
Abstract
We have previously shown that intramuscular immunization with a recombinant fragment of murine histidyl-tRNA synthetase (HRS) in the absence of exogenous adjuvant generates Ag-specific, IgG class switched Abs a murine model of myositis. Markedly diminished IgG anti-HRS auto-Ab responses in TLR4 signaling-deficient C3H/HeJ mice indicate that TLR4 is required for auto-Ab formation and/or class switching in this system. Comparative time course assessment of HRS-immunized C3H/HeOuJ (wild type) and C3H/HeJ (TLR4 mutant) mice shows here that despite significant impairment of class switched IgG anti-HRS responses in TLR4-deficient C3H/HeJ mice, production of IgM anti-HRS auto-Abs is relatively preserved—suggesting that TLR4-mediated signals modulate IgG class switching rather than auto-Ab formation in this genetic background. In C57BL/6-derived knockout mice lacking either MyD88 (B6.MyD88−/−) or TRIF (B6.TRIF−/−) adaptor molecules, immunization studies indicate that TRIF exerts a dominant role in the generation of HRS-specific IgG auto-Abs. Complementing these analyses, in vitro stimulation of unfractionated, as well as T cell-depleted, C3H/HeOuJ splenocytes with recombinant murine HRS reveals that TLR4-mediated generation of class switched auto-Abs can occur independently of T cell help. Overall, these findings support a broader role for TLR4 in the breakdown of immune tolerance and development of autoimmunity.
Introduction
The idiopathic inflammatory myopathies (IIM) encompass a group of systemic autoimmune disorders in which the immune system inappropriately targets skeletal muscle, as well as extramuscular organ systems.1–3 Subsets of IIM are associated with various nuclear and cytosolic auto-Ags that not only correlate with specific clinical characteristics, but also play a direct role in disease pathogenesis.4,5 The relationship between myositis auto-Ags and the underlying disease process is evidenced by different Ag-induced mouse models of IIM—including several based on immunization with histidyl-tRNA synthetase (HRS, also known as Jo-1),6,7 the most commonly targeted auto-Ag in IIM and the closely-linked anti-synthetase syndrome (that consists of fever, ‘mechanic’s hands’, arthritis, Raynaud’s-type vasospasm, myositis and interstitial lung disease 8 ).
Although not clearly pathogenic in all cases, well-defined myositis-specific auto-Abs reflect immune targeting of Ags, such as HRS. Because HRS and other auto-Ags often represent ubiquitous proteins contributing to normal cellular processes involved in RNA transcription and subsequent protein translation, the emergence of specific auto-Abs clearly requires a breech in immune tolerance through various mechanisms that may involve alterations in Ag structure, expression and/or presentation. Determining the relative contribution of innate and adaptive immune responses to this breakdown of immune tolerance is therefore relevant to multiple autoimmune diseases in which auto-Abs mark clinical subsets and potentially contribute to disease pathogenesis.
In general, humoral immune responses dichotomize based on the involvement of T helper cells. While T-dependent Ab formation involves integrated signals from Ag presenting cells (such as dendritic cells), T cells and B cells via specific cytokines as well as CD40/CD40 ligand engagement, T-independent B cell responses are thought to bypass the requirement for CD40/CD40 ligand co-ligation through additional signals provided by intra- or extracellular TLRs.9–11
Most conventional paradigms of autoimmunity have emphasized adaptive immunity and T-dependent B cell responses. However, emerging models of autoimmune disorders, such as systemic lupus erythematosus, have suggested that innate immune responses and TLR-signaling play a significant role in the generation of pathogenic humoral immune responses. For example, Marshak-Rothstein’s landmark studies, later extended by other investigators, demonstrated that nucleic acid could provide dual signals via the B cell receptor (BCR) and intracellular TLRs such as TLR7 or TLR9.12–14 Although the extent to which observations from these in vitro experiments apply to in vivo scenarios remains undefined, the concept that TLR-derived signals contribute to humoral immune responses has been extended to other systems where pathogen-associated structural components (such as lipoproteins, LPS, hypomethylated CpG, RNA) can trigger a range of extracellular (TLR2, TLR4) or intracellular (TLR3, TLR7, TLR9) TLRs.9,10,15,16 While these systems have not definitively been shown to operate in human autoimmune disease, known auto-Ags, such as HRS, are able to bind nucleic acid and other accessible components of necrotic cells that may serve as endogenous TLR ligands. 17
To begin exploring these concepts and the potential role of innate immunity in autoimmune diseases such as IIM, we previously established a murine model of myositis based on intramuscular immunization with recombinant HRS in the absence of additional exogenous adjuvant. 7 Among the striking features of this model (in which muscle inflammation occurred across several MHC haplotypes in multiple strains and did not depend on TCR recognition of HRS) was the highly reproducible auto-Ab response that developed despite the lack of exogenous adjuvant. Moreover, although the myositis phenotype itself was quite robust in TLR4-deficient C3H/HeJ mice and therefore independent of TLR4 signaling, class-switched IgG autoAb formation was severely impaired in this strain. 7 The latter finding not only uncoupled the humoral and cellular immune response to HRS in this model, but also underscored a key role for TLR4 signal integration in the generation of anti-HRS auto-Abs.
Based on these preliminary observations regarding humoral immune responses in our model of HRS-induced myositis, we sought to define the contribution of TLR4 and its signaling components to auto-Ab formation and class switching. Through the use of C3H/HeJ TLR4 signaling-deficient mice 18 and other C57BL/6-derived knockout strains, we investigated the relative roles of various TLR4 signaling pathways in the development of IgG class switched anti-HRS Abs. While these studies demonstrated contributions for both MyD88- and TRIF-mediated signaling pathways initiated by TLR4 engagement, the results indicated that TRIF plays a dominant role in driving Ag-specific IgG class switched Ab responses. Complementing in vivo immunization experiments, in vitro studies demonstrated that B cells could respond to HRS and produce anti-HRS Abs in the absence of T cell help—strongly implicating a T cell-independent, B cell-intrinsic mechanism for auto-Ab formation in this murine model system.
