
Abstract
Lipid A, the active moiety of LPS, exerts its effects through interaction with TLR4, triggering a signalling cascade that results in the release of pro-inflammatory cytokines. Eritoran is a lipid A analogue that competes with LPS for binding to TLR4; however, after intravenous administration, it undergoes a time-dependent deactivation as a consequence of binding to high-density lipoproteins (HDLs). The site of eritoran association with HDL remains unknown. Therefore the aim of this study was to determine if HDL-associated apolipoproteins A1, A2, serum amyloid A (SAA) and C1, inhibit the ability of eritoran to block LPS-induced TNF-α release from whole blood. Eritoran activity after LPS stimulation in human whole blood was assessed in the presence of reconstituted HDL (rHDL) containing different apos. In rHDL, the major apolipoproteins in both the healthy and septic state, A1 and SAA, caused a significant reduction in eritoran antagonistic activity and had a greater effect than minor apolipoproteins A2 and C1. Apolipoproteins associated with HDL are likely to facilitate eritoran deactivation. Apolipoproteins A1 and SAA should be of particular focus as they are the major apos found on HDL in both the healthy and septic state. Further evaluation of the physical association between apolipoproteins and eritoran should be explored.
Introduction
Sepsis, which is clinically defined when the host experiences systemic inflammatory response syndrome (SIRS) in response to an infection, causes a complex immune response often resulting in a loss of regulation that leads to vascular permeability and leakage, microthrombosis, hypotension and tissue hypoperfusion ending in organ failure and death. 1 At present, there remains an urgent clinical need to improve current approaches to treatment of sepsis in order to address the climbing rate of hospitalization and mortality from this condition seen within the last 20 years. According to a nationwide study conducted in the USA, there was a doubling in the rate of those hospitalized for severe sepsis and a 1.7-fold increase in the rate of mortality between 1993 and 2003. 2 Furthermore, in 2001, the national estimate of sepsis cases based on hospital discharge databases from seven states was 751, 000 cases per annum, with a projected average cost of $22,000 per patient. 3
Eritoran tetrasodium (E5564), a second-generation lipid A analogue, was developed to antagonize the effects of LPS, the endotoxin believed to be responsible for the activation of immune cells and subsequent release of several lipid and protein mediators seen during normal infection and Gram-negative sepsis. 4 The antagonism of LPS by eritoran is the result of its structural likeness to endotoxin and its ability to compete with LPS at the cell-surface receptor complex containing TLR4, an essential component for LPS signalling. 5 Eritoran has been shown to be highly effective at inhibiting cytokine release from responsive cells ex vivo in whole blood, and ameliorating the effects of endotoxemia in animal models and in human volunteers administered LPS.6,7 However, circulating levels of eritoran in the plasma with a half-life of approximately 50 h were disparate with the pharmacodynamics of the drug that showed a rapid time-dependent deactivation after drug administration in healthy, human volunteers. 8 This loss of activity has been attributed to the propensity of eritoran to bind rapidly to lipoproteins in plasma, predominantly high-density lipoproteins (HDL).
The role of lipoproteins in the body is to serve as transport vehicles for hydrophobic lipids in the blood, an aqueous milieu. High-density lipoproteins is distinguished from other lipoproteins by its role in reverse cholesterol transport, in which it removes excess cholesterol from peripheral tissues and transports it back to liver or steroidogenic tissues, like the adrenal glands, for excretion or the synthesis of steroid hormones. High-density lipoproteins, the smallest of all lipoproteins, are variable in size and density and in contrast to the more lipid-rich lipoproteins, very low density lipoprotein (VLDL) and low density lipoprotein (LDL), HDL particles are 50% protein by composition, have very few triglycerides and contain mostly cholesterol esters in their core. 9
A previous study conducted by Wasan et al. demonstrated that 60% of radiolabeled eritoran in normal plasma associated with HDL within five minutes and that there was no redistribution among lipoprotein classes.
10
Additionally, a similar lipoprotein distribution profile was observed after 72-h infusion of varying doses of drug into human volunteers.
