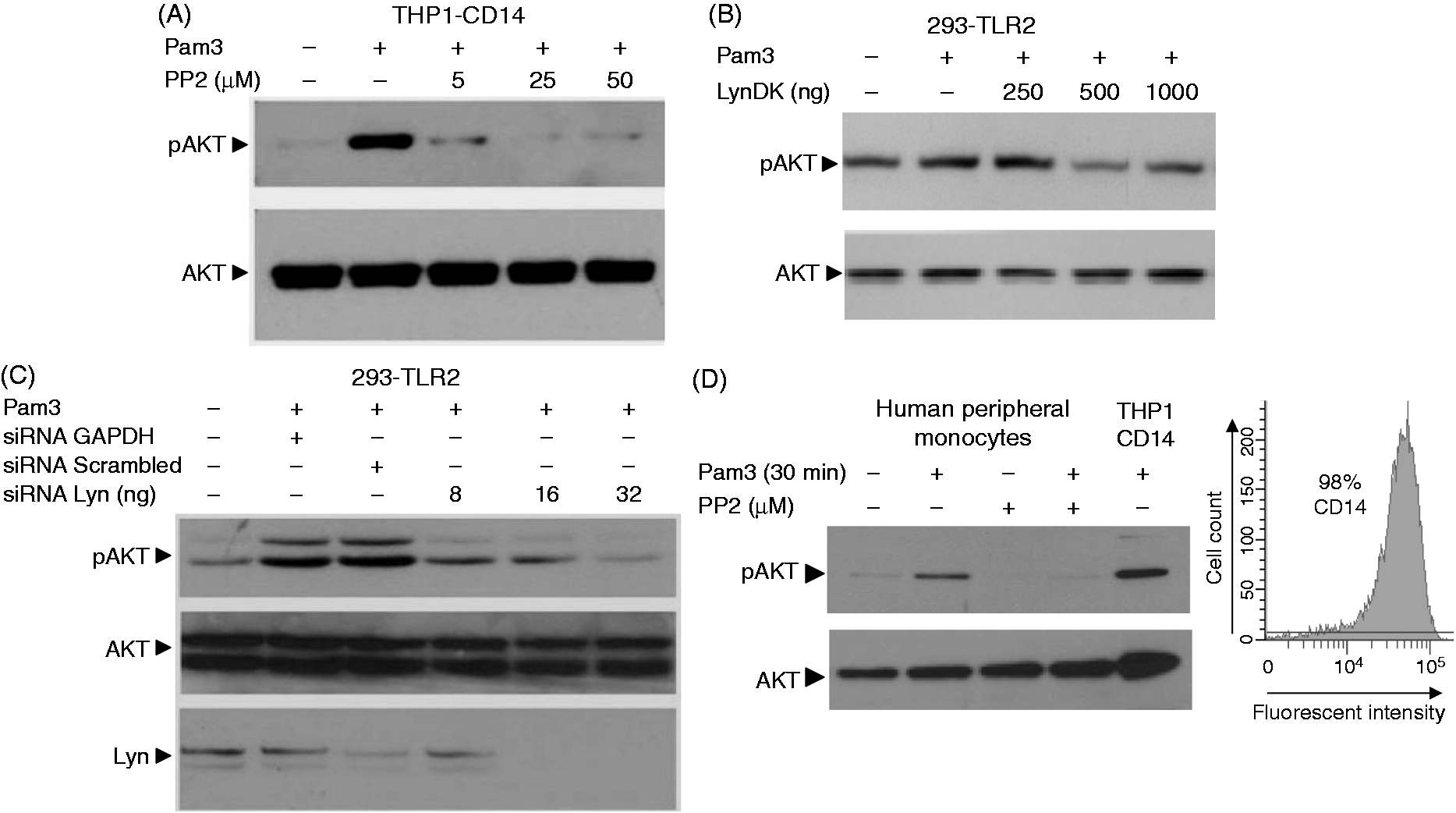
Stimulation of TLR2 has been shown to trigger the formation of a signaling cluster involving TLR2 and CD14 within the lipid rafts,28–31 and the activation of Rac upstream of PI 3-kinase and AKT.
