
Abstract
Signaling through MyD88, an adaptor utilized by all TLRs except TLR3, is pro-atherogenic; however, it is unknown whether signaling through TIR-domain-containing adaptor-inducing interferon-β (TRIF), an adaptor used only by TLRs 3 and 4, is relevant to atherosclerosis. We determined that the TRIFLps2 lack-of-function mutation was atheroprotective in hyperlipidemic low density lipoprotein (LDL) receptor knockout (LDLr−/−) mice. LDLr−/− mice were crossed with either TRIFLps2 or TLR3 knockout mice. After feeding an atherogenic diet for 10–15 wks, atherosclerotic lesions in the heart sinus and aorta were quantitated. LDLr−/− mice with TRIFLps2 were significantly protected from atherosclerosis. TRIFLps2 led to a reduction in cytokines secreted from peritoneal macrophages (Mϕ) in response to hyperlipidemia. Moreover, heart sinus valves from hyperlipidemic LDLr−/−TRIFLps2 mice had significantly fewer lesional Mϕ. However, LDLr−/− mice deficient in TLR3 showed some enhancement of disease. Collectively, these data suggest that hyperlipidemia resulting in endogenous activation of the TRIF signaling pathway from TLR4 leads to pro-atherogenic events.
Introduction
Complications from atherosclerosis are a major contributor to cardiovascular disease, the major cause of morbidity in Western society. 1 Atherosclerosis is both a lipid metabolism disorder and a chronic inflammatory disease. Because TLRs play critical roles in activating inflammatory pathways, they provide a mechanistic link between inflammation and atherosclerosis. For example, TLR2 or TLR4 activate pro-inflammatory signaling pathways required for mounting a homeostatic response to infection or sterile injury. However, inappropriate or prolonged activation of these TLRs leads to chronic inflammatory diseases, such as atherosclerosis.2,3
There is mounting evidence that certain TLRs are pro-atherogenic, although the mechanisms are unclear. Expression of TLR4 message and protein is observed in atherosclerotic plaques of humans.4,5 Administration of endotoxin, a mixture of exogenous TLR4 and TLR2 agonists from Gram-negative bacteria, 6 increases atherosclerosis in rabbits. 7 Similarly, activation of TLR2 by Pam3CSK4 promotes atherosclerosis in mice. 8 Experimental deficiency of TLR4 in apoE−/− mice, of TLR2 in LDLr−/− mice or mutation of TLR4 (as in C3H/HeJ mice), leads to less disease.8–11
Most cell types involved in atherosclerosis express TLRs 2, 3 and 4, as well as MyD88 and TIR-domain-containing adaptor-inducing interferon-β (TRIF), including at least endothelial cells (ECs), smooth muscle cells and mcrophages (Mϕ).5,12–14 Pro-inflammatory signaling initiated by TLR activation involves two intracellular signaling adaptors, MyD88 and TRIF. Most TLRs, including TLR2 and TLR4, signal through MyD88. TLR3 signals exclusively through TRIF. Only TLR4 signals through both MyD88 and TRIF. Signaling through MyD88 leads to NF-κB and MAPK activation, resulting in early up-regulation of inflammatory cytokines, such as TNF-α, whereas TRIF signaling, in addition to NF-κB activation, also activates a subset of type 1 IFN response genes, including IFN-β, regulated upon activation normal T cell-expressed and secreted (RANTES), and interferon inducible protein-10
TLRs are activated by both exogenous and endogenous ligands, both of which may play a role in atherosclerosis. Whereas exogenous agonists arise from bacteria or viruses, endogenous agonists arise from host molecules produced as a result of cell death 17 or host pathology.18–21 Hyperlipidemia results in an increase in several candidate endogenous ligands for TLRs generated from host molecules. Candidate endogenous ligands for TLR4 include: heparin sulfate fragments, HMGB1, hyaluronic acid, biglycan, minimally modified low density lipoprotein (mmLDL) and oxidized LDL (oxLDL).22–25 Hyperlipidemia provides a pro-atherogenic setting in which TLR may be activated, suggesting a mechanism for TLR activation in atherosclerosis by endogenous agonists.
To explore the role of TRIF in atherosclerosis, we studied LDL receptor knockout (LDLr−/−) mice expressing the inactive LPS2 form of the TRIF gene. 15 These mice are functionally TRIF deficient owing to a point mutation in the carboxyl terminus of the TRIF gene that results in a nonfunctional, truncated protein product. Peritoneal Mϕ isolated from these TRIF-deficient mice do not respond to exogenous TLR3 agonists and have a muted response to TLR4 agonists; however, signaling through the other TLRs, which solely depends on the MyD88 adaptor, is intact. 15 These mice are designated TRIFLPS2/LPS2. In this article, we show that this TRIF mutation was atheroprotective in hyperlipidemic LDLr−/− mice. We also tested whether mice fed a high fat diet and deficient in both LDLr and TLR3 were protected from atherosclerosis. The effect of TLR3 deficiency was weak, but, on balance, TLR3 deficiency led to more disease.
