
Abstract
Investigations have suggested degradation of native hyaluronan (HA) into small oligosaccharides as being involved in the development and progression of inflammatory diseases, particularly rheumatoid arthritis (RA). Inflammatory responses occur by modulating the TLR4 and 2, and the CD44 natural HA receptor. As reported recently, the adenosine A2 receptor (A2AR) plays an important anti-inflammatory role in arthritis. TLR4, TLR2 and CD44 stimulation activate NF-κB, which stimulates the production of pro-inflammatory cytokines and other mediators. In contrast, A2AR stimulation inhibits NF-κB activation. The aim of this study was to investigate the effect of combined treatment of HA inhibitor Pep-1 and a selective A2AR agonist (CV-1808) in collagen-induced arthritis (CIA) in mice. Arthritis was induced via intradermal injection of bovine collagen-II. Mice were treated with Pep-1 plus CV-1808 intraperitoneally daily for 20 d. CIA increased TLR4, TLR2, CD44 and A2AR mRNA expression and the related proteins in the joint cartilage of arthritic mice, where significantly increased concentrations were of TNF-α, IL-1-β, IL-17, matrix metalloprotease-13 and inducible nitric oxide synthase. Pep-1 with CV-1808 treatment significantly reduced CIA damage and all the up-regulated biochemical parameters. These reductions were supported by microscopic analysis and synovial fluid HA levels.
Introduction
Arthritis is the most common chronic joint disease. Articular cartilage is a major component of the joint and its mechanical properties depend on the integrity of the extracellular matrix (ECM), which is composed mainly of proteoglycans and collagens. 1 Degeneration of joint cartilage is a key feature of arthritis, but the disease is also associated with concomitant changes in the synovium and subchondral bone metabolism, causing inflammation of the synovial membrane in the affected joint. 2 There are no effective long-term treatments to counteract arthritis and current research is focused on understanding how the imbalance between specific ECM molecules may influence progression of the disease. Evidence is emerging to show that endogenous ECM molecules supply signals to damaged cartilage and synovial tissue in order to promote further cartilage degradation.3–5 Several reports support the conclusion that hyaluronan (HA), a major ECM component, is generally anti-inflammatory at high molecular mass, while HA at low molecular mass may function as an alarm signal to the immune system, particularly for monocytes/macrophages.4,6,7 HA deposition is greatly increased during connective tissue development, repair and regeneration, and in malignant and, particularly, inflamed tissues. 8 Catabolic HA products can act as promoters of early inflammation by enhancing cellular infiltration and the production of pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6.9–11 Following tissue injury, it has been shown that HA fragments of low molecular mass are produced as a result of hyaluronidases or by oxidation.4,9–11 In particular, HA has been shown to have a greater polydispersity in size under inflammatory conditions, with a preponderance of low molecular mass forms. 6
Cluster determinant 44 (CD44) is the best-known receptor of HA. 12 The stimulation of CD44 with HA plays a role in various physiological functions, for example cell adhesion, cell-substrate interaction and lymphocyte recruitment, as well as in pathological processes, such as inflammation and metastasis. 13 Initial studies indicated that inflammatory signaling from HA degradation products involved CD44 alone. 13 However, subsequent work reported other signalling pathways to be involved, particularly via TLRs.14,15 TLRs recognize structurally conserved molecules derived from microbes. Once these microbes cross physical barriers, such as the skin or intestinal tract mucosa, they are recognized by TLRs, and this activates the innate immune response. Although each TLR recognizes specific ligands, they may also trigger other molecules, such as small HA oligosaccharides, which are able to trigger the release of pro-inflammatory cytokines.14,15
Recent findings have shown that HA oligosaccharides can interact with both TLR4 and TLR2, as well as with CD44, thereby stimulating inflammation or increasing the inflammatory mechanism induced previously by other agents in different cell types in both in vitro and in vivo experimental models.15–18 Hence, the production of low molecular mass HA in pathologies may act as an endogenous danger signal, leading to the activation of both innate and acquired immunity.
Adenosine (ADO) is a purine nucleoside that is released from a variety of cells in response to metabolic stress and inflammatory pathologies. 19 It regulates tissue function by modulating a variety of physiological and pathophysiological processes through the activation of four well-defined G protein-coupled receptors classified as ADO A2A, A2B, A1 and A3 receptors. 20 ADO is normally found at low extracellular concentrations, but during inflammation and tissue destruction it is released into the extracellular space and acts as a negative regulator of both inflammation and immune-mediated tissue destruction. 19 Out of the ADO receptors (ARs), the A2A receptor is recognized to be a prime mediator of anti-inflammatory responses. 21 The binding of ADO, or selective agonists, through the A2A receptor induces an anti-inflammatory response, probably via a cAMP-dependent pathway. 22 Activation of the A2AR on polymorphonucleates (PMNLs) lessens the adherence of activated PMNLs to the endothelium, lowers the release of reactive oxygen species, reduces primary lysosome degranulation and decreases pro-inflammatory cytokine levels.23–25 ADO A2AR agonists have been shown to reduce the mortality associated with peritonitis in a mouse experimental model. 26
Furthermore, it has been proposed that the administration of ADO A2A agonists may be useful for treating inflammatory events and sepsis.23–25 The high-affinity A2AR is expressed in a range of cell types, including chondrocytes. 26 The selective engagement of the A2A receptor by ADO activates stimulatory G-proteins that activate adenylate cyclase, leading to increased cAMP production. 27 Although the exact signal transduction pathways used by ADO have not yet been fully elucidated, elevated levels of cAMP have been linked to the activation of protein kinase A (PKA) and the inhibition of NF-kB.19,28
One of the best documented anti-inflammatory functions of ADO is its ability to regulate cytokine synthesis. The production of pro-inflammatory cytokines, including TNF-α, IL-8, macrophage inflammatory protein-1alpha (MIP-1α), IL-2 and IL-6, are suppressed by exposure to ADO or ADO analogues. 19 Conversely, exposure to ADO increases the synthesis of IL-10, a potent regulator of macrophage function. 19 In addition, a recent study has reported that the treatment of mice with an A2AR agonist reduces the progression of arthritis in a model of collagen-induced arthritis. 29
Some years ago, a HA-binding peptide (Pep-1) was developed using the phage display technique, 30 and this peptide did not contain any known HA-binding domain sequences. Moreover, in vitro studies showed Pep-1 to bind HA in immobilized, cell-associated and soluble forms. 30
As we demonstrated previously the positive effects of combined treatment with a Pep-1 and a synthetic A2AR agonist (CGS-21680) in a model of IL-1β-induced inflammation that promotes HA fragmentation, 31 the aim of this study was to investigate, using a model of collagen-induced arthritis (CIA) in mice in which HA fragmentation is also promoted, the effect of daily chronic treatment, either separately or combined, of an exogenous selective A2AR agonist (CV-1808) and/or Pep-1. 18
Materials and methods
Animals
Male mice DBA/J1 6–7 wk old with a mean mass of 25–30 g were used in our study. Mice, purchased from Harlan Italy Srl (Correzzana, Italy), were maintained under climate-controlled conditions with a 12 h light/dark cycle. The animals were fed standard rodent chow and provided water ad libitum. The health status of the animal colony was monitored in accordance with Italian Veterinary Board guidelines. Mice were divided into the following groups: (i) control (n = 20); (ii) Pep-1 (10.0 mg/kg) (n = 20); (iii) CV-1808 (1.0 mg/kg) (n = 20); (iv) CIA (n = 30); (v) CIA + Pep-1 (5.0 mg/kg) (n = 30); (vi) CIA + Pep-1 (10 mg/kg) (n = 30); (vii) CIA + CV-1808 (0.5 mg/kg) (n = 30); (viii) CIA + CV-1808 (1.0 mg/kg) (n = 30); (ix) CIA + Pep-1 (5.0 mg/kg) + CV-1808 (0.5 mg/kg) (n = 30); and(x) CIA + Pep-1 (10.0 mg/kg) + CV-1808 (1.0 mg/kg) (n = 30). Additional sets of mice (n = 7 for each of the above groups) were included in order to evaluate HA levels in synovial fluid.
Materials
Bovine type II collagen and complete Freund’s adjuvant were obtained from MD Biosciences GmbH (Zürich, Switzerland). A selective ADO A2A agonist, 2-phenylaminoadenosine (CV-1808) (cat. P101) was purchased from Sigma-Aldrich Srl, (Milan, Italy). Abs against the CD44 receptor (cat. MABT78) to block its activity were supplied by Millipore Corporation (Billerica, MA, USA). Mouse TLR4 (cat. ABIN424269), CD44 (cat. ABIN457122) and ADO A2A (cat. ABIN428968) receptor commercial ELISA kits were provided by Antibodies-online.com GmbH, (Aachen, Germany). Mouse TLR2 (cat. E0663Mu), matrix metalloprotease (MMP)-13 (cat. E0099m) and inducible nitric oxide synthase (iNOS) (cat. E0837Mu) commercial ELISA kits were purchased from USCN Life Science Inc. (Wuhan, China). Mouse TNF-α (cat. IB49688), IL-1β (cat. IB49700) and IL-17 (cat. IB49699) commercial ELISA kits were provided by Immuno-Biological Laboratories Inc. (Minneapolis, MN, USA). HA ELISA kit (cat n° K-1200) was obtained from Echelon Biosciences Inc. (Salt Lake City, UT, USA). High molecular mass HA (HMMHA, 3,000 kDa) was purchased from Sigma-Aldrich Srl, and a previously published method was used to prepare HA at different molecular masses.
32
DMEM, FBS,
Pep-1 synthesis
Specific HA blocking peptide (Pep-1) (GAHWQFNALTVR) and scrambled control peptide (WRHGFALTAVNQ) both with an amidated GGGS linker were synthesized in our laboratory following a previously published method. 30 Briefly, Pep-1 was produced using the bacterial expression vector pGEX-sn that allows the expression of recombinant proteins as fusions to glutathione S-transferase (GST). The plasmid pGEX-SN was constructed as described previously. 33 Oligonucleotide sequences encoding the Pep-1 (GAHWQFNALTVR) were optimized for murine codon usage. DNA insert encoding for HA inhibitor peptide was obtained from Sigma-Aldrich as synthetic complementary oligonucleotides. After hybridization, the double-strand oligonucleotides insert was ligated into the pGEX-sn polylinker at BamHI/EcoRI sites in order to obtain the plasmid vector to produce the desired peptide. The same procedure described above was used to produce a scrambled control peptide (WRHGFALTAVNQ). Plasmids for in vitro production were grown in Escherichia coli DH5α and purified using Jet quick Plasmid Miniprep (Genomed Incorporated, St. Louis, MO, USA) and than transfected in E. coli AD 202 for the production of fusion proteins. After induction of the fusion proteins, these were purified from the cytoplasm of bacterial cells by affinity chromatography. 34 The HA inhibitor peptide was removed chemically using cyanogen bromide, which hydrolyses peptide bonds at the C-terminal of methionine residue used as linker between GST and HA inhibitor peptides, followed by removal of free GST by affinity chromatography.