8
To test whether Lyn interacts with TLR2 following its recruitment in the rafts we immunoprecipitated TLR2 in lysates of Pam3-stimulated HEK 293–TLR2. As detected by immunoblotting using Ab to Lyn, we observed an interaction between TLR2 and Lyn at baseline that was increased after stimulation with Pam3 (Figure 5A). Inhibition of Lyn activity with PP2 did not prevent association between Lyn and TLR2. We then investigated whether Lyn contributes to the phosphorylation of TLR2 on its tyrosine residues. As previously reported,
8
stimulation with Pam3 for 5 min induced tyrosine phosphorylation of TLR2. Inhibition of Lyn activity by PP2 or expression of LynDK did not affect phosphorylation of TLR2 after stimulation with Pam3 (Figure 5B). We then tested using pulldown experiments whether Rac was activated after Pam3 stimulation, and observed an early activation of Rac that did not differ in cells that expressed LynDK (Figure 5C) or treated with PP2 (data not shown). Although CD14 and Lyn are both known to be constitutively present in lipid rafts, their interaction has not been established. To investigate whether Lyn and CD14 are recruited to the same signaling complex after TLR2 engagement, we immunoprecipitated Pam3-stimulated THP-1 cells using anti-CD14 Abs. Whereas no constitutive association between Lyn and CD14 was detected, immunoblotting revealed an association between Lyn and CD14 3 min after stimulation with Pam3 (Figure 5D). Of note, the p85α subunit of PI 3-kinase was also recruited to the complex after stimulation. Inhibition of Lyn kinase activity with PP2 did not prevent the association of Lyn and CD14 (data not shown). Immunostaining studies using anti-Lyn and anti-CD14 Abs confirmed that stimulation with Pam3 induced colocalization of Lyn and CD14 after 3 min (Figure 5E).
Lyn kinase activity is not essential to the formation of the TLR2–Lyn–CD14 activation cluster. (A) HEK 293–TLR2 cells were stimulated with Pam3 (100 ng/ml) after pretreatment with PP2 (25 µM, 1 h) or transfection with LynDK (500 ng/ml). TLR2 was immunoprecipitated with an anti-Flag Ab and precipitates were immunoblotted with anti-Lyn Abs. CTL corresponds to DMSO conditions. (B) HEK 293–TLR2 cells were treated with PP2 (25 µM, 1 h) or