Materials and methods
Animals
All mice were of a C57BL/6 background. LDLr−/− mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA) and bred in-house. TRIF mutant (TRIFLPS2/LPS2) mice, which were generated by ethylnitroso urea mutagenesis of C57BL/6 mice, were a kind gift from Kasper Hoebe. 15 TLR3 knockout (TLR3−/−) mice on a C57BL/6 background were obtained from Richard Flavell. 26 Double-mutant LDLr−/−TRIFLPS2/LPS2 and LDLr−/−TLR3−/− mice were generated in house. Offspring displayed an apparently normal phenotype and were healthy and fertile. LDLr−/− genotyping was confirmed by PCR, as described. 27 The LPS2 point mutation in the TRIF gene was confirmed by sequencing, as described. 15 TLR3 deficiency was confirmed in LDLr−/−TLR3−/− mice by assaying for their inability to increase plasma IL-12/IL-23p40 in response to the TLR3 agonist, poly(I:C) (see below). Mice were weaned at 4 wks and given ad libitum access to standard mouse chow (7019; Harlan Teklad, Placentia, CA, USA). Age- and sex-matched cohorts were fed a high-fat diet (HFD) ad libitum at 8–10 wks of age. The diet contained 1.25% cholesterol and 15.8% fat (with no added cholate; 94059; Harlan Teklad). Blood samples were collected from fasted animals by retro-orbital puncture using heparin-coated capillary tubes. All mice were housed 4 per cage in autoclaved, filter-top cages with autoclaved water and kept on a 12-h light/dark cycle. All animal procedures were done in accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals and The Institutional Animal Care and Use Committee.
Plasma measurements/ELISAs
Blood samples were centrifuged and plasma was collected and stored at −80°C until use. Enzymatic measurement of total plasma cholesterol was performed using a colorimetric kit (Thermo Electron Corp., Waltham, MA, USA). Plasma serum amyloid A (SAA) (BioSource, San Diego, CA, USA) and IL-12/IL-23p40 (R&D Systems, Minneapolis, CA, USA) were measured by ELISA. For phenotypic assessment of TLR3 deficiency, mice were injected intraperitoneally (i.p.) with 100 μl of either 100 ng Di[3-deoxy-
Assessment of atherosclerosis
After 10 to 15 wks of consuming a HFD, mice were euthanized, perfused with PBS, fixed with formal-sucrose (4% paraformaldehyde, 5% sucrose in PBS, pH 7.4), and the aortae and hearts dissected. Atherosclerosis was assessed by measuring the en face surface area of lesions across the length of the aorta, as well as the mean lesion volume within the heart sinus valves, as described. 28 For en face lesion analysis, aortae were stained with Sudan IV and photographed. The photographs were digitized and total aortic areas and lesion areas calculated using Adobe Photoshop version 7.0 and NIH Scion Image software. The results are reported as a percentage of the total aortic area that contained lesions.
For heart sinus valve lesion analysis, lesion volume was estimated across a fixed distance of the aortic sinus. This method was used instead of lesion cross-section area analysis because the volume analysis is a more accurate method for estimating aortic sinus atherosclerosis in mice. 28 Frozen hearts were sectioned on a Leica cryostat, sections were stained with Oil Red O, counterstained with Gill hematoxylin 1 (Fisher Scientific International, Placentia, CA, USA), photographed and digitized for lesion analysis. Scoring of valve lesion areas was performed at 4 sites separated by 140 µm for each of the 3 valve cusps individually. Lesion areas found only within the valve cusp were measured. Lesion volume was calculated from an integration of the four cross-sectional areas.
Luminex assay for inflammatory cytokines
Unelicited, resident peritoneal Mϕ were harvested from mice fed HFD for 6 wks and cultured overnight at identical densities (2 × 106 cells/ml) in 6-well dishes (Corning 3516; Corning, NY, USA). 0.5 ml of medium (RPMI, 10% FBS) was added to each well. Cell supernatants were collected 24 h later. Cytokines and chemokines from cell supernatants were quantified using a Bioplex 200 Suspension Array and Procarta assay kits (Panomics/Affymetrix) as per the manufacturer’s instructions.
Immunofluorescence of heart sections
Serial heart sections adjacent to selected sections that represented the mean of the volumetric analysis for lesion burden were chosen for immunofluorescent staining. Sections were stained with 1:200 rabbit anti-mouse CD68 IgG (Serotec, Raleigh, NC, USA) and 1:800 DAPI nuclear stain (Invitrogen, Carlsbad, CA, USA). Alexa-Fluor 488 conjugated to goat anti-rabbit IgG (Invitrogen) was applied at 1:500 to visualize the anti-CD68 primary Ab. Slides were examined by confocal microscopy. ImageJ and ImagePro Plus 7.0 software was employed to quantitate CD68-positive staining area and the number of CD68-positive cells that co-localized with DAPI. All quantitations were normalized to the total area of three heart sinus valves per heart section.
Assessment of necrosis in heart lesions
Sections were stained with Harris hematoxylin and eosin (H&E), and images were viewed and captured with a Nikon Labophot 2 microscope equipped with an Olympus DP25 color digital camera attached to a computerized imaging system with Image-Pro-Plus software (version 3.0; Mediacybernetics Bethesda, MD, USA). Total intimal lesion area (from internal elastic lamina to the lumen) and acellular/anuclear areas (negative for hematoxylin-positive nuclei) per cross-section were quantified by taking the average of 3 sections spaced 10 µm apart, beginning at the base of the aortic root. The necrotic core was defined as a clear area that was H&E free. Boundary lines were drawn around these regions and the area measurements were obtained by image analysis software. A 3,000-µm2 threshold was implemented to avoid counting very small clear areas frequently observed in H&E-stained sections that likely do not represent substantial areas of necrosis.
Statistical analysis
All scatter plots are expressed with the mean ± standard error. Atherosclerosis data were analyzed by the unpaired Student’s t-test. A value of P < 0.05 was considered significant.