Induction of CIA and arthritis evaluation
Mice were injected intradermally at the tail base with 100 µl of 2.0 mg/ml type II collagen emulsion in Freund’s complete adjuvant containing 2.5 mg/ml heat-killed Mycobacterium tuberculosis H37Ra. Mice were immunized a second time 7 d later.
Evaluation of joint inflammation was carried out blindly by an independent observer with no knowledge of the treatment protocol. The severity of the arthritis in each limb was graded daily on a scale of 0–4 as follows: 0, no macroscopic signs of arthritis; 1, swelling of one group of joints (i.e. knee or ankle joints); 2, two groups of swollen joints; 3, three groups of swollen joints; and 4, swelling of the entire limb. The maximum score for each mouse was 16. Clinical severity was also assessed by quantifying changes in limb volume. Measurements were performed using a dial gauge caliper. At the end of the experimental period (d 35) mice were anaesthetized with ethyl ether and then euthanized to remove their hind limbs.
Pep-1 and CV-1808 treatment
On d 20, to coincide approximately with CIA onset, the animals were randomized to receive the treatments listed above. Pep-1 was dissolved in saline solution (0.9% NaCl), while CV-1808 was dissolved in a solution of ethanol:water 25%, v:v, and administered intraperitoneally using a volume of 1.0 ml/kg body mass, once daily up to d 35. Pep-1 was administered at doses of 5.0 and 10.0 mg/kg, and CV-1808 was administered at doses of 0.5 and 1.0 mg/kg.
Cartilage isolation from mice joints
At the end of the experimental phase (d 35) mice were euthanized by cervical dislocation and the whole hind limbs isolated from the body. One knee joint was conserved for histological examination; the remaining knee joint and the two paw joints were dissected and the whole cartilage was separated from bone and muscular tissue, washed in ice-cold 10 mM Tris-HCl, pH 7.4 and blotted on absorbent paper. These operations were performed on a cooled plate refrigerated on dry ice. The whole cartilage was stored immediately on liquid nitrogen until biochemical assay.
RNA isolation, cDNA synthesis and real-time quantitative PCR amplification
Total RNA was isolated from cartilage samples for reverse-PCR real-time analysis of TLR4, TLR2, CD44, A2AR, TNF-α, IL-1β, IL-17, MMP-13 and iNOS (RealTime PCR system, Mod. 7500; Applied Biosystems Inc., Foster City, CA, USA) using an Omnizol Reagent Kit (Euroclone Ltd, West York, UK). The first strand of cDNA was synthesized from 1.0 µg total RNA using a high capacity cDNA Archive kit (Applied Biosystems). β-Actin mRNA was used as an endogenous control to allow the relative quantification of TLR4, TLR2, CD44, A2AR, TNF-α, IL-1β, IL-17, MMP-13 and iNOS PCR. RealTime was performed by means of ready-to-use assays (assays on demand ready to use; Applied Biosystems Inc) on both targets and endogenous controls (TLR4: reporter 5’ 6-FAM- assay Mm00445274_m1- Ex. 2-3; TLR2: reporter 5’ 6-FAM- assay Mm00442346_m1- Ex. 2-3; CD44: reporter 5’ 6-FAM- assay Mm01277163_m1- Ex. 2-3; A2AR: reporter 5’ 6-FAM- assay Mm00802075_m1- Ex. 2-3; TNF-α: reporter 5’ 6-FAM- assay Mm00443259_g1- Ex. 1-2; IL-1β: reporter 5’ 6-FAM- assay Mm1336189_m1- Ex. 1-2; IL-17: reporter 5’ 6-FAM- assay Mm00439619_m1- Ex. 2-3; MMP-13: reporter 5’ 6-FAM- assay Mm00439491_m1- Ex. 4-5; iNOS: reporter 5’ 6-FAM- assay Mm00440502_m1- Ex. 21-22; β-actin: reporter 5’ VIC – assay code 4352341E). The amplified PCR products were quantified by measuring the calculated cycle thresholds (CT) of TLR4, TLR2, CD44, A2AR, TNF-α, IL-1β, IL-17, MMP-13, iNOS and β-actin mRNA. The CT values were plotted against the log input RNA concentration in serially diluted total RNA of cartilage samples and used to generate standard curves for all mRNAs analysed. The amounts of specific mRNA in samples were calculated using the 2 −ΔΔCT method. The mean value of normal cartilage target levels became the calibrator (one per sample) and the results are expressed as the n-fold difference relative to normal controls (relative expression levels).
NF-kB p50/65 transcription factor assay
NF-κB p50/65 DNA binding activity was evaluated in nuclear extracts of cartilage tissue in order to measure the degree of NF-κB activation. The analysis was performed following the manufacturer’s protocol for a commercial kit (NF-kB p50/65 EZ-TFA Transcription Factor Assay Colorimetric, cat. n°70-510; Millipore, Billerica, MA, USA). Cytosolic and nuclear extraction was performed by lysing the cell membrane with an apposite hypotonic lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1.0 mM EDTA, 1.0 mM EGTA, 1.0% Triton, 2.5 mM Na4P2O7. 1.0 mM β-glycerophosphate, 1.0 mM Na3VO4) containing protease inhibitor cocktail and tributylphosphine (TBP) as reducing agent. After centrifugation at 8000 g, the supernatant containing the cytosolic fraction was stored at −70℃, while the pellet containing the nuclear portion was then re-suspended in the apposite extraction buffer (25 mM HEPES pH 7.5, 500 mM NaCl, 1.0 mM DTT, 10 mM NaF, 10% glycerol, 0.2% NP40, 5.0 mM MgCl2) and the nuclei were disrupted by a series of drawing and ejecting actions. The nuclei suspension was then centrifuged at 16,000 g. The supernatant fraction was the nuclear extract. After the determination of protein concentration, this extract was stored in aliquots at −80℃ for the subsequent NF-κB assay. After incubation with primary and secondary Abs, colour development was observed following the addition of the substrate TMB/E. Finally, the absorbance of the samples was measured using a spectrophotometric microplate reader set at λ 450 nm. Values are expressed as relative OD/mg protein.
CD44, TLR4, TLR2, A2AR, MMP-13 and iNOS ELISA
Samples of cartilage, obtained from mice hind limbs in the presence of 1.0 nM phenylmethylsulfonyl fluoride (PMSF) and protease inhibitor cocktail, were first lysed using an opportune hypotonic lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1.0 mM EDTA, 1.0 mM EGTA, 1.0% Triton, 2.5 mM Na4P2O7. 1.0 mM β-glycerophosphate, 1.0 mM Na3VO4, 0.2% TBP) and then centrifuged at 10,000 g at 4℃ for 10 min. The analysis of CD44, TLR4, TLR2, A2AR, MMP-13 and iNOS was carried out using a specific commercial kit. In brief, 100 µl of standards, blanks or samples were added to each well of each specifically-coated microplate. The microplates were then covered with the plate sealer and incubated at 37℃ for 2 h. After incubation, the liquid from the wells was discarded and 100 µl of biotin-antibody was added to the unwashed wells. After covering with plate sealer, the microplates were further incubated at 37℃ for 60 min. At the end of incubation, the liquid was aspirated from the microplates, which were then washed three times with Wash Buffer. After adding 100 µl of HRP-avidin to each well, the microplates were again covered with plate sealer and incubated at 37℃ for 60 min. After washing again 3 times, and adding 90 µl of TMB substrate solution to each well, the microplates were left to incubate, protected from light, for a further 30 min at 37℃. After incubation and the addition of 50 µl of stop solution, the absorbance of each well was read spectrophotometrically at λ450 nm. CD44, A2AR and MMP-13 values are expressed as pg/mg protein, TLR4 and TLR2 values are expressed as ng/mg protein and iNOS values are expressed as U/mg protein.
TNF-α, IL-1β and IL-17 ELISA
Samples of cartilage, obtained from mice hind limbs in the presence of 1.0 nM PMSF and protease inhibitor cocktail, were first plotted using an automatic plotter (Mod.8533 Automatic Plotters, B. Braun Melsungen AG, Germany) in order to isolate cartilage cells, then lysed using an opportune hypotonic lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1.0 mM EDTA, 1.0 mM EGTA, 1.0% Triton, 2.5 mM Na4P2O7. 1.0 mM β-glycerophosphate, 1.0 mM Na3VO4, 0.2% TBP) and then centrifuged at 10,000 g at 4℃ for 10 min. The analysis of TNF-α, IL-1β and IL-17 was carried out using a specific commercial kit. Briefly, 50 µl of standards, samples and controls were added to each well of the coated microplate. Fifty microlitres of each specific biotin-conjugate Ab was then added to each well. After 120 min incubation at 20–22℃, the liquid from the wells was discarded, the wells were washed 3 times and 100 µl of streptavidin–HRP was added. After further incubation for 60 min and washing the wells again, 100 µl of a substrate chromogen solution was added. After 10 min incubation and the addition of 100 µl of stop solution, the absorbance of each well was read spectrophotometrically at λ450 nm. TNF-α and IL-1β values are expressed as ng/mg protein, while IL-17 values are expressed as pg/mg protein.
Light microscopy
At the end of the experimental phase, knee joints were removed from the hind limbs of the animals and fixed with 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4, at 4℃ for 4 h, and then washed with 0.2 M phosphate buffer, pH 7.4. The specimens were decalcified with a ready-to-use decalcifying solution (Cal-Ex; Thermo Fisher Scientific, Waltham, MA, USA) for 6 h, dehydrated in graded ethanol and embedded in a special tissue embedding media (Paraplast; McCormick Scientific, Richmond, IL, USA). Traditional histological sections (7 µM) were cut with a microtome (mod. RM 2125 RT; Leica Camera AG, Solms, Germany). Sections were deparaffinized, rehydrated using xylene and a graded series of ethanol, stained with haematoxylin and eosin and Safranin O-Fast Green stains, and viewed and photographed using a light microscope (mod. Primo Star, Carl Zeiss Inc., Oberkochen, Germany).
HA assay
HA analysis was performed on synovial fluid samples obtained at the end of the experiment (d 35). Synovial fluid was drawn from mice knee joints using a 25-μl microsyringe (33-gauge needle) (mod. 1702; Hamilton Company, Reno, NV, USA). The HA analysis assay was carried out using a specific enzyme-linked binding protein assay test kit. The total volume of synovial fluid obtained from the knee joints of each mouse was approximately 75.0–85.0 µl, and samples were diluted with the kit diluent to reach a volume of 100.0 µl, as required by the assay. In brief, 100 µl of samples and standards were added to an uncoated 96-well polystyrene plate, including diluent buffer as blank controls. Fifty µl of detector buffer was then added to all samples and standards except the blanks. After mixing gently, the plate was covered with plate sealer and incubated at 37℃ for 30 min. Following incubation, 100 µl of controls and samples was added to the corresponding well of the Detection Plate. After mixing contents, the plate was covered with sealer and incubated at 37℃ for 30 min. After discarding the liquid from the wells, and washing 4 times with the wash buffer solution, 100 µl of working enzyme was added to each well. The contents were mixed, covered and incubated at 37℃ for 30 min. After washing the detection plate 4 times with the wash buffer, 100 µl of working substrate was added to each well. After a brief incubation in the dark at 20–25℃, the reaction was stopped by adding 50 µl of stop solution to each well. Absorbance was read, within 15 min, at 405 nm using a microplate reader (DAS Srl, Rome, Italy). The concentration of HA in each sample was determined by interpolation from a standard curve ranging from 0.0 to 100.0 µg/ml. Synovial HA values are expressed as µg/mg protein.