LynDK (500 ng/ml) and stimulated with Pam3 (100 ng/ml) at different time-points. Lysates were then immunoprecipitated with 4G10 anti-phosphotyrosine Abs and probed with anti-TLR2 Abs. Controls correspond to DMSO or pcDNA empty vector. (C) HEK 293–TLR2 transfected with LynDK (500 ng/ml) or pcDNA empty vector were stimulated with Pam3 (100 ng/ml). Time-dependent activation of Rac was quantified by the presence of Rac–GTP detected by pull-down with Rac–PBD beads. Rac1 expression in whole lysates was used as control loading. (D) THP1-CD14 cells were stimulated with Pam3 (100 ng/ml) and lysates were immunoprecipitated with anti-CD14 mAbs. Immunoprecipitates were subjected to Western blot with anti-Lyn or anti-p85α Abs. (E) THP1–CD14 cells were stimulated with Pam3 and then fixed with 4% paraformaldehyde and washed with glycin 0.1 M. Cells were then permeabilized with PBS–BSA 1% containing 0.05% saponin and incubated with anti-CD14 and anti-Lyn Abs. Cells were subsequently labeled with Alexa Fluor 488 and Texas Red conjugated secondary Abs. Cells were washed with PBS and mounted on slides. All images were obtained using a confocal Leica DMIRE2 microscope and analyzed using ImageJ 1.40. Overlay was represented by yellow staining. These results are representative of three independent experiments.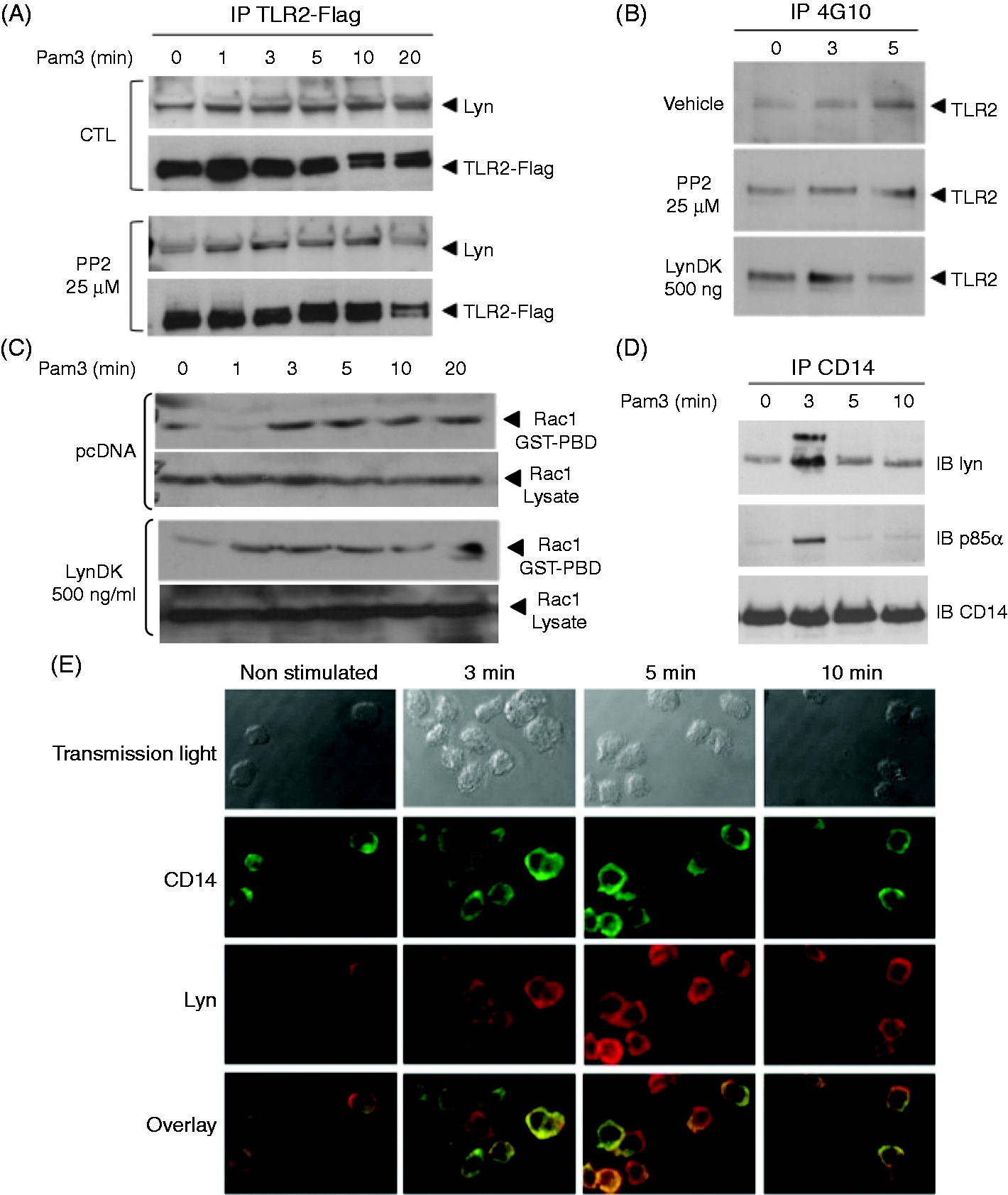
In addition to the recruitment and activation of Rac, activation of PI 3-kinase and formation of phosphatidyl-inositol 3-phosphate is required for membrane recruitment and activation of AKT. As Lyn was found to be essential to AKT activation, we investigated the consequences of Lyn inhibition with PP2 or LynDK on PI 3-kinase activation by dynamic cellular imaging of AKT translocation to the plasma membrane. A construct encoding a fusion protein between the PH domain of AKT and GFP was transfected in HEK 293–TLR2 cells pre-treated with PP2 or expressing Lyn DK. Whereas stimulation with Pam3 in control cells induced after 15 min a significant translocation of the probe from the cytosol to the cell membrane, fluorescence remained within the cytosolic compartment when Lyn was inhibited after treatment with PP2 or when transfecting HEK 293–TLR2 cells with LynDK (Figure 6A and Supplementary Video).