Results
Attenuated TLR-induced inflammation in normolipidemic LDLr−/−TRIFLPS2/LPS2 mice
We confirmed the phenotype of the chow-fed LDLr−/−TRIFLPS2/LPS2 mice by assessing levels of plasma inflammatory markers in response to the TLR4 or TLR3 exogenous agonists, Kdo2-LA or poly(I:C) respectively. The plasma levels of SAA 24 h after i.p. injection of 100 ng Kdo2-LA were significantly lower in LDLr−/−TRIFLPS2/LPS2 mice compared with LDLr−/− mice, P < 0.01 (Figure 1A). Because TLR3 also signals through TRIF, we examined the effects of exogenous TLR3 activation on IL-12/IL-23p40 production, activated in response to TLR3 agonist administration. Normolipidemic LDLr−/− or LDLr−/−TRIFLPS2/LPS2 mice were i.p. injected with 20 µg of poly(I:C) and plasma was collected 4 h later. As expected, poly(I:C) injection increased plasma levels of IL12/IL23p40 in LDLr−/− mice but had no effect on LDLr−/− TRIFLPS2/LPS2 mice (Figure 1B). Thus, the TRIF pathway was functionally deficient in the LDLr−/−TRIFLPS2/LPS2 mice that were used in these studies.
LDLr−/−TRIFLPS2/LPS2 mice plasma response to exogenous TLR4 or TLR3 agonists. Groups of chow-fed male LDLr−/− (n = 4) or LDLr−/−TRIFLPS2/LPS2 mice (n = 9) were i.p. injected with saline, 100 ng of Kdo2-LA (TLR4 agonist, A) or 20 µg poly(I:C) (TLR3 agonist, B). Plasma was collected at baseline, 1, 6, and 24 h post-injection for Kdo2-LA, and 4 h post-injection for poly(I:C). Total plasma SAA in responses to Kdo2-LA (A), or total plasma IL-12/IL-23p40 in responses to poly(I:C) (B), were measured by ELISA.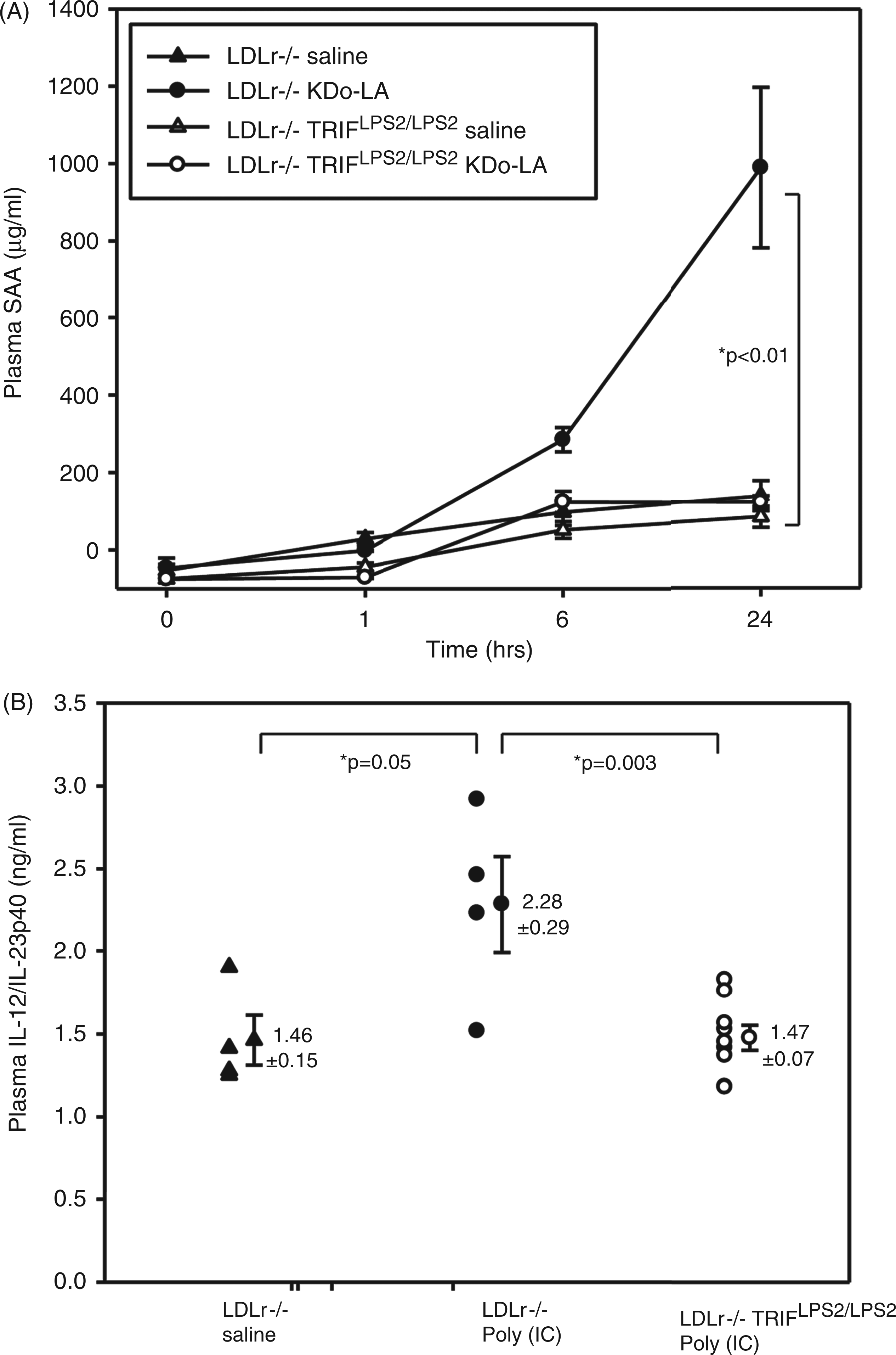
Reduced inflammation in peritoneal Mϕ from hyperlipidemic LDLr−/−TRIFLPS2/LPS2 mice
Because TRIF is involved in stimulating inflammatory pathways, we predicted inflammation might be suppressed in LDLr−/−TRIFLPS2/LPS2 mice in response to hyperlipidemia. Supernatants of unelicited, resident Mϕ harvested from LDLr−/− and LDLr−/−TRIFLPS2/LPS2 mice fed HFD for 6 wks were examined for inflammatory cytokines. No exogenous agonist was administered to these HFD-fed mice. As expected, hyperlipidemia was induced in these mice, as indicated by elevated levels of total plasma cholesterol, which were similar in both LDLr−/− and LDLr−/−TRIFLPS2/LPS2 mice (1073 ± 32 vs 1028 ± 64 mg/dl). All the cytokines assessed were markedly suppressed in macrophage supernatants from HFD-fed LDLr−/−TRIFLPS2/LPS2 mice (Figure 2). These results indicate that inflammation observed in response to HFD is attenuated in resident peritoneal Mϕ of LDLr−/−TRIFLPS2/LPS2 mice.