Materials and methods
Mice
Eight-to-ten-week-old female mice of the following strains were used in immunization protocols approved by the University of Miami Institutional Animal Care and Use Committee: C3H/HeJ (functional TLR4−/−), C3H/HeOuJ [wild type (WT)], C57BL/6 (B6), B6.MyD88−/− (heterozygous human DR4 (+/−) and mouse Ia (+/−) MHC loci), and B6.TRIF−/−. All mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA) except for B6. MyD88−/− mice that were derived from B6.DR4 and B6.MyD88−/− mice maintained in the University of Miami animal facility.
Generation of recombinant Ags
As previously described, 6 the immunodominant amino terminal fragment of mHRS was produced as a maltose binding protein fusion protein (MA/MBP = aa 1-151 of mHRS linked to the carboxy terminal end of MBP) by first subcloning the appropriate full length mHRS sequence into the bacterial expression vector pMALc2 (New England Biolabs, Ipswich, MA, USA) and then introducing a premature stop codon after base pair 453 via in situ mutagenesis (Stratagene, La Jolla, CA, USA). Using a similar approach, we generated a series of additional MBP fusion proteins containing amino terminal subfragments of human HRS (hHRS) that were designated as HA5/MBP (aa 1–151), HA4/MBP (aa 1–120), HA3/MBP (aa 1–90), and HA2/MBP (aa 1–60). Bacterially-expressed fusion proteins were purified with amylose resin as per the manufacturer's protocol (New England Biolabs), dialyzed against PBS, filter sterilized and assessed for TLR4 signaling capacity [through in vitro stimulation of HEK293 cells transfected with murine TLR4 (InvivoGen, San Diego, CA, USA)] prior to intramuscular immunization of mice. Although Limulus amebocyte lysate testing revealed residual endotoxin-like activity in all purified protein preparations, the uniformity of relative TLR4 signaling capacity demonstrated by multiple independent lots of each protein construct ensured the reliability/reproducibility of reported results.
Immunization procedure
Experimental mice received intramuscular injections of soluble MA/MBP protein (4–5 mg/ml) administered to both hamstrings in a total volume of 100 µl (50 µl/side). At designated time points ranging from 4 to 14 d, mice underwent retro-orbital vein phlebotomy for subsequent isolation of serum. Terminal bleeding was performed at d 17, an established endpoint for the development of myositis in this model. 7
Determination of serum Ab levels
IgM and IgG isotype anti-MJo-1 Ab levels in the sera of mice immunized with MA/MBP were measured using standard solid phase ELISA according to the following protocol. Ninety six-well microtiter plates (Nunc, Rochester, NY, USA) were coated with recombinant mHRS (full length murine HRS, 1.0 µg/ml) in carbonate buffer (50 mM NaHCO3/Na2CO3, pH 9.6) and incubated overnight at 4°C. Plates were then washed 4 × with PBS containing 0.05% Tween-20. After blocking wells with PBS/Tween containing 1% BSA, diluted serum samples (1 : 1000) from immunized mice were added for 2 h. Following a 60 min incubation with HRP-conjugated secondary Abs recognizing IgG, IgG1, IgG2a, or IgM (0.04 mg/ml; Santa Cruz Biotechnology, Santa Cruz, CA, USA), enzymatic reactions were visualized using 3,3,5,5-tetramethylbenzidine (TMB) (Sigma-Aldrich, St Louis, MO, USA) and subsequently terminated with 1 N H2SO4. Color development was measured at 450 nm by a Wallac 1420 multilabel counter (PerkinElmer, Wellesley, MA, USA), and values were plotted as OD450 substrate Ag minus OD450 no Ag.
Assessment of in vitro anti-HRS Ab production
Splenocytes harvested from naïve, eight-wk-old C3H/HeJ or C3H/HeOuJ mice were subjected to osmotic RBC lysis, washed in PBS and labeled with anti-CD3e microbeads (Miltenyi Biotec, Boston, MA, USA) according to the manufacturer’s instructions. AutoMACS separation then yielded T cell-depleted splenocyte fractions (<1% Thy 1.2+/CD3+ T cells) for subsequent in vitro stimulation. Unfractionated versus T cell-depleted splenocytes were plated in 48 well plates at a density of 2 × 106 cells/ml (400 µl total volume/well) and stimulated with either no Ag or varying concentrations of MA/MBP (0.1–1.0 µg/ml) for 4–11 d. Culture supernatants obtained at designated time points were diluted 1 : 4 in PBS prior to ELISA-based assessment of IgM and IgG anti-HRS Ab levels using the protocol outlined for serum.
Statistical analysis
ELISA-based OD450 readings from individual mice were compared using the Mann-Whitney U-test, while group mean OD450 values were assessed using the unpaired, two sample t test. Significance for each type of analysis was based on a two-tailed P value <0.05.
Results
Loss of TLR4 signaling reduces HRS-induction of class switched autoAbs
Based on previous work implicating TLR4 signals in the production of IgG class switched Abs in our murine model of HRS-induced myositis,
7
we compared anti-HRS Ab titers in C3H/HeJ (TLR4 signaling-deficient) and C3H/HeOuJ (WT) mice following IM immunization with recombinant MA/MBP (immunodominant amino terminal fragment of murine HRS fused to MBP) in the absence of additional exogenous adjuvant. As shown in the dilutional analysis of Figure 1, TLR4 signaling blockade in C3H/HeJ mice reduced the levels of IgG class switched anti-HRS Ab levels by more than a log order of magnitude at an established time point for the development of myositis (d 17). Stimulation assays involving TLR4-transfected HEK293 cells provided additional support for the role of this receptor in HRS-driven Ab responses, as the ability of various HRS fusion proteins to promote in vitro TLR4 signal induction correlated very directly with their capacity to generate Ag-specific class switched IgG Abs in vivo (Supplementary Figure 1). This apparent dependence on TLR4 and innate immune signaling was consistent with the rapid kinetics of class switched Ab production occurring as early as 4 d post-MA/MBP immunization in WT C3H/HeOuJ mice (Figure 2, lower panel).