HA size analysis
Purification of HA from a culture medium sample was carried out as described previously. 35 HA size was evaluated by agarose and polyacrylamide gel electrophoresis (1.5–4.0%, 1 × TBE buffer, pH 7.9, 40–400 V) using a modified version of a previously published method.36,37 After electrophoresis, the gel was removed from the apparatus, stained with Stains-All dye (0.005% w/v in ethanol) and then illuminated in a dark room connected to a digital camera (mod. Coolpix 4500; Coolpix, Tokyo, Japan) Data were acquired and processed by the computer using a specific data acquisition program (Nikon view 5).
Protein analysis
The amount of protein was determined using the Bio-Rad protein assay system (Bio-Rad Lab Inc., Richmond, CA, USA) with BSA as a standard in accordance with the published method. 38
Statistical analysis
Data are expressed as means ± SD of no fewer than seven experiments for each test. Statistical analysis was performed by ANOVA followed by the Student–Newman–Keuls test. The statistical significance of differences was set at P < 0.05.
Statement of animal care
The studies reported in this manuscript were carried out in accordance with the Helsinki declaration and the National Institutes of Health guidelines for the Care and Use of Laboratory Animals.
Results
Effects of Pep-1 and CV-1808 treatment on clinical signs of CIA
Animals began to show evidence of clinical inflammation in one or more hind paws around d 20. The first manifestation of disease was erythema of one or more ankle joints, followed by the metatarsal and interphalangeal joints. Figure 1A shows the incidence of CIA at the end of the 35-d study period. The initial signs of arthritis were evident on d 25 in all mice treated with CIA. In the CIA group not treated with Pep-1 and/or CV-1808, the percentage of mice developing arthritis reached 100% on d 25 and the same percentage was maintained up until the end of the experiment. Treatment with Pep-1 significantly attenuated the development of CIA at both doses used—21% with the dose of 5.0 mg/kg; 30% with the dose of 10 mg/kg. CV-1808 also reduced CIA incidence, but to a lesser extent—18% with the dose of 0.5 mg/kg, and 26% with the dose of 1.0 mg/kg. The combined treatment of Pep-1 plus CV-1808 markedly decreased CIA incidence—44% with the lower doses and 57% with the higher doses.
Effect of Pep-1 and/or CV-1808 treatment on the development (A) and progression (B) of arthritis at the end of experiment (d 35). Evaluation was performed on d 35. Values are the mean ± SD of no fewer than seven experiments. °P < 0.001 vs CTRL; *P < 0.005 and **P < 0.001 vs collagen-induced arthritis (CIA).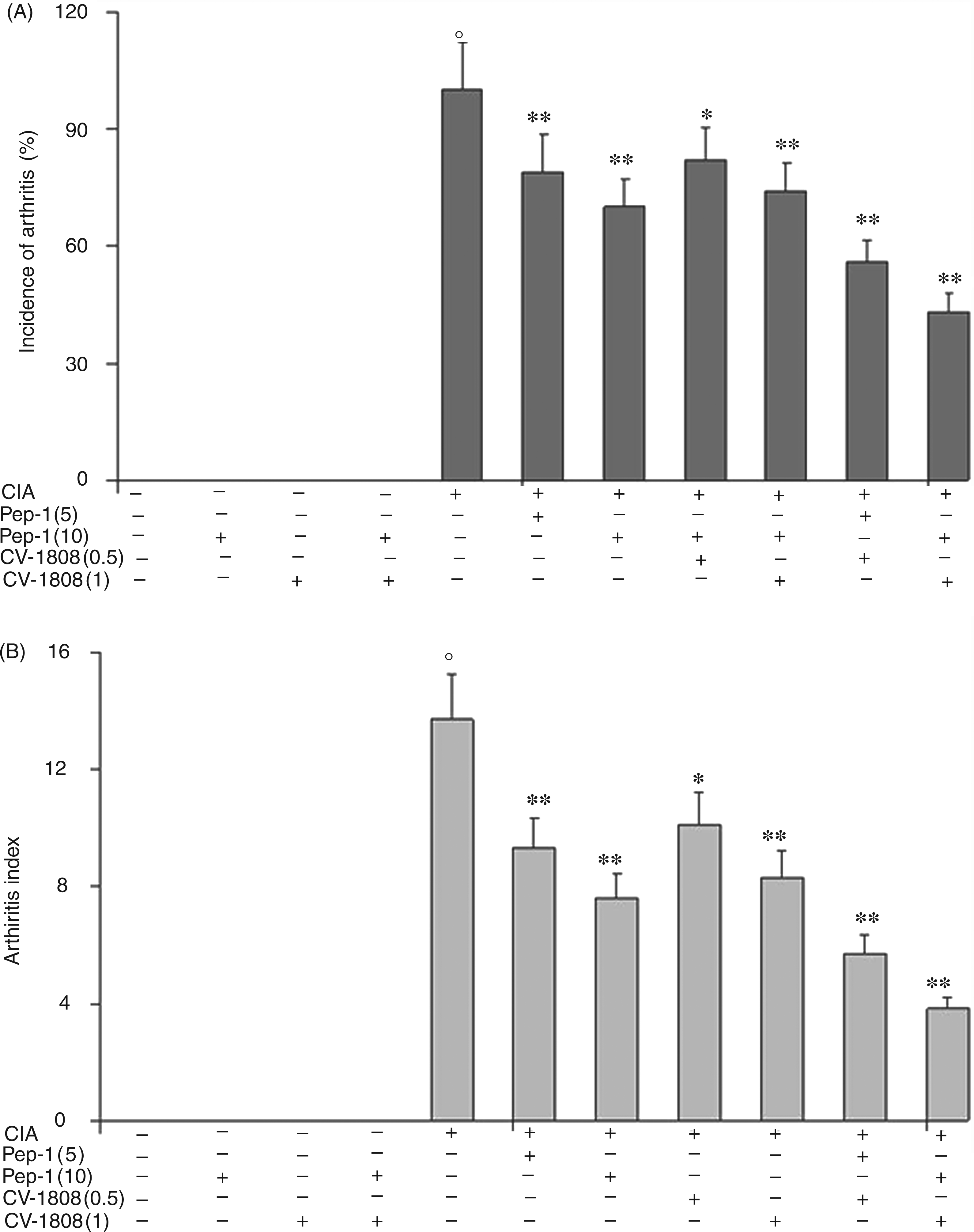
Figure 1B shows the final stage of the disease, as assessed by the arthritis evaluation scale. By d 25 all animals showed evidence of the disease, predominantly in the hind paws. The clinical signs progressed continuously with joint recruitment following an identical pattern: tarsal, metatarsophalangeal followed by interphalangeal. The interphalangeal joints were never affected alone, and inflammation in these joints was invariably associated with inflammation in the tarsal joint. The arthritis score in CIA mice grew progressively from d 25 and reached an arthritis index score of about 13.7 ± 2.2 in the final week. The same changes were observed for the hind paw diameter of CIA animals. Administration of Pep-1 and CV-1808 significantly attenuated all clinical signs of arthritis in terms of arthritic score as follows: Pep-1 = 9.3 ± 1.1 at a dose of 5.0 mg/kg; Pep-1 = 7.6 ± 1.0 at a dose of 10.0 mg/kg; CV-1808 = 10.1 ± 1.2 at a dose of 0.5 mg/kg; CV-1808 = 8.3 ± 1.0 at a dose of 1.0 mg/kg; Pep-1 + CV-1808 = 5.7 ± 0.9 with the lower doses and 3.8 ± 0.8 with the higher doses.
TLR4, TLR2, CD44, and A2AR mRNA expression and protein levels
TLR4, TLR2, CD44 and A2AR mRNA levels (Figure 2 A, B, E, F) and the related protein concentrations (Figure 2 C, D, G, H) were evaluated in order to estimate the degree of TLR4, TLR2, CD44 and A2AR activation in the articular cartilage after CIA and/or Pep-1 and/or CV-1808 treatment. The results showed a significant increase in the expression and protein synthesis of the TLR4, TLR2, CD44 and A2A receptors in arthritic mice (Figure 2). No changes in TLR4, TLR2 and CD44 receptors were found in normal mice treated with Pep-1 and/or CV-1808. Conversely, a marked increment in A2AR expression and protein production was obtained in normal mice receiving the ADO A2AR agonist CV-1808. Arthritic mice receiving Pep-1 showed a significant reduction in TLR4, TLR2 and CD44 expression, while no significant effect on A2AR expression was exerted, although a slight reduction was observed. Mice receiving CV-1808 after CIA treatment showed no changes in TLR4, TLR2 and CD44 expression compared with the results for arthritic mice. Interestingly, the evaluation of A2AR in arthritic mice treated with CV-1808 showed an additional increment compared with the results for either CIA mice or CV-1808-treated control mice. Control mice treated with Pep-1 plus CV-1808 showed no effect on TLR4, TLR2 and CD44 expression, while the effect for A2AR was comparable with the one obtained with CV-1808 alone. Finally, the treatment of arthritic mice with Pep-1 plus CV-1808 showed a reduction in TLR4, TLR2 and CD44 expression and protein levels similar to those obtained when using arthritic mice plus Pep-1, while, for A2AR, the increment in expression was comparable to that obtained by treating the arthritic mice with CV-1808.