Cytokines secreted by resident peritoneal Mϕ obtained from HFD-fed mice. Cohorts of male LDLr−/− (n = 8) or LDLr−/−TRIFLPS2/LPS2 (n = 13) mice were fed the HFD for 6 wks. Peritoneal Mϕ were harvested, plated at identical densities and cultured overnight. Supernatants were collected and assayed for multiple cytokines by Luminex assay. Cytokines from both MyD88 (MCP-1, IL-6, IL-12p40, TNF-α, top panel) and TRIF (IL1-β, IP-10, RANTES, IFN-γ, bottom panel) pathways were examined.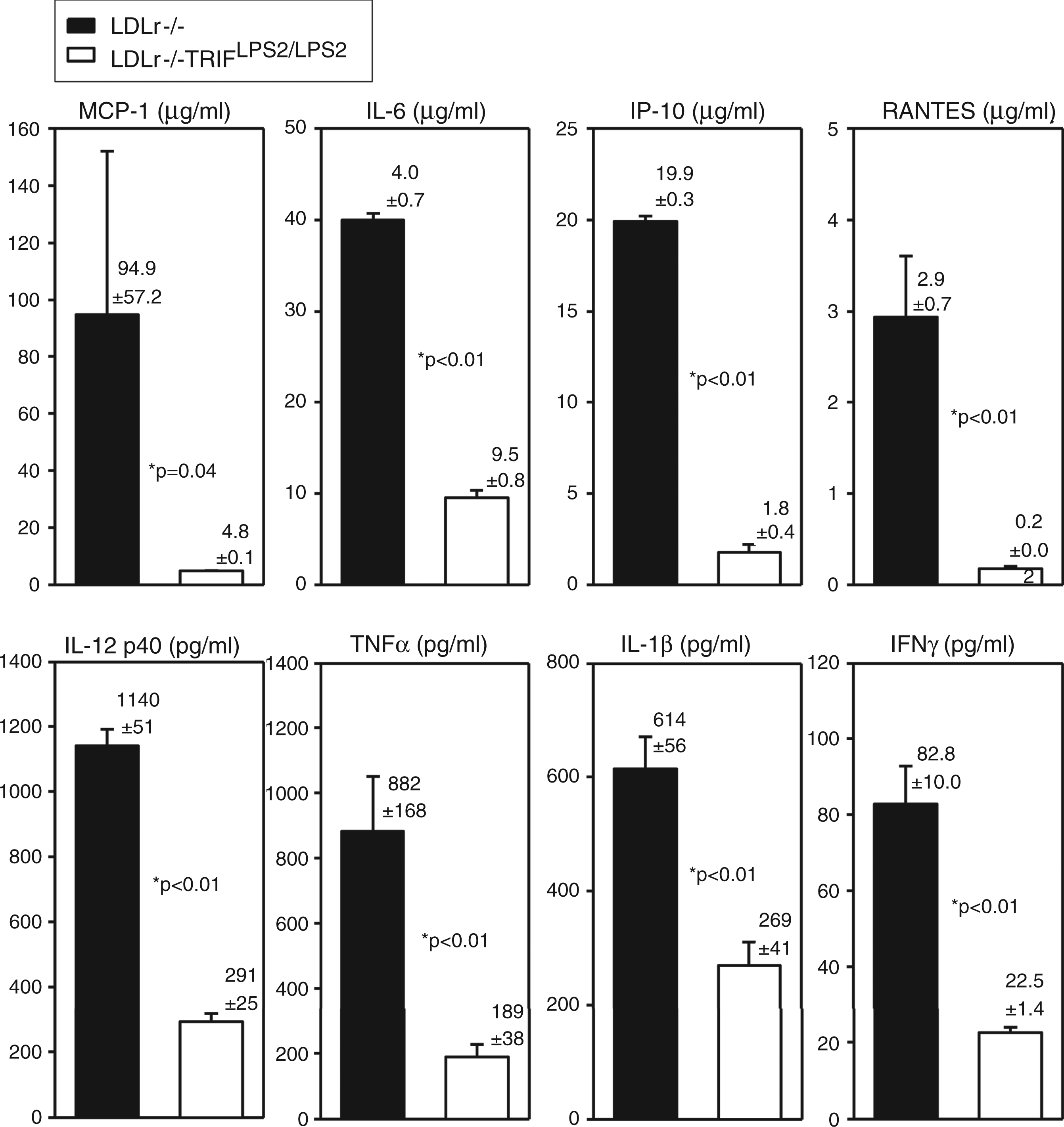
Reduced lesion burden in LDLr−/−TRIFLPS2/LPS2 mice
Because TRIF activates inflammatory pathways and chronic inflammation can be pro-atherogenic, we predicted that TRIF inactivation would result in reduced atherosclerosis. To determine the effects of TRIFLPS2 mutation on atherosclerosis, we assessed lesion burden in cohorts of age-matched LDLr−/− or LDLr−/−TRIFLPS2/LPS2 males fed a HFD for 12 or 15 wks.