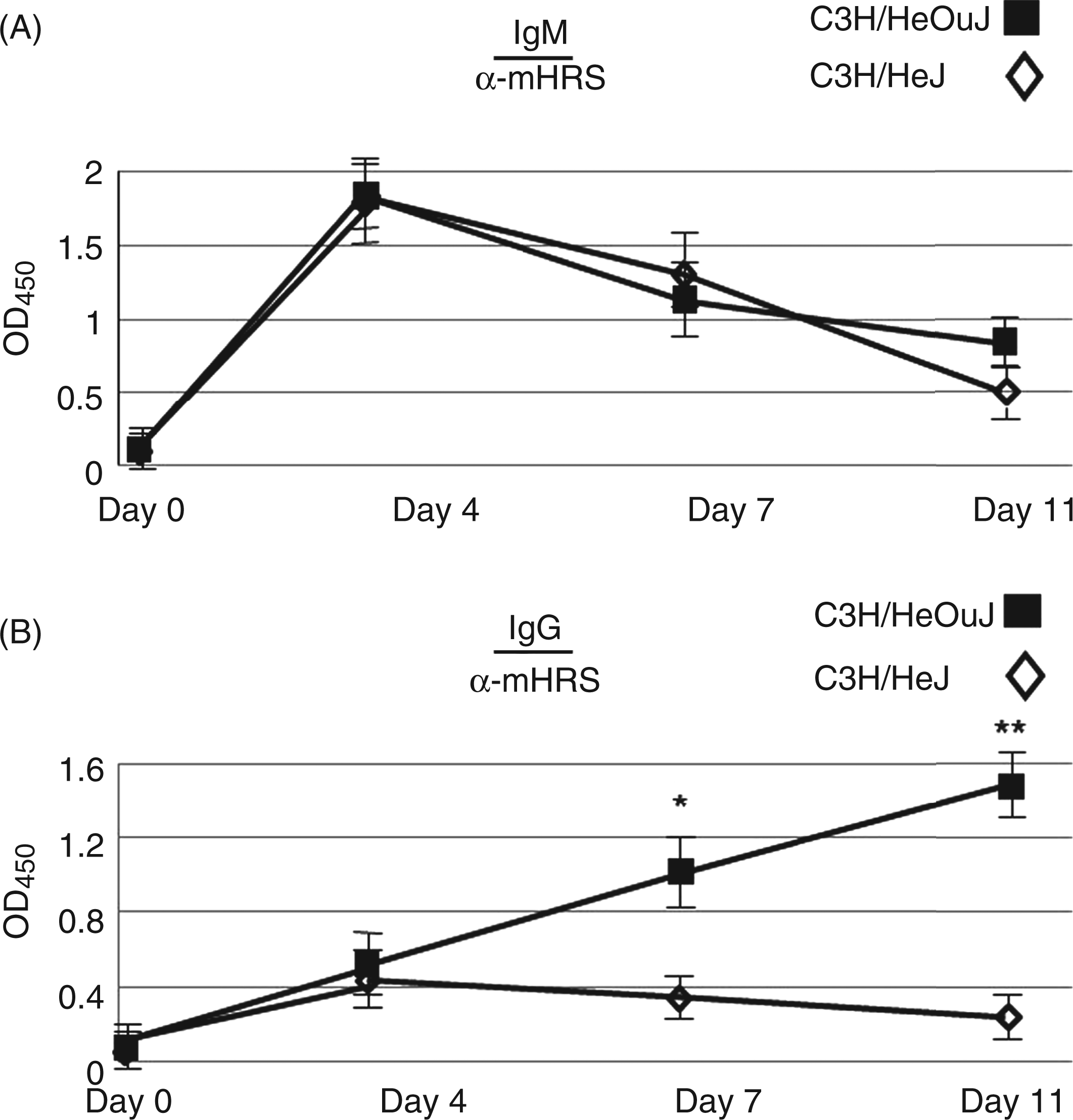
Relative production of IgG class switched Abs in C3H/HeJ versus C3H/HeOuJ mice immunized with recombinant murine HRS. Serial dilutions of sera obtained from mice 17 d post-IM immunization with MA/MBP were tested by ELISA (using anti-IgG secondary Ab) for recognition of recombinant full length murine HRS (mHRS, 1.0 µg/ml). The resulting curves depict adjusted OD450 values on the y-axis [OD450 mHRS - OD450 No Ag (no antigen)] and serum dilutions on the x-axis. Individual data points designate the mean OD450 value of three independent serum samples derived from C3H/HeOuJ (WT; squares) or C3H/HeJ (functionally deficient TLR4 signaling; open diamonds) mice, with error bars representing SEM. *P < 0.001; **P < 0.005 by two sample unpaired t test. Impaired TLR4 signaling negatively impacts IgG class switching in C3H/HeJ mice. In this graph demonstrating the time course of IgM (A) and IgG (B) anti-mHRS Ab production in Ce3H/HeJ (TLR4 signaling deficient) and C3H/HeOuJ (WT) mice, mean OD450 values representing ELISA-based recognition of mHRS (1 µg/ml) are plotted against time post-IM immunization with MA/MBP. Each data point designates the mean OD450 value (adjusted for No Ag background) ± SEM generated by sera (1 : 1000 dilution) obtained from 5 mice. *P < 0.005; **P < 0.0001 by two sample unpaired t test.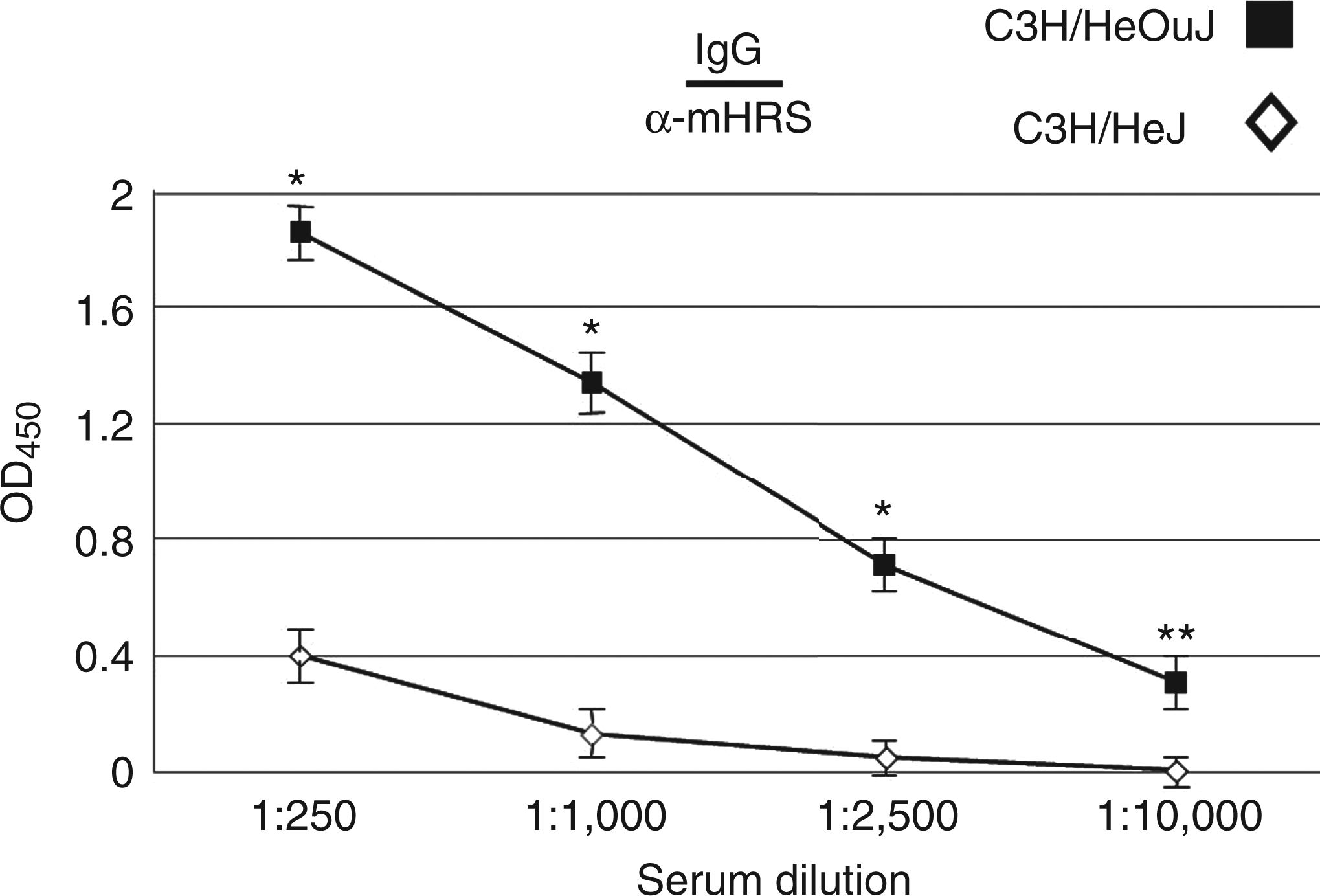
TLR4 signaling is required for Ab class switching, but not Ab formation, in response to HRS immunization