Effect of Pep-1 and/or CV-1808 treatment on normal and arthritic mice articular tissue TLR4, TLR2, CD44 and A2AR mRNA expression (A, B, E and F respectively) and related protein production (C, D, G and H respectively). Values are the mean ± SD of no fewer than seven experiments and are expressed as the n-fold increase with respect to the controls (A, B, E, F) and as ng/mg protein (C, D) for the TLR4 and TLR2 protein levels, and as pg/mg protein (G, H) for the CD44 and A2AR protein levels. °P < 0.001 vs CTRL; §P < 0.001 vs CTRL; #P < 0.05 vs collagen-induced arthritis (CIA); ##P < 0.01 vs CIA; *P < 0.001 vs CIA.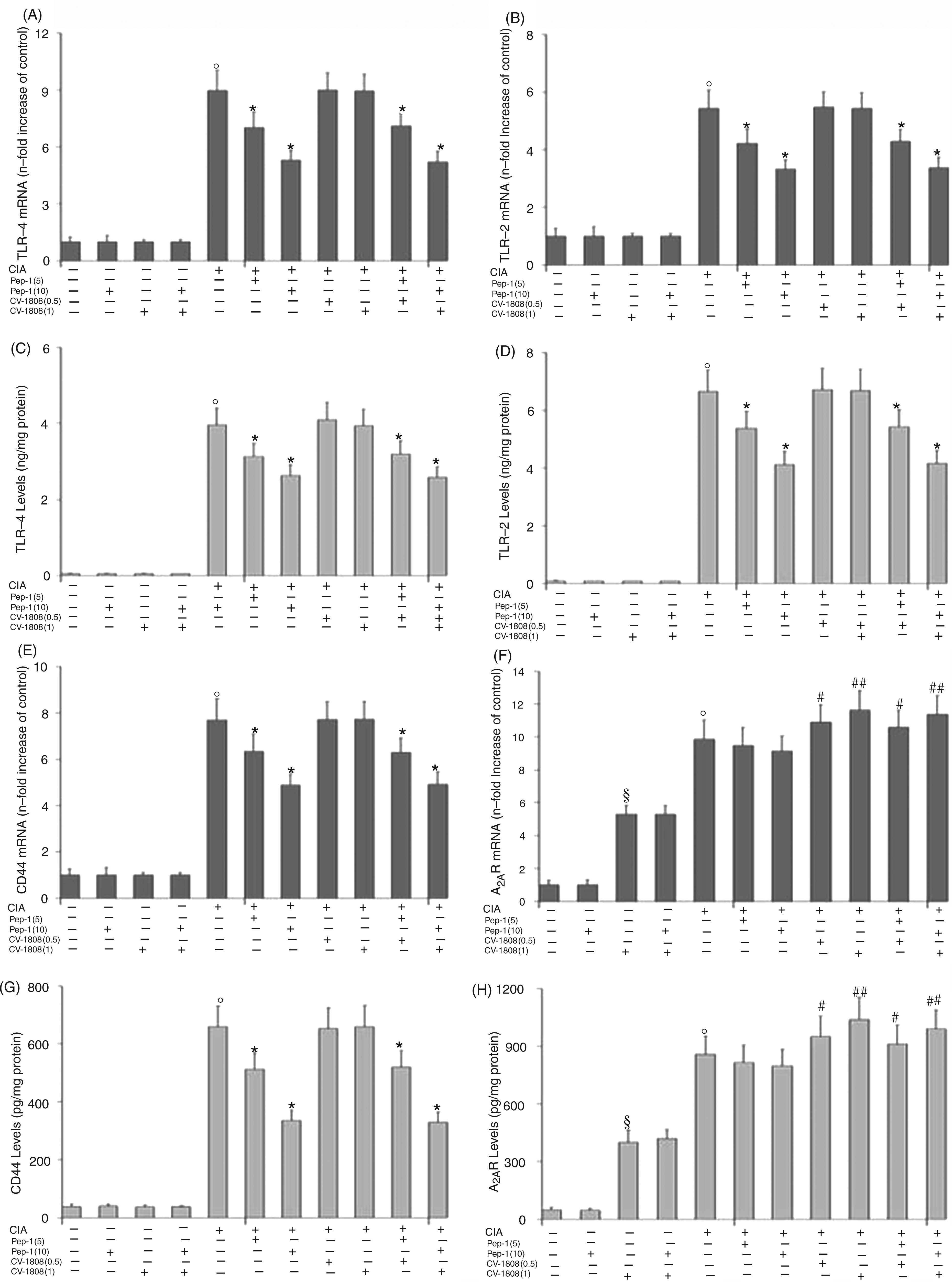
NF-κB activation
Figure 3 shows the changes in the NF-kB p50/p65 heterodimer translocation over the course of the experiment in CIA mice treated with Pep-1 and/or CV-1808. CIA treatment generated a marked increase in NF-kB activation. The treatment with Pep-1 and/or CV-1808 alone had no effect on NF-κB activation. The treatment of arthritic mice with Pep-1 was able to decrease NF-κB activation significantly at both doses. The treatment of CIA mice with CV-1808 was also able to reduce NF-κB activation, but to a lesser extent compared to Pep-1. Of interest was the evidence that CV-1808 treatment reduced, in particular, the p/65 subunit of the NF-κB heterodimer, while Pep- reduced both heterodimer subunits. The combined treatment of normal mice with both Pep-1 plus CV-1808 had no effect on NF-κB. Conversely, the combined treatment of arthritic mice with both Pep-1 plus CV-1808 showed a further significant reduction in NF-κB activation in comparison to the results for cells treated with Pep-1 or CV-1808 alone.
Effect of Pep-1 and/or CV-1808 treatment on normal and arthritic mice articular tissue NF-κB p50/65 transcription factor DNA binding activity. White bars represent the p/50 subunit; black bars represent the p/65 subunit. Values are the mean ± SD of no fewer than seven experiments and are expressed as OD at λ450 nm/mg protein of nuclear extract. °P < 0.001 vs CTRL; *P < 0.005 vs collagen-induced arthritis (CIA); **P < 0.001 vs CIA.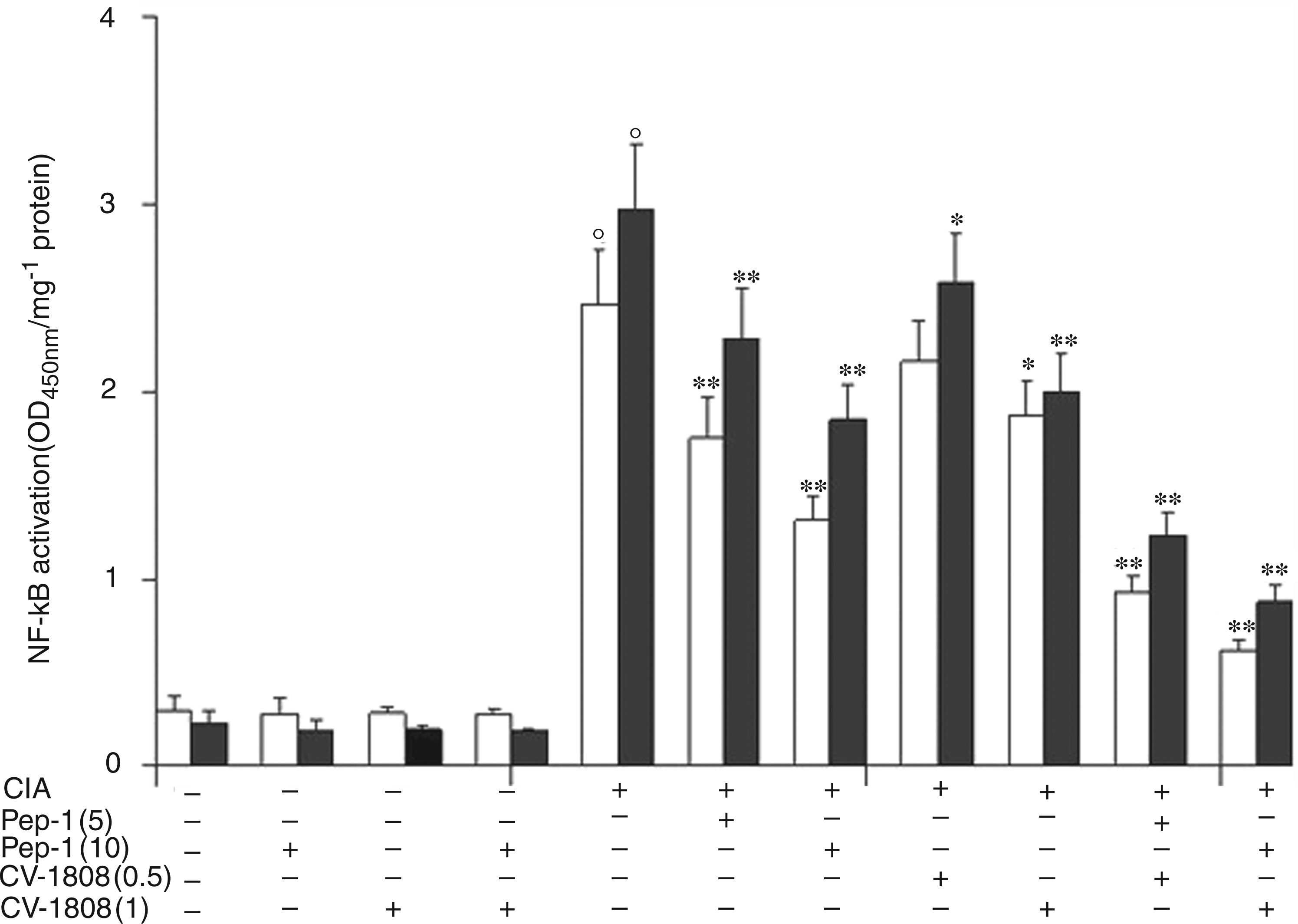
TNF-α, IL-1β, IL-17, MMP-13, and iNOS mRNA expression and protein levels
Effect of Pep-1 and/or CV-1808 treatment on normal and arthritic mice articular tissue TNF-α mRNA expression (A) and related protein production (B).
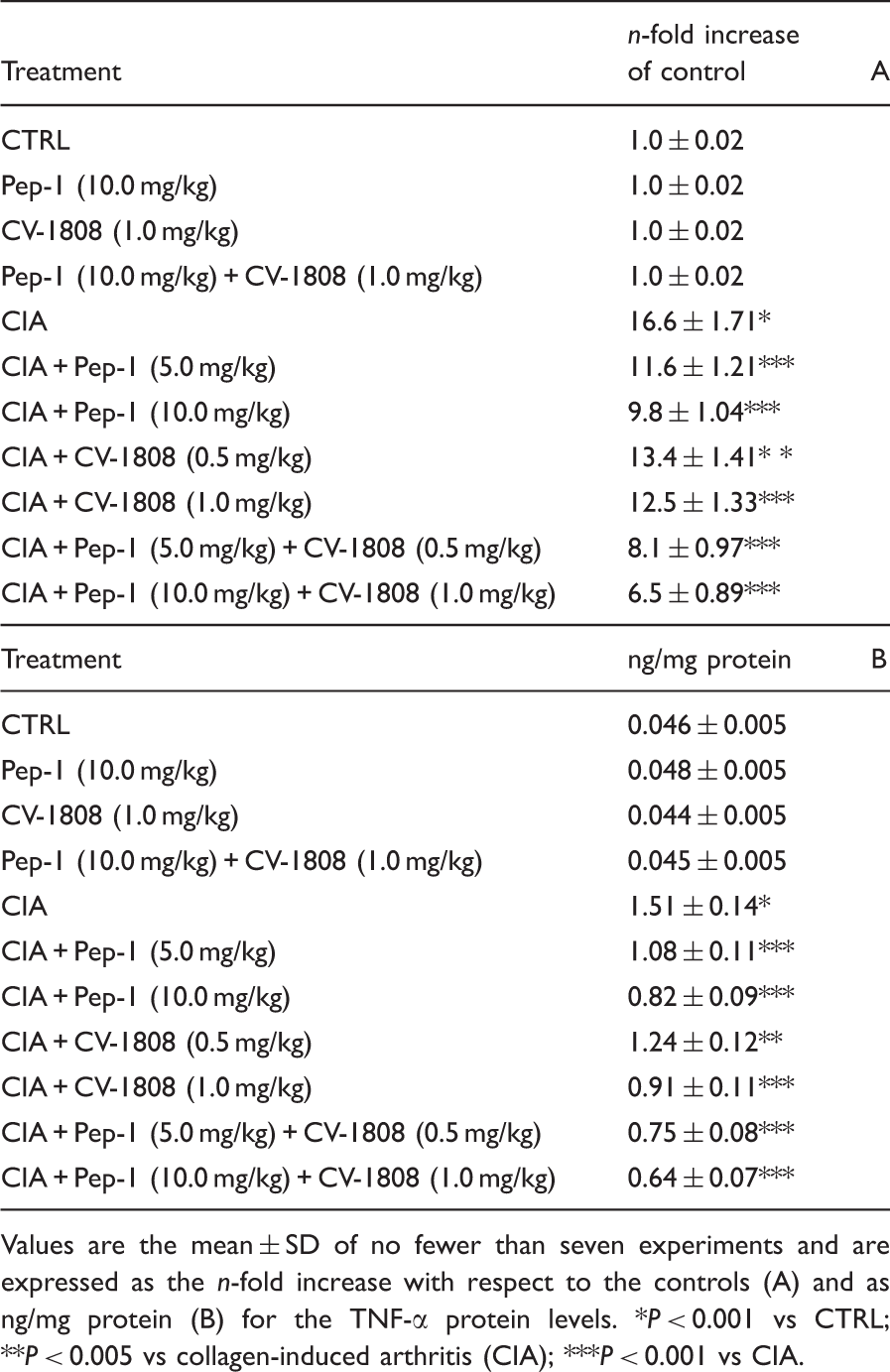
Values are the mean ± SD of no fewer than seven experiments and are expressed as the n-fold increase with respect to the controls (A) and as ng/mg protein (B) for the TNF-α protein levels. *P < 0.001 vs CTRL; **P < 0.005 vs collagen-induced arthritis (CIA); ***P < 0.001 vs CIA.