Lyn is essential to the activation of PI 3-kinase through phosphorylation of its p110 subunit. (A; Supplementary movies M1–M4) HEK 293–TLR2 cells were transfected with a fusion protein combining GFP and the PH domain of AKT. Transfected cells were either co-transfected with LynDK (500 ng/ml) or incubated with PP2. Cells were then stimulated with Pam3 (100 ng/ml) and the kinetics of the AKT–PH construct was observed by microvideoscopy using Zeiss Axiovert inverted microscope equipped with the Metafluor imaging system. Presented here are the images corresponding to 15 min of Pam3 stimulation. Controls correspond to cells incubated with DMSO and pcDNA empty vector, or cells incubated with LY294002 (25 µM), a specific inhibitor of PI 3-kinase. (B) HEK 293–TLR2 cells were transfected with pcDNA vector (vehicle) or LynDK (500 ng/ml) and stimulated with Pam3 (100 ng/ml). Lysates were immunoprecipitated with anti-Flag Abs and recruitment of PI 3-kinase to TLR2 was observed by Western blot with anti-p85α Abs. (C) Tyrosine phosphorylation of the p85α subunit was evaluated by Western blot in HEK 293–TLR2 transfected with LynDK (500 ng/ml) and stimulated with Pam3 (100 ng/ml). Anti-phosphotyrosine (4G10 clone) and anti-p85α Abs were used for immunoprecipitation and Western blot. (D) HEK 293–TLR2 were transfected with LynDK (500 ng/ml) and stimulated with Pam3 (100 ng/ml) and lysates were immunoprecipitated with either 4G10 or anti-p110 Abs. Phosphorylation of p110 was then revealed by Western blot with anti-p110 or 4G10 Abs. Controls correspond to cells transfected with pcDNA empty vector. (E) THP1–CD14 cells were incubated with PP2 (25µM) or DMSO, stimulated with Pam3 (100 ng/ml) and lysed. Phosphorylation of p110 catalytic subunit of PI 3-kinase was revealed by Western blot of THP1–CD14 lysates immunoprecipitated with either 4G10 or anti-p110 Abs. Controls correspond to cells treated with DMSO. These results are representative of three independent experiments.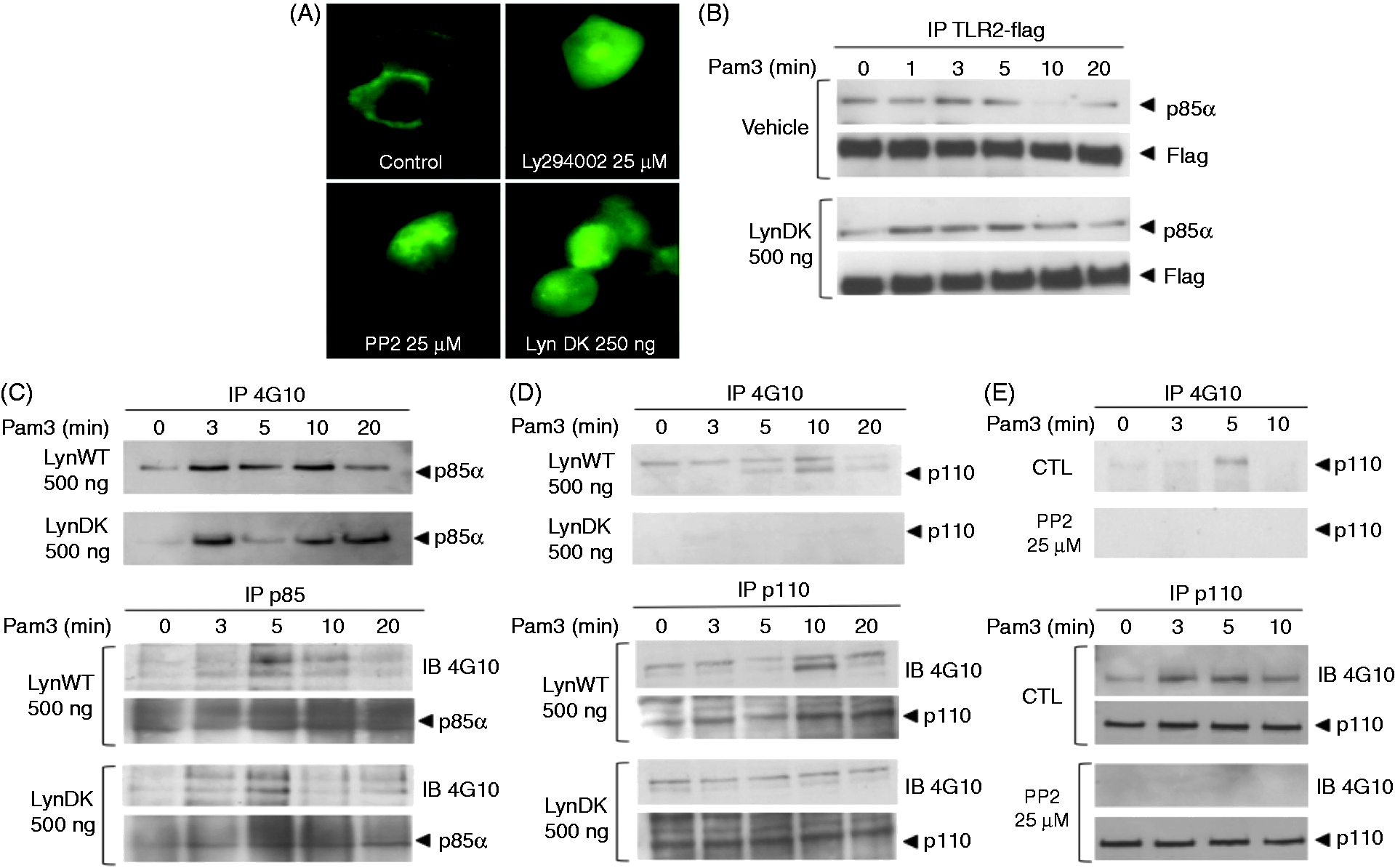
Lyn phosphorylates the p110 subunit of PI 3-kinase upon stimulation of TLR2