To better assess the relative contribution of TLR4 signaling to auto-Ab formation versus class switching, we measured anti-HRS IgM and IgG levels at various time points following MA/MBP immunization of C3H/HeJ and C3H/HeOuJ mice. As shown by the time course experiment depicted in Figure 2, the impairment of TLR4 signaling found in C3H/HeJ mice had no impact on development of IgM anti-HRS Abs. In contrast, C3H/HeJ mice failed to develop the significant IgG anti-HRS Ab responses evident in TLR4-intact C3H/HeOuJ mice at successive time points, highlighting a block in class switching associated with impaired TLR4 signaling.
TRIF-dependent signaling pathways play a critical role in the generation of class switched anti-HRS auto-Abs
Because MyD88 and TRIF regulate the two primary signaling pathways triggered by TLR4 engagement,
19
we further explored the contribution of these adaptor proteins to TLR4-directed anti-HRS auto-Ab formation through MA/MBP immunization of B6.MyD88−/− and B6.TRIF−/− mice. MyD88 was not absolutely required for the production of IgG anti-HRS Abs, as demonstrated by the near WT levels of IgG anti-HRS Abs that developed in B6.MyD88−/− mice relative to C57BL/6 (B6) mice (Figure 3). In contrast, immunization of B6.TRIF−/− mice resulted in significantly lower titers of class switched IgG anti-HRS Abs compared to B6.MyD88−/− and C57BL/6 (B6) mice (Figure 3), suggesting that TLR4 signals directed through TRIF play a critical role in the development of class switched anti-HRS IgG auto-Abs following IM immunization of B6-derived mice with MA/MBP.