Although masses of 9-wk-old, age-matched, male LDLr−/− mice were similar to LDLr−/−TRIFLPS2/LPS2 mice at baseline, the LDLr−/− mice gained mass faster than LDLr−/−TRIFLPS2/LPS2 mice, as evidenced by a significant mass difference at 6, 12 and 15 wks of HFD (Supplementary Table 1). However, there were no differences between total plasma cholesterol following consumption of the HFD, regardless of the time point examined (Supplementary Table 1).
There were significant reductions in lesion burden in aortae of LDLr−/−TRIFLPS2/LPS2 male mice at 15 wks (Figure 3B) and in the sinus valves of the hearts of LDLr−/−TRIFLPS2/LPS2 males at both 12 and 15 wks (Figure 3B). Careful examination of potential relationships between lesion burden in hearts or aortae and mass revealed no significant correlations at either time point (Supplementary Fig. 1).
Atherosclerosis in LDLr−/−TRIFLPS2/LPS2 mice. (A) Representative aortae (left) and heart sections (right) from each group. Lesion is stained with Sudan IV (aortae) or Oil Red O (hearts). (B) Quantitation of lesion burden. Aortic lesion area (left) was calculated as a percentage of total aortae surface area covered by lesion. Heart sinus valve lesion volume (right) was calculated from an integration of lesion area in cross sections from the proximal 500 µm of the sinus.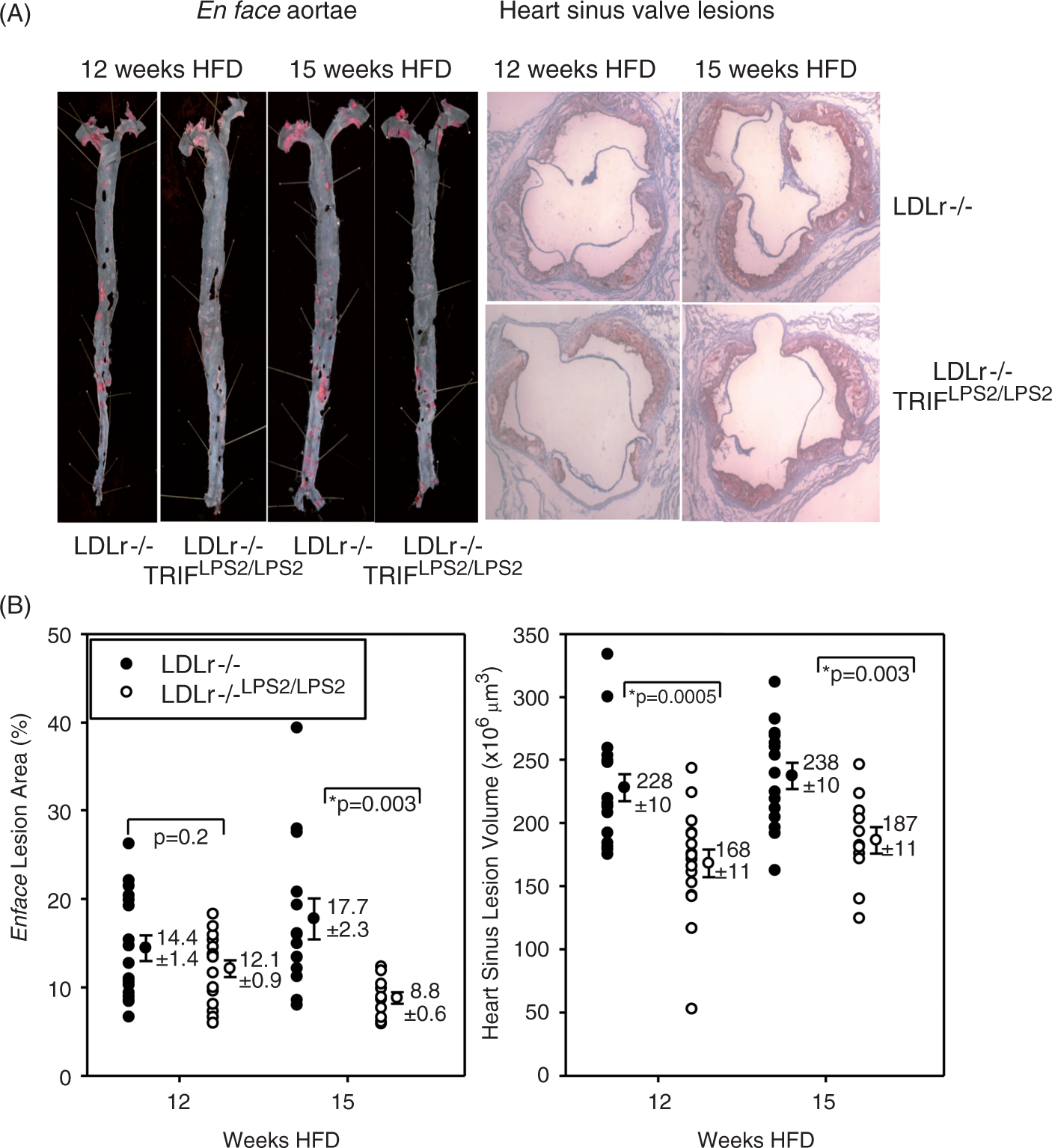
Decreased accumulation of Mϕ in heart sinus valves lesions in hyperlipidemic LDLr−/−TRIFLPS2/LPS2 mice
Next, we assessed the effect of the LPS2 mutation on Mϕ accumulation in the heart sinus valves of LDLr−/− mice fed HFD for 12 wks. The central section of the heart sinus analyzed for lesion burden was selected for analysis of Mϕ accumulation. The selected sections were stained with anti-mouse CD68 to identify Mϕ, and DAPI, a nuclear marker, to identify individual cells. A significant decrease in both CD68-positive staining area and CD68-positive staining cell number was observed in hearts of HFD-fed LDLr−/−TRIFLPS2/LPS2 mice compared with LDLr−/− mice (Figure 4). Notably, the LDLr−/− TRIFLPS2/LPS2 hearts also had significantly less lesion burden (Figure 3). Morphometric lesion analysis revealed no significant differences in percent necrotic core area between LDLr−/− and LDLr−/−TRIFLPS2/LPS2 hearts (Supplementary Figure 2), suggesting no overall changes in the levels of Mϕ cell death between LDLr−/− and LDLr−/−TRIFLPS2/LPS2 sinus valve lesions.