Effect of Pep-1 and/or CV-1808 treatment on normal and arthritic mice articular tissue IL-1β mRNA expression (A) and related protein production (B).
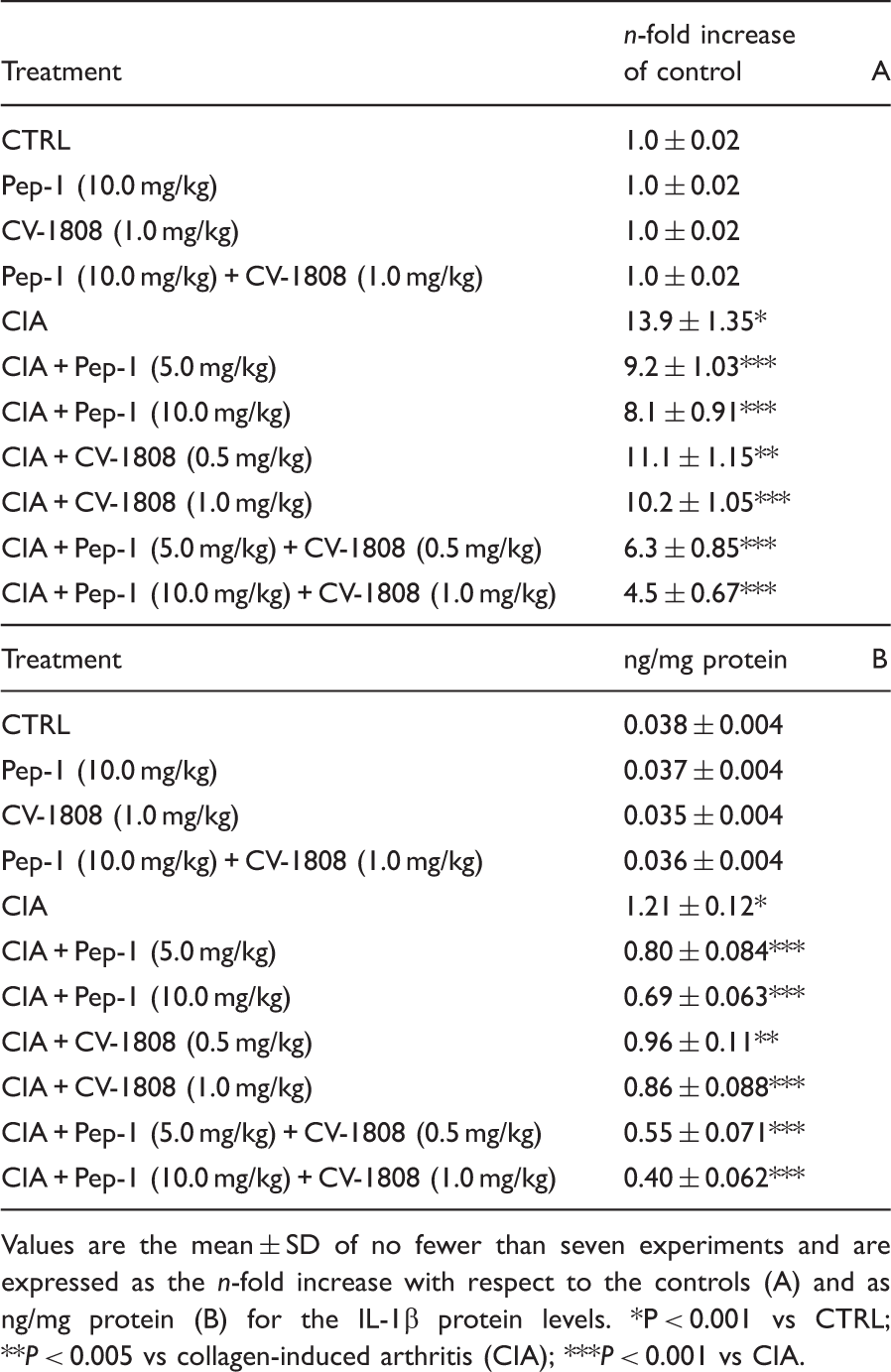
Values are the mean ± SD of no fewer than seven experiments and are expressed as the n-fold increase with respect to the controls (A) and as ng/mg protein (B) for the IL-1β protein levels. *P < 0.001 vs CTRL; **P < 0.005 vs collagen-induced arthritis (CIA); ***P < 0.001 vs CIA.
Effect of Pep-1 and/or CV-1808 treatment on normal and arthritic mice articular tissue IL-17 mRNA expression (A) and related protein production (B).
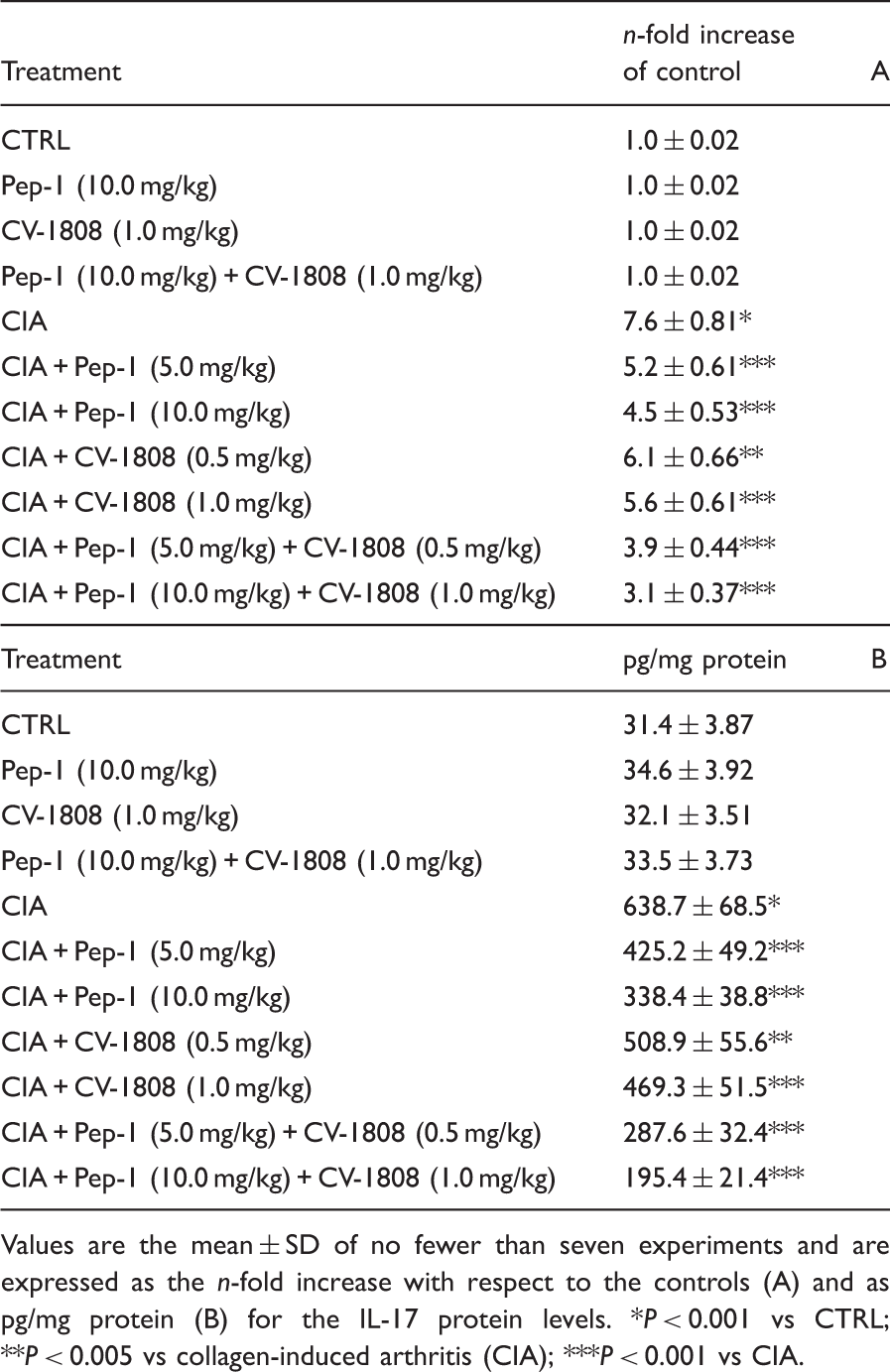
Values are the mean ± SD of no fewer than seven experiments and are expressed as the n-fold increase with respect to the controls (A) and as pg/mg protein (B) for the IL-17 protein levels. *P < 0.001 vs CTRL; **P < 0.005 vs collagen-induced arthritis (CIA); ***P < 0.001 vs CIA.
Effect of Pep-1 and/or CV-1808 treatment on normal and arthritic mice articular tissue MMP-13 mRNA expression (A) and related protein production (B).