Abrogation of TRIF signaling impairs TLR-mediated induction of IgG class switched Abs in response to IM immunization with recombinant murine HRS. The dot plot shows adjusted OD450 values resulting from ELISA analysis of serum samples obtained 17 d following immunization of C57BL/6 (B6), B6.TRIF−/−, or B6.MyD88−/− mice with recombinant MA/MBP. All sera were diluted 1 : 1000 and tested in wells containing no antigen or full length recombinant mHRS (1 µg/ml), permitting calculation of corrected OD450 values (OD450 mHRS-OD450 No Ag). Each data point represents serum anti-mHRS IgG Ab levels obtained from individual mice, while the horizontal bars indicate mean OD450 values for each strain. As stated in the ‘Materials and methods’ section, all mice were 8–10 wks old at the time of immunization, with the exception of B6.MyD88−/− mice that ranged up to 32 wks of age. P values are based on the Mann-Whitney U rank sum test.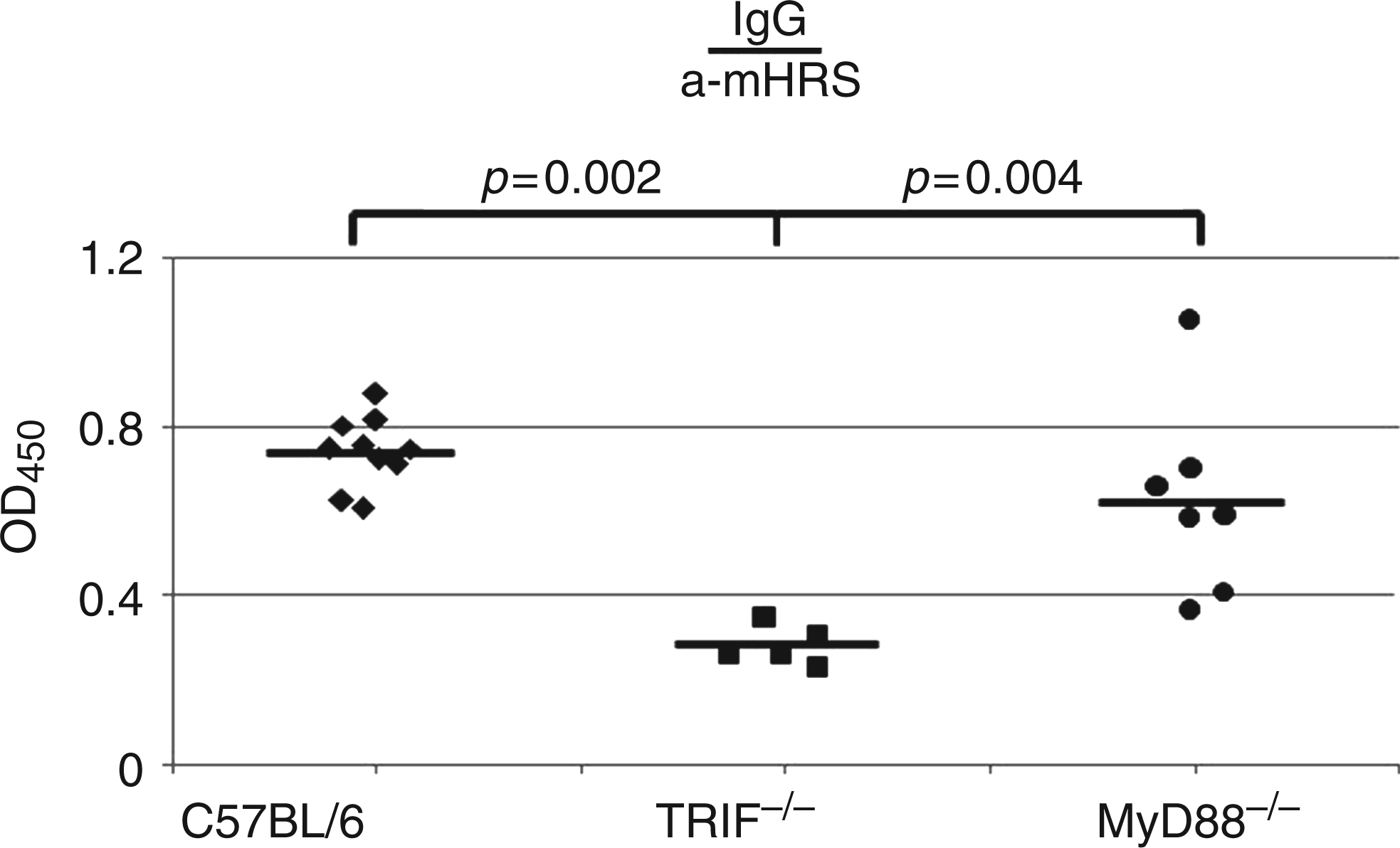
Evaluation of relative anti-HRS Ab titers in B6 and B6.TRIF−/− mice at various time points post-immunization clearly showed impairment of both IgM and IgG auto-Ab production in the absence of TRIF (Figure 4A). While this finding indicated a potential role for TRIF in auto-Ab formation, more detailed assessment of IgG anti-HRS Ab responses normalized by HRS-specific IgM levels revealed preferential impairment of IgG class switching at early time points in B6.TRIF−/− mice (Figure 4B). Coupled with the relative preservation of anti-HRS IgG Ab levels in B6.MyD88−/− mice immunized with MA/MBP (Figure 3), this analysis provided additional support for the predominant role of TRIF-directed signals in mediating HRS-induced auto-Ab class switching within the B6 genetic background.
Comparative analysis of anti-mHRS IgM and IgG Ab formation in the absence of TRIF-directed signaling. 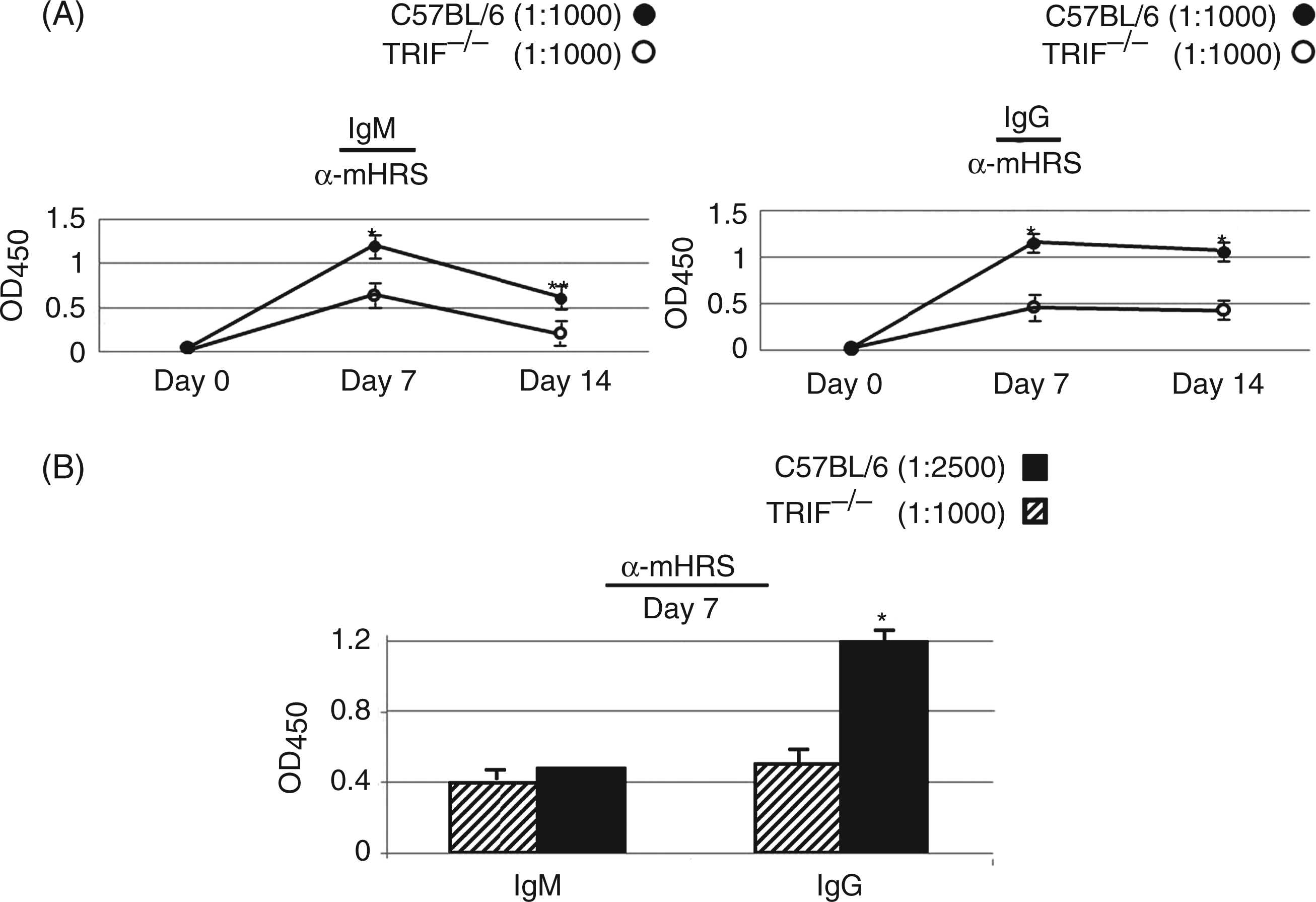
TRIF and MyD88 influence IgG isotype profile