Recruitment of PI 3-kinase to the TLR2 activation complex is required to initiate the transactivation pathway to NF-κB. We thus investigated whether inhibition of Lyn altered the recruitment of the p85α subunit of PI 3-kinase to this receptor-signaling complex. TLR2 immunoprecipitation in HEK 293–TLR2 cell lysates and immunobloting of p85α subunit of PI 3-kinase showed that neither PP2 (data not shown) nor LynDK expression prevented Pam3-induced physical association between TLR2 and PI 3-kinase (Figure 6B). These data suggest that Lyn controls the activity of PI 3-kinase independently of its recruitment to the TLR2 activation cluster. As several reports indicate that PI 3-kinase activity depends upon its tyrosine-phosphorylation status,32–34 we tested whether Lyn activates PI 3-kinase through phosphorylation of one of its subunits. As shown by immunoprecipitation with 4G10 or anti-p85α Abs, stimulation with Pam3 resulted in phosphorylation of p85α after 3 min independently of Lyn kinase activity (Figure 6C). To assess the effect of Lyn kinase activity on the tyrosine phosphorylation of the p110 subunit of PI 3-kinase, we immunoprecipitated lysates of Pam3-stimulated HEK 293–TLR2 with 4G10 anti-phosphotyrosine or anti-p110 Abs, and submitted cell extracts to Western blot analysis. Immunostaining with anti-p110 or 4G10 Abs revealed maximal phosphorylation of the p110 subunit 10 min after stimulation with Pam3 (Figure 6D). Inhibition of Lyn kinase activity by lynDK overexpression blocked phosphorylation of the p110 subunit of PI 3-kinase. We also tested the ability of PP2 to modulate the phosphorylation of p110 subunit of PI 3-kinase after stimulation of THP1 cells. Whereas stimulation with Pam3-induced phosphorylation of p110, treatment with PP2 decreased drastically the phosphorylation of p110 in THP1 cells (Figure 6E). Altogether, these data suggest that Lyn controls the PI 3-kinase pathway to TLR2-dependent NF-κB activation through phosphorylation of its p110 subunit.
Discussion
In the present study, we report that in human cell models (i) Lyn is essential for the transactivation of the P65 subunit of NF-κB after binding of acylated bacterial lipoproteins to TLR2, (ii) Lyn regulates PI 3-kinase activity upstream of AKT, independently of the tyrosine phosphorylation of TLR2 and of the recruitment of PI 3-kinase to TLR2, and (iii) Lyn activates PI 3-kinase through phosphorylation of its p110 subunit. Whereas PI 3-kinase has been well characterized in monocyte/macrophage adhesion and spreading in response to the engagement of TLRs,35,36 the involvement of a Lyn-dependent activation of PI 3-kinase to activate NF-κB signaling has not been previously described.