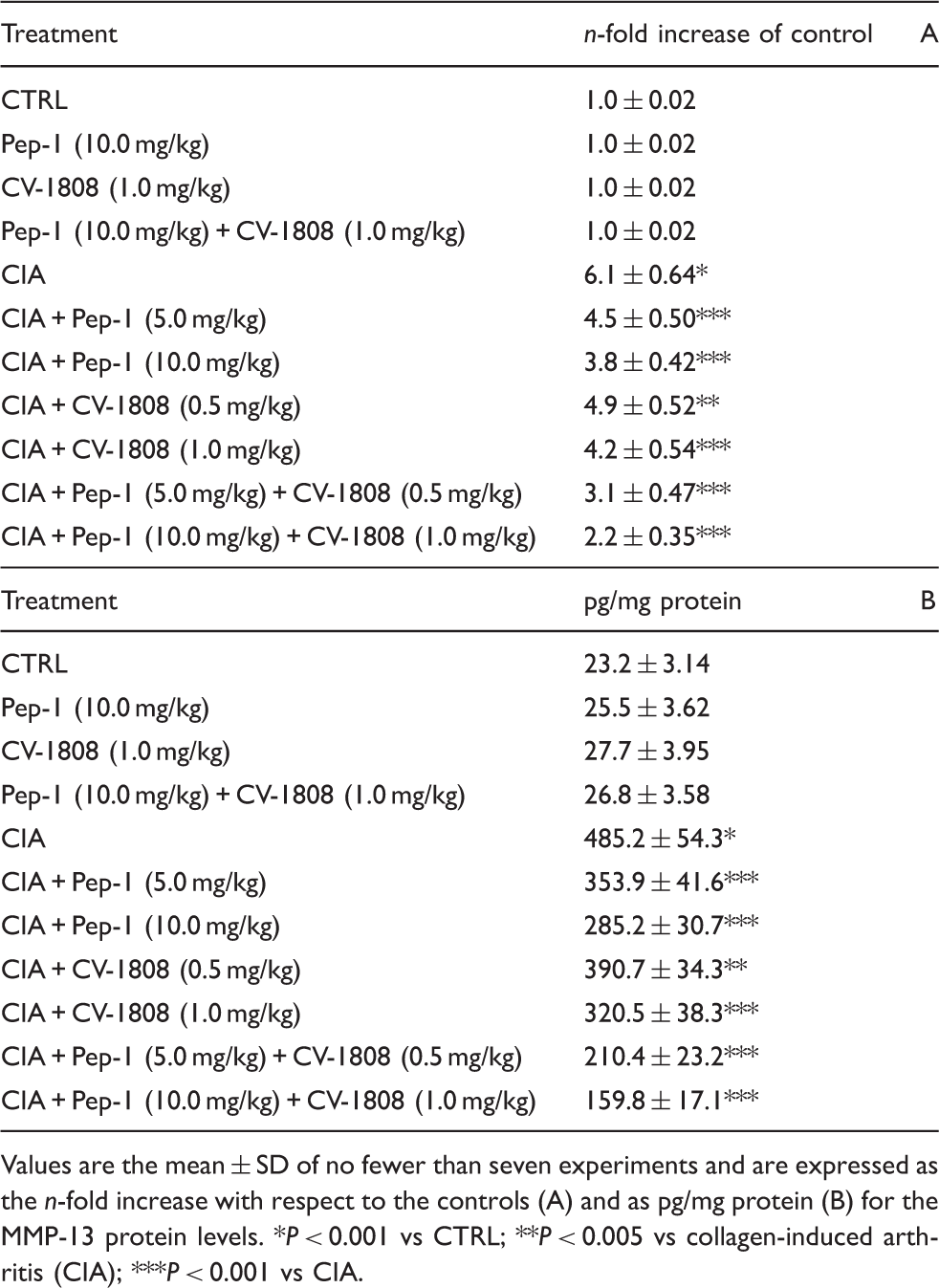
Values are the mean ± SD of no fewer than seven experiments and are expressed as the n-fold increase with respect to the controls (A) and as pg/mg protein (B) for the MMP-13 protein levels. *P < 0.001 vs CTRL; **P < 0.005 vs collagen-induced arthritis (CIA); ***P < 0.001 vs CIA.
Effect of Pep-1 and/or CV-1808 treatment on normal and arthritic mice articular tissue iNOS mRNA expression (A) and related protein production (B).
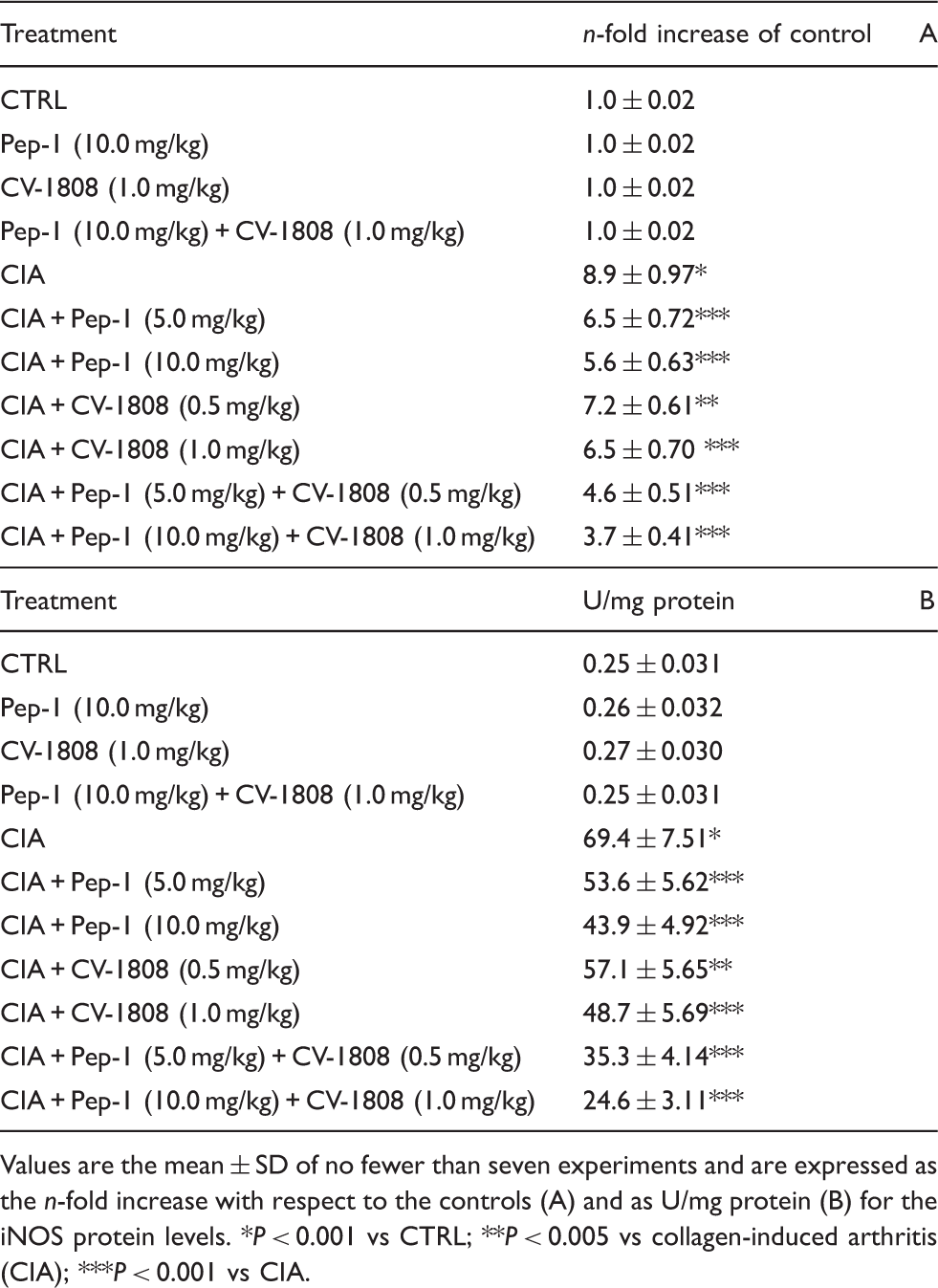
Values are the mean ± SD of no fewer than seven experiments and are expressed as the n-fold increase with respect to the controls (A) and as U/mg protein (B) for the iNOS protein levels. *P < 0.001 vs CTRL; **P < 0.005 vs collagen-induced arthritis (CIA); ***P < 0.001 vs CIA.
Morphological data
Figure 4 shows histopathology of a joint that is representative of all the experimental groups. In a typical section of the control mice knee joint (Figure 4 A) both the growth plate and the articular cartilage could be observed under a light microscope. When viewed at higher magnification (Figure 4, A, boxed area magnification), it clearly showed chondrocytes surrounded by the Safranin O-positive pericellular matrix. When the articular cartilage of mice treated either with PEP-1 or with CV-1808 was examined (Figure 4 B, C), no changes could be detected in the cellular organization and in the staining features of the pericellular matrix. In contrast, complete cartilage erosion occurred in arthritic mice (Figure 4 D). When arthritic mice were treated with PEP-1 at a dose of 5.0 mg/kg (Figure 4 E), isolated areas of chondrophytic remodelling were present under the fibrotic deposits, while, at the higher dose (10.0 mg/kg) (Figure 4 F), the Safranin O-positive cartilage reached the articular surface, thus filling the arthritic erosions. Similar results were demonstrable from the administration of CV-1808: at the dose of 0.5 mg/kg (Figure 4 G), small clusters of chondrocytes were present under the fibrous tissue, while, at the dose of 1.0 mg/kg (Figure 4 H), a rudimental articular cartilage was demonstrated. When both PEP-1 and CV-1808 were administered at the lower doses (5.0 mg/kg and 0.5 mg/kg respectively) (Figure 4 I), a faintly Safranin O-positive continuous cartilage was present over the articular surface. Lastly, when both PEP-1 and CV-1808 were administered at the higher doses (10.0 mg/kg and 1.0 mg/kg respectively) (Figure 4 L), a Safranin O-positive articular cartilage with normal morphology was demonstrated.