Anti-HRS IgG isotype profile a
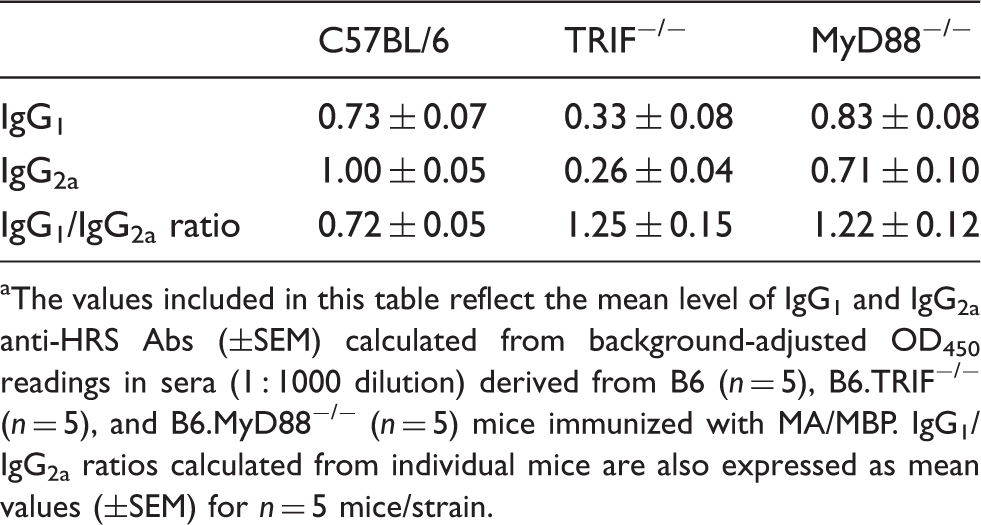
The values included in this table reflect the mean level of IgG1 and IgG2a anti-HRS Abs (±SEM) calculated from background-adjusted OD450 readings in sera (1 : 1000 dilution) derived from B6 (n = 5), B6.TRIF−/− (n = 5), and B6.MyD88−/− (n = 5) mice immunized with MA/MBP. IgG1/IgG2a ratios calculated from individual mice are also expressed as mean values (±SEM) for n = 5 mice/strain.
TLR4-directed anti-HRS Ab formation can occur independently of T cell help
The relatively rapid kinetics of class switched Ab production following IM immunization with soluble MA/MBP (see Figure 2) suggested an underlying innate immune mechanism bypassing the need for T cell help. We therefore established an in vitro tissue culture system that permitted assessment of cellular elements contributing to the processes of Ab formation and class switching. As shown in Figure 5, stimulation of naïve, unfractionated C3H/HeOuJ splenocytes with varying concentrations of MA/MBP ranging from 0.1 to 1.0 µg/ml promoted IgG anti-HRS Ab secretion into culture supernatants as early as d 4 (Figure 5A). Replicating findings from in vivo immunization studies, the magnitude of this class switched Ab response clearly exceeded that generated by HRS stimulation of TLR4 signaling-deficient C3H/HeJ splenocytes (Figure 5B). Of note, depletion of T cell subpopulations through magnetic cell separation exerted only a modest impact on the ability of MA/MBP to induce class switched anti-HRS Abs in C3H/HeOuJ-derived splenocyte cultures (Figure 5B), substantiating a T cell-independent component of this humoral immune response. At the same time, the failure to generate significant IgG anti-HRS Abs in either whole or T cell-depleted C3H/HeJ splenocyte cultures (Figure 5B and data not shown) clearly demonstrated the significance of intact TLR4 signaling pathways in this process.