Several studies have recently suggested the activation role of Lyn in the TLR-mediated innate immune response. A Lyn gain-of-function mouse model displayed enhanced sensitivity to LPS with high levels of pro-inflammatory and low anti-inflammatory cytokines in the serum, and enhanced leukocyte pulmonary recruitment and TNF-α production in response to LPS inhalation. 37 In HEK 293–TLR4/MD-2 stable transfectants, stimulation with LPS induces the recruitment of Lyn to TLR4, whereas LPS tolerance is associated with an inhibition of the association between Lyn and TLR4. 38 Lyn also plays a positive role in TLR4-dependent production of TNF-α in murine mast cells through the modulation of the TRAF-6/TAK-1 protein complex. 39 Stimulation of macrophages with bacterial DNA, a TLR9 agonist, results in increased cytokine production in a Lyn- and Hck-dependent manner. 17 Recent data also implicate Lyn in TLR2-mediated events. Kannan et al. reported that Lyn is crucial for internalization into lung epithelial cells of P. aeruginosa,16,40 a pathogen that signals through TLR4, TLR5 and TLR2. 41 In triple knock-out mice deficient for Src kinases Lyn, Hck and Fgr, antimicrobial control of Streptococcus pneumoniae meningitis, a TLR2-mediated process, 42 was inefficient with decreased production of reactive oxygen species. 43
In contrast, Keck et al. recently reported that Lyn negatively regulates TLR signaling in murine bone marrow-derived macrophages (BMDMs) through the activation of PI3-kinase. 18 However, this study based on a murine model provided no mechanistic insights into the role of Lyn and PI 3-kinase in TLR2-mediated signaling. PI 3-kinase and Lyn are known to act as either a positive or a negative regulator of signal transduction depending on many cellular and extracellular parameters. Therefore, these discrepancies could possibly be attributed to the cell type and its developmental stage, as well as the method used to generate murine BMDM. Besides, a recent study showed that inflammatory responses to sepsis in mouse models correlate poorly with the corresponding human diseases, suggesting significant discrepancies between species regarding signal transduction. 44 In this setting, studies on TLR signaling involving human primary cells showed that Lyn had a positive role as chemical inhibition of Lyn affects LPS-activated pathways in primary human macrophages. 45 Lyn was also part of a TLR9-DOCK8-Pyk2 cluster and was essential to the activation of STAT3 cascade and subsequent human B cell proliferation and differentiation. 46 Using human primary monocytes and two different human cellular models, as well as complementary inhibition strategies to inhibit Lyn, we report that Lyn is necessary to transactivate the P65 subunit of NF-κB. In our model, the activation of NF-κB involved the PI 3-kinase pathway in a Lyn-dependent manner, tightly linking these molecules to the pro-inflammatory component of TLR2-mediated host defense.
An interesting finding of our study is that Lyn contributes to PI 3-kinase activation through tyrosine phosphorylation of its p110 subunit. In B cells, PI 3-kinase activity is regulated by a number of mechanisms, including binding of Src-kinase family members to the p85 regulatory subunit through its SH2 and SH3 domains. 12 Interactions of p85–SH2 domains with tyrosine-phosphorylated receptors have been reported. 47 In addition to this mechanism, SH3 domains of Src-kinases Fyn and Lyn bind to the proline-rich region of p85, leading to an increase in PI 3-kinase activity. 48 Several reports suggested a relationship between tyrosine phosphorylation of p85 and PI 3-kinase activation in lymphocytes or neutrophils.32,33,49 Other studies reported tyrosine phosphorylation of the p110 subunit following engagement of the BCR, of CD4 or of the NK cell receptor.34,50,51 Interestingly, these phosphorylation events seemed to be dependent on the activity of Src-kinases or tyrosine-kinase Syk. In our model, consistent with the time-course of PI 3-kinase activation, we observed tyrosine phosphorylation of both p85 and p110 5 – 10 min after the engagement of TLR2. Interestingly, only phosphorylation of p110 was dependent on Lyn kinase activity, which led to PI 3-kinase activation. Hence, the present study is the first to suggest the role of p110 tyrosine phosphorylation in PI 3-kinase activation after TLR2 stimulation.