CD68-positive staining of Mϕs in heart sinus valves. Serial sections from hearts stained with Oil Red O (A, top panel) were selected for staining of Mϕ (anti-CD68) and nuclei (DAPI) (A, bottom panel). (B) CD68+ area (left) and number of CD68+ cells that co-localized with DAPI (right), normalized to the total valve area.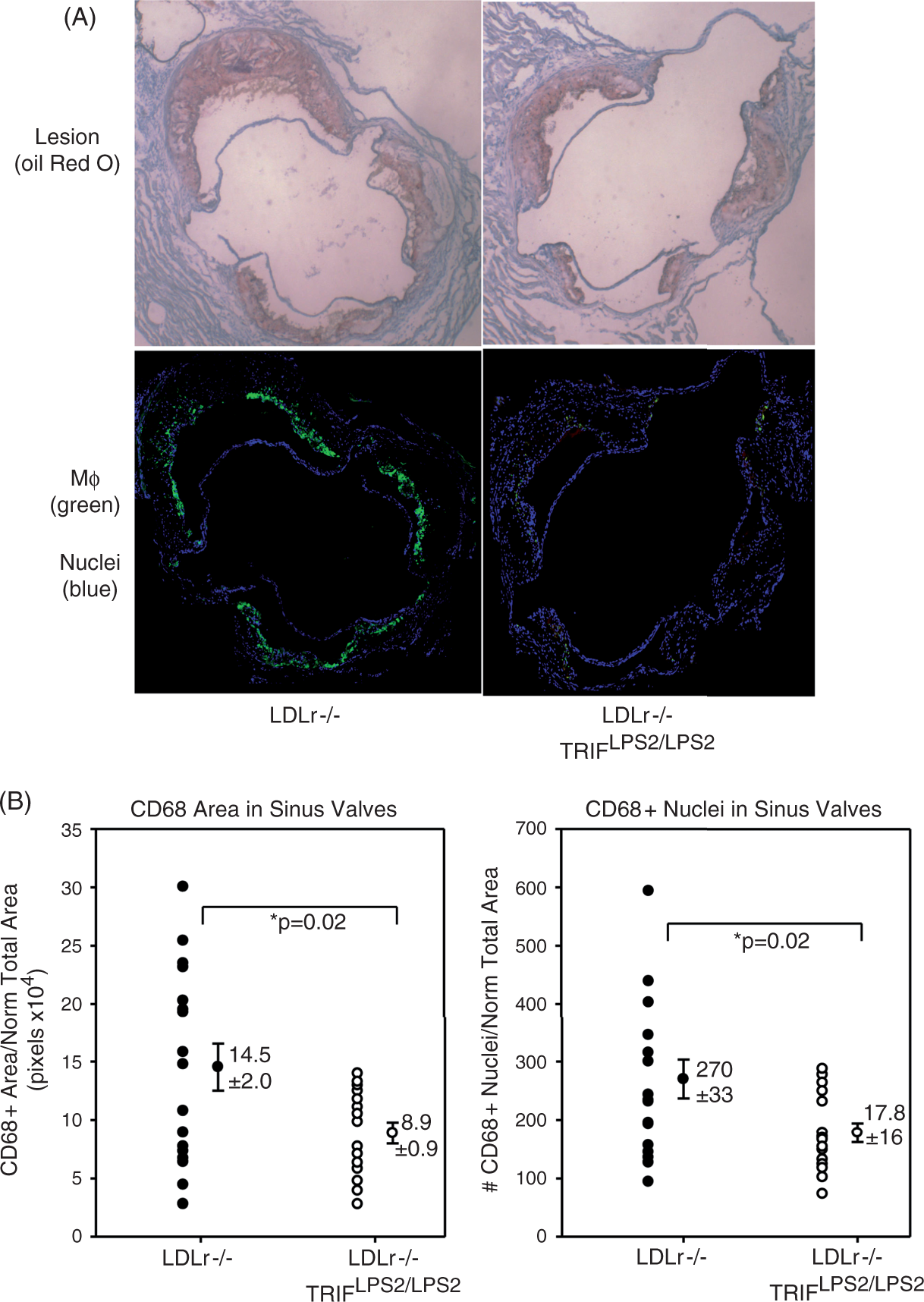
Atherosclerosis in LDLr−/−TLR3−/− mice
Because TRIF signals through TLR3, we determined if deletion of TLR3 in LDLr−/− mice was atheroprotective in response to hyperlipidemia. To phenotype the LDLr−/−TLR3−/− mice, plasma from cohorts of chow-fed, age-matched males was assayed for plasma IL-12/IL-23p40 levels in response to i.p. administration of 20 µg poly(I:C), an exogenous TLR3 agonist. LDLr−/− mice treated with poly(I:C) had increased plasma levels of IL-12/IL-23p40 compared with mice treated with saline (Supplementary Figure 3). As expected, LDLr−/−TLR3−/− mice treated with poly(I:C) did not exhibit increased plasma levels of IL-12/IL-23p40 compared with LDLr−/−TLR3−/− mice treated with saline.