Effect of Pep-1 and/or CV-1808 treatment on structural organization of the articular surface of the knee joint in the different groups examined. Evaluation was performed at the end of the experiment, on d 35. (A) In control mice both the growth plate (GP) and the articular cartilage (AC) can be observed. At higher magnification of the boxed area, clearly evident chondrocytes surrounded by Safranin O-positive pericellular matrix are shown. (B, C) No changes are evident in the cellular organization and in the staining features of the pericellular matrix of the articular cartilage of control mice treated either with PEP-1 or CV-1808. (D) Complete cartilage erosion (arrow) in arthritic mice. (E) Isolated areas of chondrophytic remodelling (c) under the fibrotic deposits are evident in arthritic mice treated with PEP-1 at a dose of 5.0 mg/kg. (F) Safranin O-positive cartilage (c) is placed on the articular surface in arthritic mice treated with PEP-1 at a dose of 10.0 mg/kg. (G) Small clusters of chondrocytes (c) are present under the fibrous tissue (F) in arthritic mice treated with CV-1808 at a dose of 0.5 mg/kg. (H) A rudimental, but discontinuous, articular cartilage (c) is present in arthritic mice treated with CV-1808 at a dose of 1.0 mg/kg. (I) A faintly Safranin O-positive continuous cartilage (c) is present over the articular surface in arthritic mice treated with both PEP-1 and CV-1808 at lower doses (5.0 mg/kg and 0.5 mg/kg respectively). (L) A Safranin O-positive articular cartilage of normal morphology is evident over the articular surface in arthritic mice treated with both PEP-1 and CV-1808 at higher doses (10.0 mg/kg and 1.0 mg/kg respectively). Scale bar: A = 50 µm; A inset, B–L = 30 µm.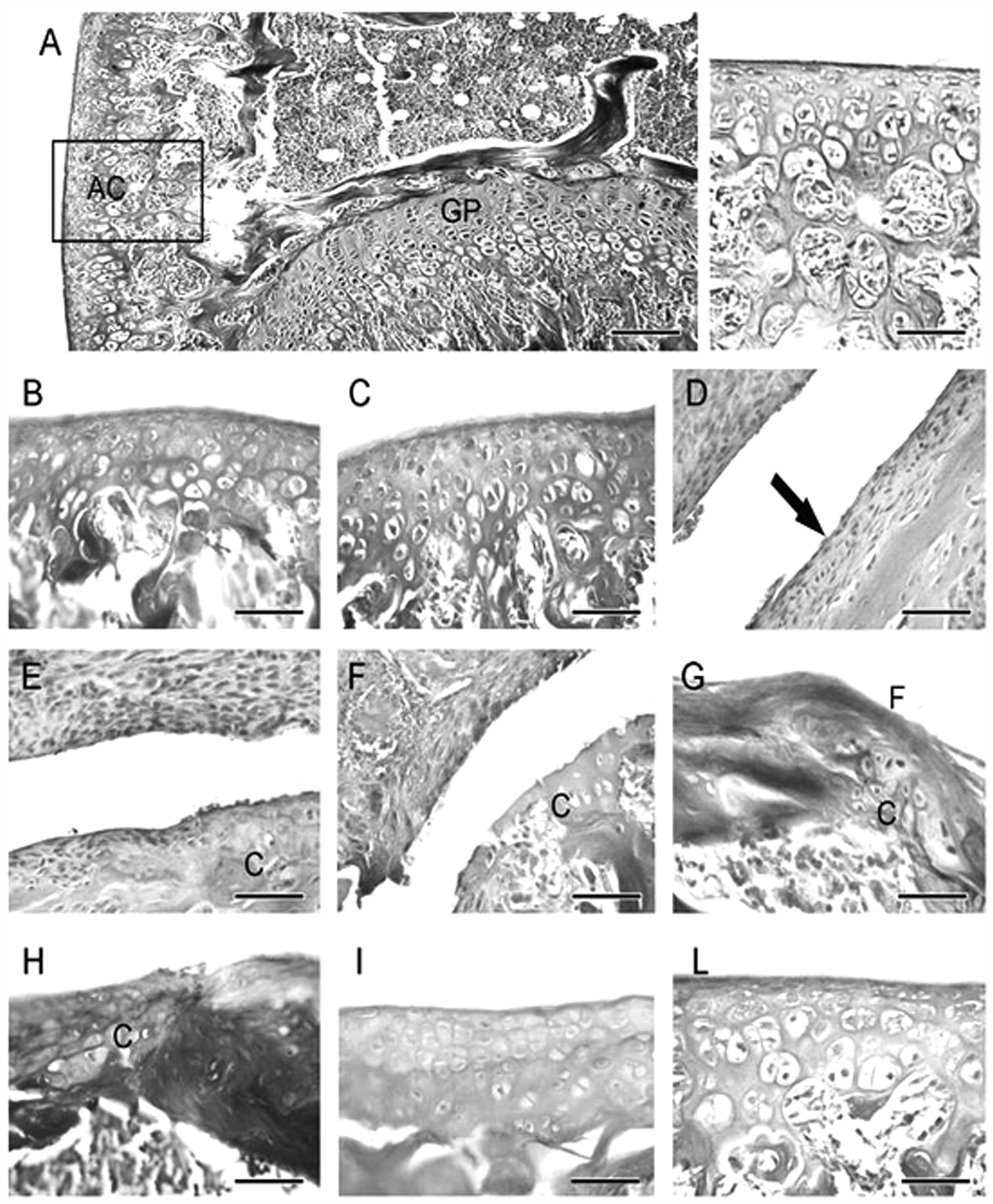
Synovial HA levels and molecular size
HA concentrations were measured in the synovium of normal and arthritic mice in order to verify and confirm the inductive effects on HA levels associated with CIA treatment (Figure 5 A). In mice not subjected to CIA, HA concentration in controls was 4.3 ± 0.2 µg/mg protein. CIA treatment produced a significant increment in synovial HA levels with respect to controls (6.2 ± 0.14 µg/mg protein). The addition of Pep-1 and/or CV-1808 in arthritic mice did not modify HA concentrations with respect to the untreated arthritic mice.
Effect of Pep-1 and/or CV-1808 treatment on normal and arthritic mice synovial hyaluronan (HA) levels (A) and synovial HA molecular size (B). Values are the mean ± SD of no fewer than seven experiments and are expressed as µg/mg protein for HA levels, and as percentages (%) for HA molecular size. White bars indicate HMMHA (>500 kDa); grey bars indicate LMMHA (<500 kDa). °P < 0.001 vs CTRL; $P < 0.001 vs CTRL; #P < 0.001 vs CTRL; *P < 0.001 vs collagen-induced arthritis (CIA).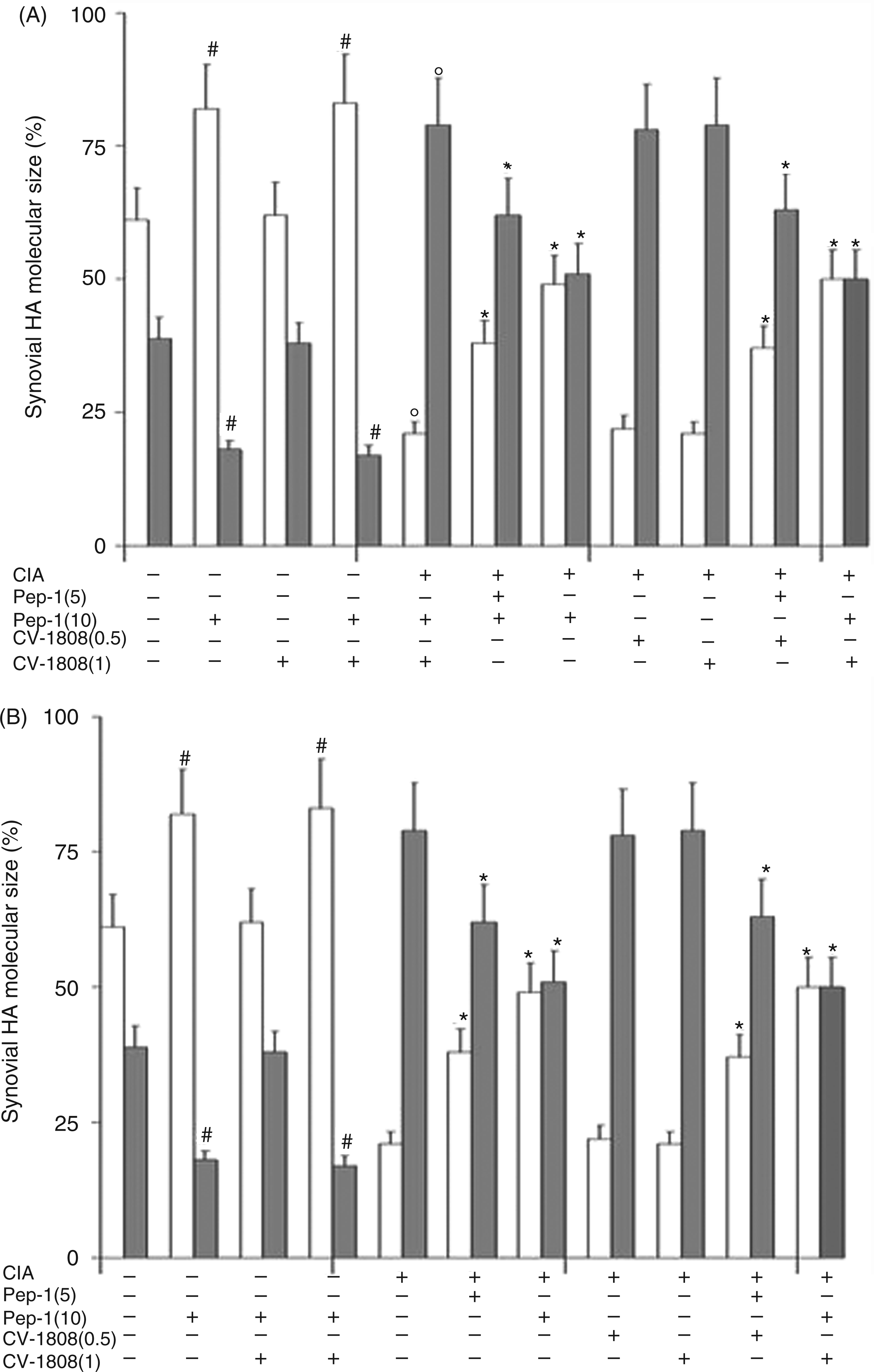
Figure 5 B shows the evaluation of HA molecular size in all groups of mice expressed as percentages. We considered HA at high molecular mass (HMMHA) when its size was >500 kDa, and at low molecular mass (LMMHA) when its size was <500 kDa. Control non-arthritic mice presented roughly comparable amounts of LMMHA and HMMHA, although the latter predominated (HMMHA = 61%; LMMHA = 39%). In normal mice, i.e. untreated with CIA, the administration of Pep-1 produced a marked increment in the percentage of HMMHA with respect to that of LMMHA (HMWHA = 82%; LMWHA = 18%) owing to the formation of the complex Pep-1-HA. Conversely, CIA treatment produced an inversion of the HMMHA/LMMHA ratio (HMWHA = 38%; LMWHA = 62%). The treatment of arthritic mice with Pep-1, at the two doses used, was able to limit this inversion of the HMMHA/LMMHA ratio by blocking small HA fragment formation.
Pep-1 control scramble treatment
In order to verify the specific effect of Pep-1, a control non-active peptide was also synthesized and administered to a separate group of mice. Figure 6 shows the comparison of the effects of the Pep-1 and control Pep-1 scramble on the incidence of arthritis, TLR4 and TNF-α mRNA expression. As reported, the Pep-1 control scramble did not exert any effect on these parameters, while Pep-1 was able to bind HA, thus confirming the results obtained.