Activation of most TLRs initiates both MyD88-dependent and MyD88-independent signaling pathways to mount a sequentially coordinated response. 1 Although the existence of strictly MyD88-independent signaling is yet not well established for TLR2, a Rac1/PI 3-kinase-dependent pathway has been identified as critical to transactivation of the P65 subunit of NF-κB. The pivotal role of Lyn following TLR2 stimulation clearly highlights the physiological relevance of this Rac1/PI 3-kinase pathway. Indeed, inhibition of Lyn by pharmacologic methods, dominant negative expression of Lyn or gene silencing blocked PI 3-kinase activity without disrupting its binding to TLR2 or its tyrosine phosphorylation. Inactivation of Lyn kinase activity was also associated with a decrease in the phosphorylated forms of AKT and P65, and thus with the inhibition of NF-κB activity and pro-inflammatory cytokine release. Consistent with previous work by Arbibe et al., 8 these results suggest that Lyn kinase activity is required to activate the PI 3-kinase/AKT pathway upon stimulation of TLR2. Although several reports suggested that PI 3-kinase and AKT can be involved in modulating activation of MAP kinases,9,27 blocking Lyn kinase activity did not alter the activation profile of P38, ERK and JNK downstream of TLR2, lending further support to the specific contribution of the PI 3-kinase pathway to NF-κB activation. However, other signaling molecules might also be involved in PI 3-kinase activation and subsequent AKT and P65 phosphorylation, such as MyD88 Adapter-like (Mal)/TIRAP and TRAF6, as previously suggested. 10 Of note, several studies have also highlighted the contribution of PI 3-kinase to other TLR2-dependent immune responses such as maturation of dendritic cells.52,53 The immune response depends on the type of TLR2 ligand and innate immune cells. 54 Whereas a few studies have shown that Lyn is also involved in the maturation of DCs,20,21 the contribution of Lyn-dependent phosphorylation of the p110 subunit of PI3K downstream of TLR2 has not been investigated thus far.
We propose a model in which engagement of TLR2 triggers its recruitment in lipid rafts to form a signaling cluster with CD14, Lyn, Rac and PI 3-kinase (Figure 7). After tyrosine phosphorylation of the cytoplasmic domain of TLR2, the p85 subunit of PI 3-kinase is recruited to the receptor and phosphorylated on tyrosine. In addition, Lyn is required for tyrosine phosphorylation of the p110 subunit. Phosphorylation of p110 could either directly increase its catalytic activity or recruit SH2-containing proteins that may, in turn, regulate its intrinsic activity. In addition, the exact p110 isoform (alpha, beta or delta)