These cohorts of mice were then fed the HFD beginning at 9 wks of age to examine differences in lesion burden. The mass of the two groups was similar at baseline and following 10 or 14 wks of HFD consumption (Supplementary Table 2). Total cholesterol levels of LDLr−/−TLR3−/− mice were similar to that of LDLr−/− mice at baseline and following HFD feeding, except for a reduction in LDLr−/−TLR3−/− mice at 10 wks (Supplementary Table 2). Compared with LDLr−/− mice, lesion burden was certainly not decreased in male LDLr−/−TLR3−/− mice fed a HFD for 10 or 14 wks in either heart sinus valves or aortae (Figure 5). Depending on the duration of HFD consumption, there was a slight, but significant, increase in lesion burden in the aortae of the LDLr−/− TLR3−/− mice at both time points, as well as an increase in lesion volume in the hearts of the mice.
Atherosclerosis in LDLr−/−TLR3−/− mice. Aortic lesion area (left) was calculated as a percent of total surface area covered by lesion (Sudan IV). Heart sinus valve lesion volume (right) was calculated from an integration of lesion area in cross sections from the proximal 500 µm of the sinus (Oil Red O staining).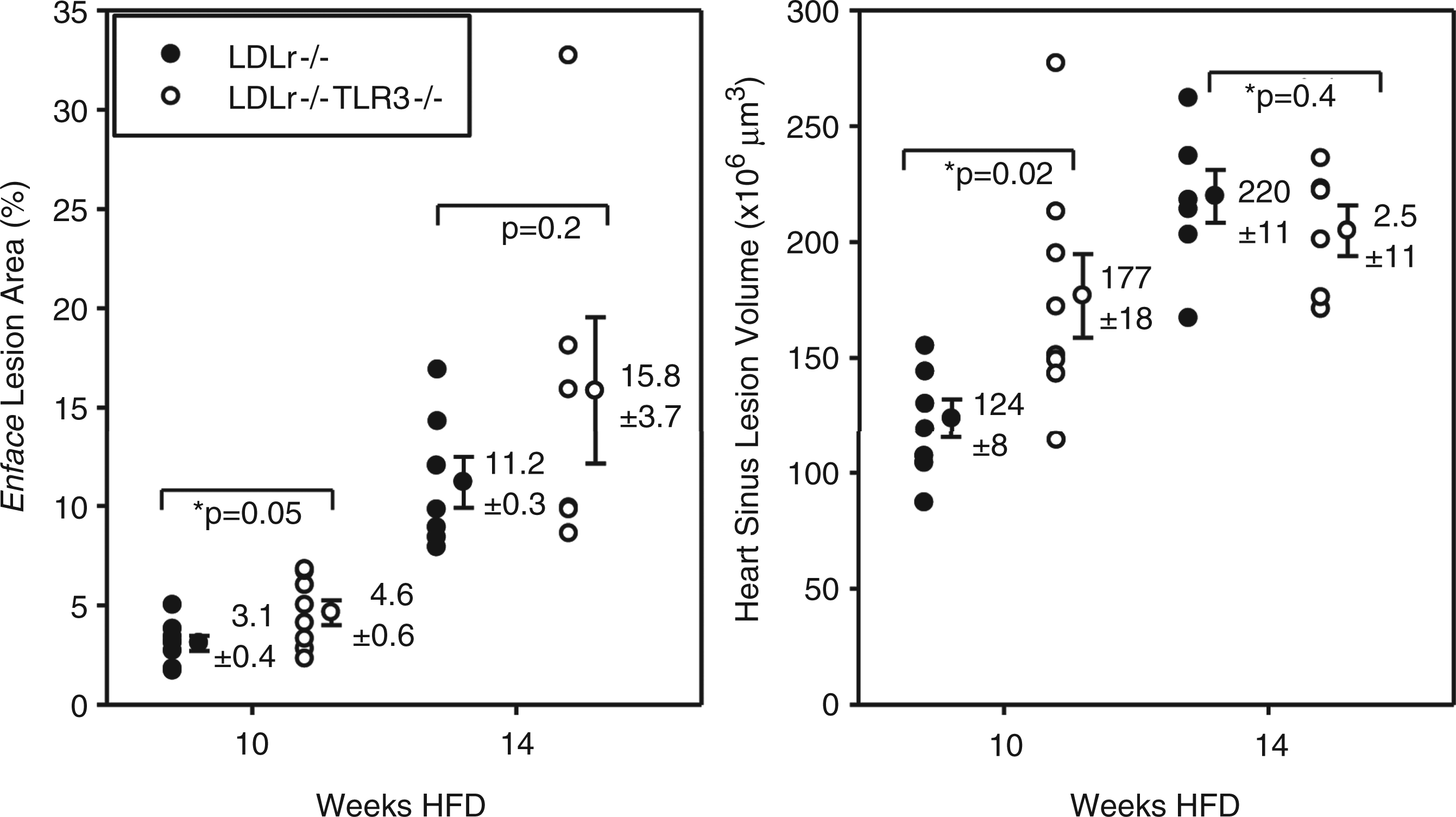
Discussion
TRIF deficiency diminishes atherosclerosis in the context of a high fat diet
Our principal conclusion from this work is that in the context of LDLr−/− mice consuming a high fat diet for 12–15 wks in the absence of an administered TLR agonist, a deficiency of TRIF diminishes aortic and heart sinus lesion development.