In vitro production of IgG anti-mHRS Abs requires TLR4 signaling, but can occur in the absence of T cell help. Cultures of unfractionated versus T cell-depleted splenocytes derived from C3H/HeOuJ (WT) and C3H/HeJ (functional TLR4 deficiency) mice were stimulated with no Ag (No Ag) or various concentrations of recombinant MA/MBP (MA), as outlined in the ‘Materials and methods’ section. While panel (A) demonstrates levels of IgG anti-mHRS Abs produced at indicated time points following in vitro stimulation of unfractionated C3H/HeOuJ splenocytes with MA/MBP, panel (B) depicts relative anti-mHRS IgG secretion from unfractionated versus T cell-depleted C3H/HeOuJ and C3H/HeJ splenocytes after 5 d of in vitro stimulation. Data points indicate average IgG anti-mHRS Ab levels [based on adjusted OD450 values (OD450 mHRS-OD450 No Ag) from duplicate wells] in diluted (1 : 4) culture supernatants generated by cell fractions from designated strains. Each plot is representative of at least two independent experiments. Negligible error bars have been omitted for ease of interpretation.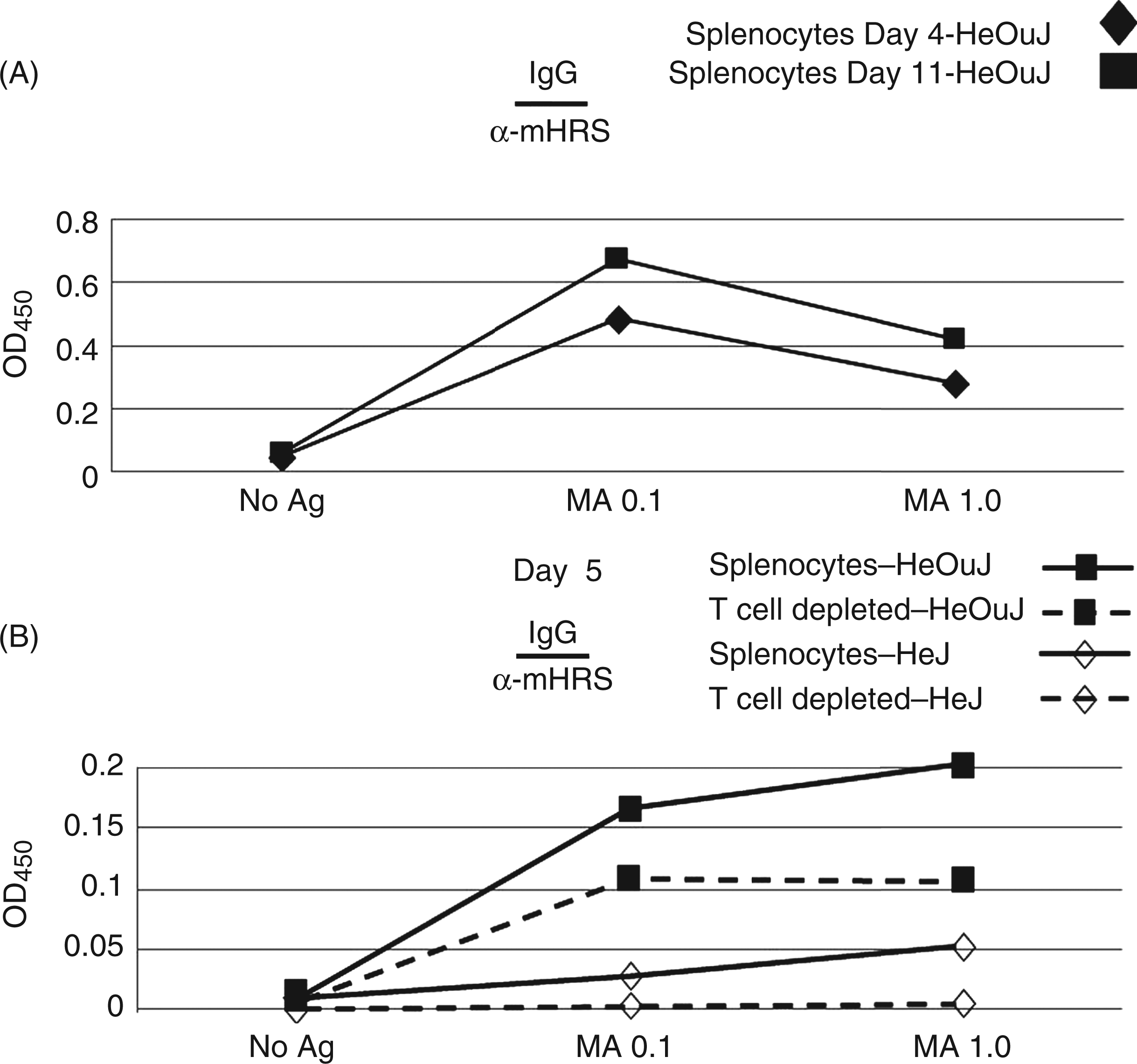
Discussion
As a model system exploring potential interactions between innate and humoral immune responses, these experiments demonstrate that TLR4 signaling plays a critical role in the development of IgG anti-HRS Abs following IM immunization with the recombinant HRS fragment, MA/MBP. More detailed analysis reveals that MyD88 is not absolutely required for anti-HRS Ab formation or class switching, though in the absence of this TLR adaptor protein, the IgG isotype profile is skewed in favor of IgG1. While TRIF knockout mice also demonstrate similar IgG isotype profiles, they clearly manifest a more global quantitative impairment in IgG class switching—indicating that TRIF is the major driver influencing the development of class switched Abs targeting HRS in our model system of autoimmune inflammatory myopathy. Parallel experiments in C3H/HeJ and C3H/HeOuJ mice confirm the importance of these pathways in generating class switched auto-Abs, as IgG anti-HRS levels are highly divergent (significantly reduced in the TLR4 signaling-deficient strain, C3H/HeJ) in the context of unimpaired IgM anti-HRS responses. In vitro studies comparing Ab production in MA/MBP-stimulated cultures of unfractionated and T cell-depleted splenocytes (derived from C3H/HeJ and C3H/HeOuJ mice) support these observations and further demonstrate that the TLR4-dependent process of anti-HRS Ab class switching can occur in the absence of T cell help. Viewed together, these experiments provide direct evidence that TLR4-associated adaptor proteins, particularly TRIF, control both qualitative and quantitative aspects of IgG class switching responsible for shaping the Ab repertoire induced by the auto-Ag, HRS.