Effect of Pep-1 control scramble treatment on normal and arthritic mice for incidence of arthritis (A), articular tissue TLR4 (B) and TNF-α mRNA expression (C). Values are the mean ± SD of no fewer than seven experiments and are expressed as a percentage (%) for the incidence of arthritis, and n-fold increase with respect to the controls for TLR4 and TNF-α *P < 0.001 vs collagen-induced arthritis (CIA).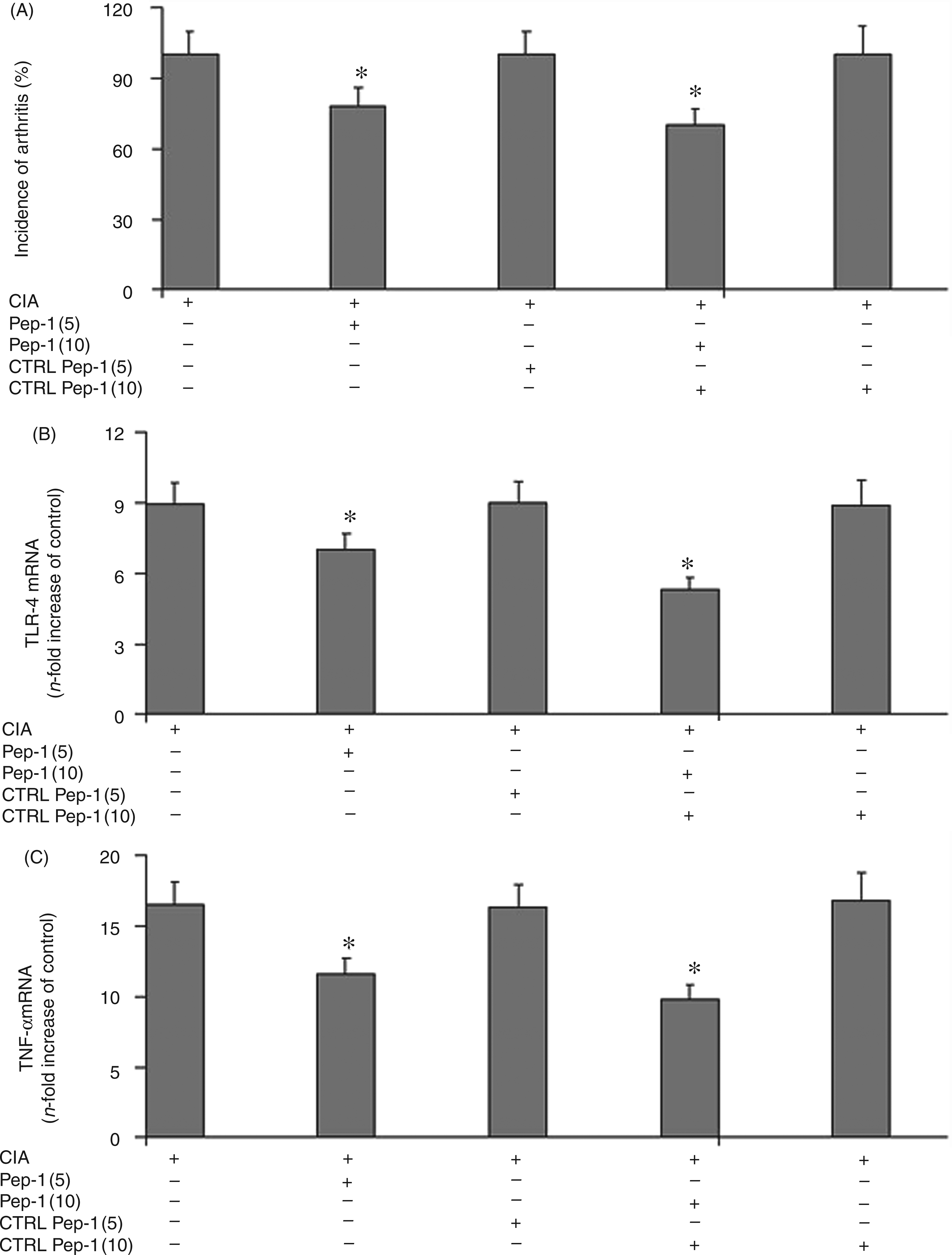
Discussion
Approximately 1% of the world population is affected by RA, with women being about three times more often affected than men. Infections, toxins and drugs have been implicated, as well as an interplay between genetic and hormonal background, environmental and immunological factors. 39 The main features of this crippling disease are symmetric inflammatory polyarthritis, chronic synovitis associated with synovial proliferation and secretion of high levels of pro-inflammatory mediators, including cytokines, matrix metalloproteases and growth factors leading to joint destruction. 40 The pro-inflammatory mediators released act on different cell populations, including synoviocytes, osteoclasts and chondrocytes by inducing the continuance of inflammation, and by eroding cartilage and bone. 41
Owing to its destructive course, RA has a considerable effect on both direct and indirect social costs. The progressive clarification of the pathogenesis of RA and subsequent biopharmaceutical discoveries has led to more effective drugs. Recently, the therapeutic approach has undergone a series of innovative inputs directed towards earlier and more aggressive treatment, particularly combined therapies that are able to block cytokine pathway and inflammatory receptor activation. This has profoundly changed the therapeutic scene and the strategy currently adopted to tackle RA is aimed at achieving clinical remission as quickly as possible and at slowing down the pathological signs. 42
There is now increasing evidence to suggest not only the involvement of HA fragments in stimulating TLR4 and CD44 inflammatory pathways, but also the anti-inflammatory response in RA possibly produced by the ADO pathway through its different receptors.4,16,43–45
In the present study, the treatment of arthritic mice with two different doses of Pep-1 and/or CV-1808 was able to reduce inflammation and cartilage erosion induced by CIA. This study examined the effects of the prolonged intraperitoneal administration of the two compounds on TLR4, TLR2, CD44 and A2AR modulation in arthritic mice. Our findings suggested all four receptors to be involved in this experimental disease, and that Pep-1 reduced TLR4, TLR2 and CD44 expression and the inflammatory mechanism primed by their activation, while CV-1808 stimulated the A2AR expression that limited inflammation by reducing NF-κB activation. Indeed, the data obtained show that both compounds, and particularly their combined administration, were able to reduce the NF-κB activation of a series of mediators of inflammation and cartilage degradation. The effects are well validated by the dose-dependent efficacy exerted by both compounds.
Our data show that treatment of arthritic mice with Pep-1 and/or CV-1808 significantly attenuated the incidence of arthritis, in the first instance by reducing all clinical signs of the disease. Furthermore, we found a significant increase in the activity of the TLR4, TLR2, CD44 and A2A receptors in CIA mice. Arthritic mice receiving Pep-1 showed a significant reduction in TLR4, TLR2 and CD44 expression, and mice receiving CV-1808 after CIA treatment showed further enhanced expression of the A2AR. The treatment of arthritic mice with Pep-1 plus CV-1808 showed a decrease in levels of TLR4, TLR2 and CD44 expression similar to those obtained when using arthritic mice plus Pep-1, while for A2AR, the increment in its expression was comparable to that obtained from treating the arthritic mice with CV-1808 alone.
The results for NF-kB showed that arthritis generated a marked increase in the activation of NF-κB. The treatment of arthritic mice with Pep-1 was able to decrease NF-κB activation significantly, as also seen for the administration of CV-1808, but to a lesser extent compared with Pep-1. The combined treatment of arthritic mice with both Pep-1 plus CV-1808 showed a further significant reduction in NF-κB activation in comparison to the results for cells treated with Pep-1 or CV-1808 alone.
The results obtained by assaying the inflammatory cytokines and the other inflammation intermediates confirmed the data reported above for the NF-κB factor. CIA treatment produced a marked increment in the activity of all considered inflammation mediators. Based on the NF-κB results, treatment with Pep-1 and/or CV-1808 had no effect on TNF-α, IL-1β, IL-17, MMP-13 and iNOS activity in non-arthritic mice. Conversely, the administration of Pep-1 to CIA mice significantly reduced the levels of all pro-inflammatory mediators. Similarly, CIA mice given CV-1808 showed a reduction in all inflammatory parameters considered. As found for NF-κB, the concomitant treatment of CIA mice with both Pep-1 and CV-1808 produced the greatest effect in decreasing the levels of all pro-inflammatory mediators.
The histological evaluation confirmed the biochemical parameters; in CIA mice we found severe knee joint inflammation and a loss of the colouration staining the cartilage layers, as well as the complete disruption of the original cartilage architecture. The treatment with Pep-1 and/or CV-1808 showed a gradual reduction in joint damage and cartilage erosion. The greatest protection was produced by concomitant treatment with Pep-1 and CV-1808. The quantification of cartilage thickness and surface erosion in the same sections also confirmed the above data.
HA levels were measured in the synovium of normal and arthritic mice in order to verify the inductive effects of CIA treatment on HA increment and further confirmed the results obtained, while the addition of Pep-1 and/or CV-1808 in arthritic mice did not affect HA concentrations compared to untreated CIA mice.
Of particular interest are the findings concerning HA molecular size. Normal mice presented roughly similar amounts of HA at low and at high molecular mass. Pep-1 treatment by itself produced a significant increment in the proportion of HMMHA with respect to that of LMMHA owing to the formation of the complex Pep-1-HA. Conversely, arthritis produced an inversion of the HMMHA/LMMHA ratio due to native HA degradation. The administration of Pep-1 to CIA mice was able to limit free HA at low molecular mass thus reducing its inflammatory effects, as demonstrated by the reduction in the percentage of LMMHA and the increase in the percentage of HMMHA due to the formation of the Pep-1-HA complex. Indeed, the larger molecule obtained by binding Pep-1 to LMMHA may have had slower mobility during electrophoresis migration in the gel and, consequently, this proportion was included together with HMMHA. Therefore, the increase in LMMHA was correlated positively with the corresponding CIA treatment. Hence, the change in the HA ratio, and particularly the reduction of LMMHA exerted by Pep-1, can be linked to the reduction in all inflammatory parameters and thus in the limitation of the propagation of inflammation.
The results presented here underline that, as degraded HA is able to interact with a wide range of biological molecules, the activation of NF-κB during inflammation needs to be taken into account in order to better understand the mechanism of inflammation. Similarly, the anti-inflammatory mechanism exerted by ADO needs to be better elucidated in order to steer pharmacological strategies in this direction. The intricate and complex inflammation mechanism seems to be due to a delicate equilibrium in which there co-exist pro-inflammatory pathways induced by agents such as HA oligosaccharides derived from native HA degradation during inflammation, together with anti-inflammatory pathways induced by molecules such as the extracellular endogenous ADO released during inflammatory diseases. The present in vivo results confirm our previous in vitro study on articular chondrocytes 30 and further support the evidence that the modulation of these pathways could be a useful tool for modulating inflammation in arthritis. Therefore, we believe that by acting on pathways degrading HA with the appropriate tools, together with the use of other opportune and specific tools acting on ADO pathways, the combined action would enable the intricate mechanism of inflammation to be clarified and better understood. At the same time, this would allow new anti-inflammatory strategies to be developed in order to tackle inflammation more effectively than the currently available therapeutic drugs.
However, further studies are needed to verify and fully confirm this complex mechanism.
Footnotes
Funding
This study was, in part, supported by a grant PRIN (Programs of Scientific Research of Relevant National Interest) (COFIN 2009) 20 094C2H2M_003 of the MIUR, Italy.
Conflict of interest
The authors declare that there is no conflict of interest