TRIF signaling and production of inflammatory cytokines
TRIF deficiency was atheroprotective without changes in total plasma cholesterol. Although there was reduced body mass in LDLr−/−TRIFLPS2/LPS2 mice upon consumption of the HFD, there was no correlation between body mass and lesion burden in either hearts or aortae, suggesting that the decrease in lesion burden was not dependent on mass differences (Supplementary Figure 1). However, levels of inflammatory cytokines secreted by unelicited, peritoneal Mϕ from hyperlipidemic LDLr−/−TRIFLPS2/LPS2 mice were significantly lower (Figure 2). The decreased inflammatory phenotype of LDLr−/−TRIFLPS2/LPS2 mice fed the HFD provides a plausible mechanism for the decrease we observed in lesion burden.
Cytokine expression was suppressed in LDLr−/−TRIFLPS2/LPS2 mice despite wild-type expression of MyD88. In addition to up-regulation of type I IFNs, the TRIF signaling pathway also activates the NF-κB pathway. 29 Therefore, it is likely that these NF-κB-dependent genes require both TRIF and MyD88 for full activation, at least in response to the endogenous ligands produced by hyperlipidemia. The requirement of both MyD88 and TRIF to mount a full inflammatory response may explain why SAA levels were inhibited in plasma from LDLr−/−TRIFLPS2/LPS2 mice injected with Kdo2-LA (Figure 1).
Deficiency of MyD88 also causes elimination of atherosclerosis.9,16 These reports show a greater effect of MyD88 deficiency than we observe, but the fact that their experiments were done using apoE−/− MyD88−/− mice cofounds a direct comparison.
TRIF deficiency decreased macrophage accumulation in heart sinus lesions
We observed a decrease in CD68+ Mϕ in the sinus lesions of hyperlipidemic, LDLr−/−TRIFLPS2/LPS2 mice. This correlated with a significant decrease in lesion burden, suggesting that the decrease in Mϕ in the heart contributes to reduced atherosclerosis. Decreased recruitment of monocytes to lesion-prone areas seems the likely explanation for the lesser number of Mϕ in the heart. Increased cell death could also lead to the reduction of Mϕ in heart sinus lesions. This possibility is less likely, however, because no differences in the amount of necrosis in heart lesions were observed between the LDLr−/− and LDLr−/− TRIFLPS2/LPS2 mice (Supplementary Figure 4).
TRIF promotes atherosclerosis via a TLR3-independent mechanism
Collectively, our data show that in HFD-fed, LDLr−/− mice, signaling through TRIF by endogenous agonists are pro-atherogenic. As far as is known, TRIF signaling is activated only in response to TLR4 and TLR3 ligands. Because we found that TLR3 deletion did not reduce lesion burden, we conclude that TRIF signaling through another receptor, most likely TLR4, mediates TRIF’s atherogenic activity in hyperlipidemic LDLr−/− mice.
Conclusion
Because atherosclerosis is a chronic inflammatory disease of the vasculature, defining the pathways by which that inflammation is initiated is important for a basic understanding of the disease, as well as future strategies that might ameliorate the condition. In this work we have focused on the role of a particular inflammatory pathway initiated by activation of TLR4 and TLR3—specifically, the TRIF pathway. Using mice deficient in TRIF, we observed a diminution of atherosclerotic disease in the disease model using mice also deficient in the LDLr fed a HFD. Using the same model we also found that mice deficient in TLR3 did not have less disease; in fact, they showed a slight increase in disease. Thus, we conclude that signaling initiated by TLR4 transmitted intracellularly through TRIF promotes pro-atherosclerotic inflammatory events. Whether it is possible to ameliorate atherosclerosis in mice or humans through manipulation of this pathway cannot be tested inasmuch as there are no drugs, as yet, which target this specific pathway.
While there is no other study of TRIF deficiency in atherosclerosis, the role of TLR3 has been reported in two publications, with variable approaches and variable results. Cole et al. studied ApoE−/− mice fed a standard chow diet. 30 They observed that TLR3 deficiency led to diminished lesions in the aortic sinus. They also observed that administration of the TLR3 agonist poly(I:C) diminished neointimal thickening in a TLR3 dependent manner. Zimmer et al. also studied ApoE−/− mice, but fed them an atherogenic Western diet containing 21% fat, 19.5% casein and 1.5% cholesterol. 31 They observed that administration of poly(I:C) enhanced aortic sinus lesion formation. They also observed a number of pathologic effects of poly(I:C) on endothelial cell function that were dependent on intact TLR3 function. We tentatively hypothesize that the differences in results observed by Cole et al., Zimmer et al. and ourselves are probably attributable to the difference in diets fed, as well as the strains of mice used. Further work will be needed to identify the relevant variables and to understand the mechanisms involved.
Footnotes
Funding
This study was supported by Fellowship AHA 0825013F (to M.R. Richards), NIH grant HL088093 (to L.K. Curtiss), CIHR Fellowship-FAH (to C. Woo), and NIH grants HL087123 and HL075662 (to I. Tabas).
Acknowledgements
The authors wish to thank Karen McKeon for technical assistance, Kasper Hoebe for the TRIF-deficient mice, and Anna Meyers and Barbara Parker for administrative assistance. This is the Scripps Research Institute manuscript no. 20466 (Department of Immunology & Microbiology Science).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.