After examining the role of TLR4 in the humoral immune response to bacterial pathogens, several investigators have noted similar uncoupling of MyD88- and TRIF-associated signaling pathways in the generation of protective Abs.20,21 However, these previous studies involving antimicrobial (rather than autoimmune) responses focused primarily on IgG and IgG isotype levels without distinguishing the relative contribution of MyD88- and TRIF-directed signals to Ab formation versus Ab class switching.20,21 Of note, the one study that used human papilloma virus (HPV) Ag to specifically examine the role of MyD88 and TRIF in TLR4-mediated Ab class switch recombination concluded that MyD88, rather than TRIF, was indispensable for this process. 22 While this major difference from the current study (in which TRIF plays a predominant role in anti-HRS IgG class switching) could stem from involvement of distinct cell types/cellular compartments in the two systems,9,10 the possibility that both TLR4-dependent Ab responses involve overlapping cell types raises the issue of how different Ags might direct divergent intracellular signaling pathways following TLR4 engagement. Ultimately, juxtaposition of these experimental systems suggests an element of redundancy in MyD88- and TRIF-directed signals, possibly reflecting downstream NF-κB activation/nuclear translocation that occurs in both pathways.16,19,23,24
Beyond the specific mechanisms involved in the generation of class switched Abs, these studies demonstrate the importance of the innate immune system in shaping Ab responses—in some cases, without a requirement for T cell help. Of note, results from our model system indicate that TLR4-directed pathways may play a significant role in autoimmunity and the breakdown of B cell tolerance to specific Ags such as HRS. This assertion is supported by the persistence of relatively high titer IgG anti-HRS Abs in mice up to 7 wks after a single IM immunization with soluble Ag (data not shown)—an observation suggesting that TLR4-dependent (and potentially T cell-independent) pathways induced by HRS lead to the generation of long-lived plasma cells rather than transient stimulation of B1 or marginal zone B cells. 25
The extent to which this process applies to other auto Ags or reflects associations with bacterially derived products remains undefined. However, even in cases where IM immunization of alternative fusion proteins does generate some degree of humoral immune response, the level of this response is generally far less than that induced by MA/MBP and its human orthologue, HA5/MBP (Supplementary Figure 1; data not shown). The latter observation indicates that MA/MBP-induced production of IgG anti-HRS Abs does not simply represent a universal property of all bacterially-produced MBP fusion proteins but, instead, stems from heightened immunogenicity of HRS and/or enhanced physical association of HRS with extrinsic TLR4 ligands. Apart from bacterial substances such as LPS, potential contributors to TLR4-mediated anti-HRS auto-Ab production include HMGB1, a host-derived alarmin that has previously been linked to human idiopathic inflammatory myopathy. 26 Whatever property of HRS ultimately governs this response is clearly tied to a well defined region of this auto-Ag (amino acids 60–90) that dictates both immunogenicity and the capacity to engage TLR4 (Supplementary Figure 1).
Although the precise role of TLR4-mediated autoimmunity in this model of HRS-induced myositis is unclear given the discordance between auto-Ab production and tissue phenotype in C3H/HeJ mice (that develop florid T cell infiltration of muscle in the setting of diminished IgG anti-HRS Ab levels), our previously established model of the anti-synthetase syndrome induced by HRS/CFA (complete Freund's adjuvant) immunization has demonstrated a very strong correlation between species-specific humoral immune responses against murine HRS and extra-muscular organ (lung) involvement. Collectively, these data indicate that the balance between cell mediated and humorally-driven immune responses may vary in different aspects of HRS-induced myositis and interstitial lung disease, highlighting the differential role of specific innate immune pathways that may be operative in subsets of inflammatory myopathy (including dermatomyositis), as well as other autoimmune disorders that are more dependent on humorally mediated disease.4,27–29 In fact, previous in vitro and in vivo autoimmune disease models indicate that endogenous TLR ligands, possibly produced via apoptosis or cell necrosis, could directly or indirectly utilize parallel mechanisms to facilitate auto-Ab production and/or class switch recombination more central to disease pathogenesis.30–37 As shown in several systems potentially applicable to rheumatoid arthritis and systemic lupus erythematosus, for example, RNA or DNA nucleotides encompassed by immune complexes may ultimately engage intracellular/endosomal TLRs that include TLR3, TLR7, and TLR912–14,38,39—directly or indirectly (via plasmacytoid dendritic cell-derived IFN-α) promoting Ab production from auto-reactive B cells. While a clear role for extracellular, TLR4-mediated signaling pathways in human autoimmune disease(s), such as idiopathic inflammatory myopathy, is less well established, this study illustrates yet another potential link between innate immunity and the breakdown of self-tolerance.
Overall, this work provides a novel demonstration of the differential roles played by TLR4-directed MyD88 and TRIF signaling pathways in the generation of class switched Abs linked to a model of autoimmune disease, advancing earlier models based on antimicrobial and/or vaccine responses. Although TLR4 is not constitutively expressed on human B cells (but can be induced in subsets of human B cells/plasma cells),16,40,41 signaling through this receptor has been clearly shown to activate various subsets of Ag presenting cells that may then prime B cells through T cell-dependent and T cell-independent pathways.9,21,42 Alternatively, auto-Ags, such as HRS, may activate endosomal TLRs if complexed with RNA ligands, as has been shown in vitro when HRS Ab-positive sera is coupled with necrotic cell debris in the stimulation of PBMC. 17 Ultimately, the relevance of these paradigms and the specific role of MyD88- and/or TRIF-directed TLR signaling in humoral autoimmunity must be assessed through additional in vivo models, particularly those based on Ags such as Mi-2, signal recognition particle, U1RNP, and proteinase 3 where associated auto-Abs may contribute more directly to disease pathogenesis.5,43–48 In turn, establishing pathogenic connections between TLR activation and such models of humorally-mediated autoimmunity will set the stage for much needed, pathway-specific therapeutic intervention.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.