55
phosphorylated remains to be defined in order to characterize fully the exact role of the tyrosine phosphorylation of p110 on the PI 3-kinase activation required to allow membrane recruitment and phosphorylation of AKT.
Lyn controls the PI 3-kinase pathway through phosphorylation of its p110 subunit after TLR2 engagement. The presence of bacteria-derived acylated lipoproteins results in the heterodimerization of TLR2 with TLR1/6 in membrane microdomains. MyD88/IRAK are recruited to the receptor and lead to degradation of IκB and translocation of NF-κB subunits. A Rac/PI 3-kinase-dependent signaling pathway has also been described, including CD14 and Lyn that contribute to the activation cluster. After tyrosine phosphorylation of the cytoplasmic domain of TLR2, the p85α subunit of PI 3-kinase is recruited to the receptor and phosphorylated on tyrosine. A Lyn-dependent tyrosine-phosphorylation is required to activate the PI 3-kinase catalytic subunit p110 that allows for recruitment of AKT to the inner membrane, precluding a cascade that results in transactivation of the p65 subunit of NF-κB. This leads to the nuclear translocation of a functional p50/p65 NF-κB heterodimer that results in the gene expression of pro-inflammatory cytokines.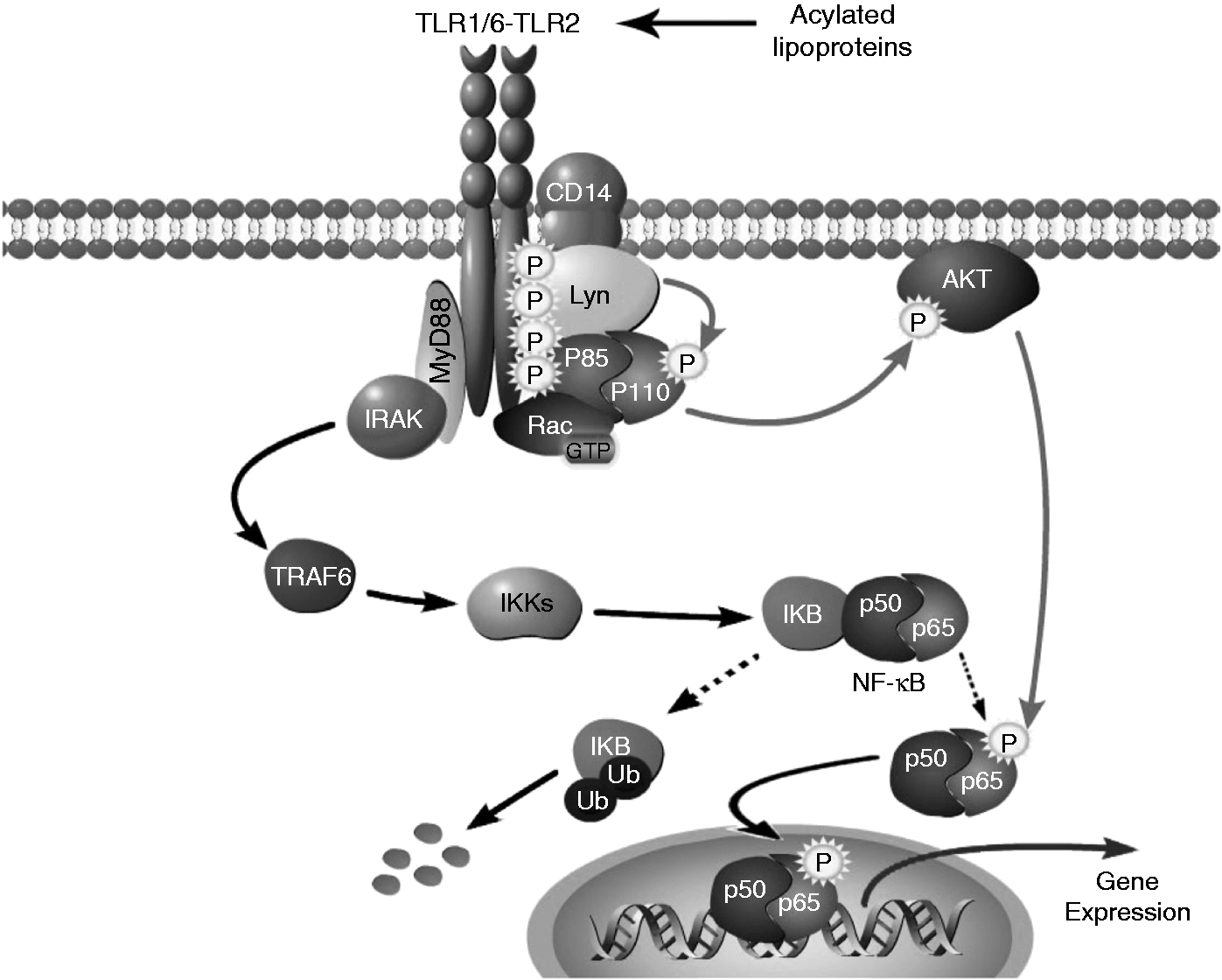
In summary, we demonstrate that in human cells Lyn plays a major role in the activation of NF-κB upon initiation of TLR2 signaling, and therefore in the innate immune response after pathogen recognition by TLR2. Lyn binds TLR2 and PI 3-kinase independently of its kinase activity, and activates PI 3-kinase upstream of Akt to result in NF-κB activation and subsequent production of pro-inflammatory cytokines. By identifying Lyn as an important actor of the innate immune response, this study suggests a potentially interesting new therapeutic target to modulate the immune response during sepsis.
Footnotes
Funding
This work was supported by grants from INSERM and the Cochin Association for Research on Inflammation, Sepsis and Molecular Advances (CARISMA). JT was supported by grants from APHP/CNRS/CEA, Fondation pour la Recherche Médicale, and ESICM (ESICM Levi Montalcini Award).
Conflict of interest
The authors do not have any potential conflicts of interest to declare.