11
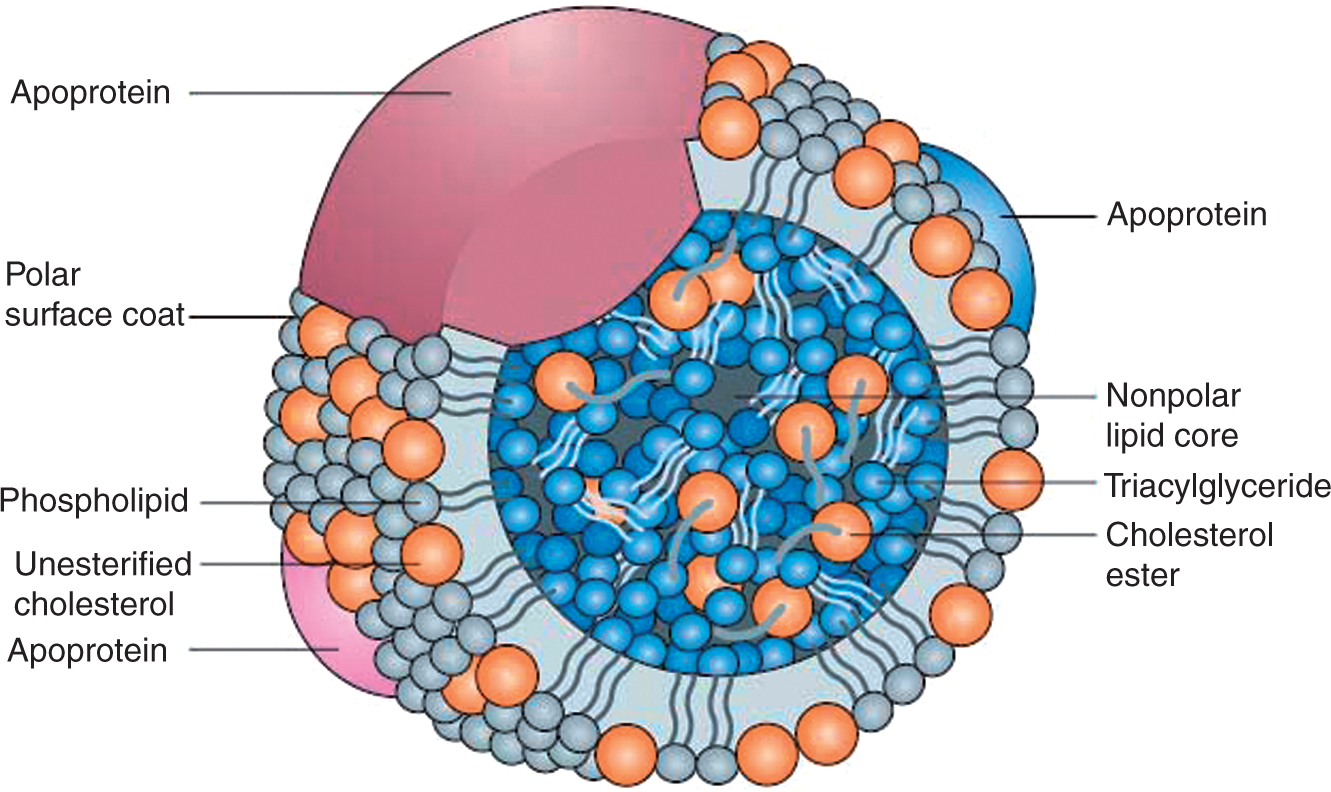
The general structure of a lipoprotein is a phospholipid monolayer intercalated by solubilizing apolipoproteins and cholesterol that enclose a core of neutral lipids, which include triglycerides and cholesterol esters (Figure 1). Previous research has postulated that distinctive components of HDLs, namely shell-forming phospholipids and core cholesterols, should serve as regions of LPS binding and neutralization.
12
However, there is evidence that HDL-associated apolipoproteins, which are important in maintaining particle integrity and have specific functions in cholesterol and lipoprotein metabolism, may facilitate this deactivation event, as they are largely affected during sepsis.
General structure of a lipoprotein. Lipoproteins are soluble complexes of protein and lipid that act as carriers of hydrophobic molecules, namely cholesterol, throughout the blood. They are composed of a phospholipid shell containing unesterified, free cholesterol that is held together by apolipoproteins and enclose a hydrophobic core of neutral lipids including triglycerides and cholesterol esters. There are four main classes of lipoproteins: chylomicrons, very low-density lipoprotein (VLDL), low-density lipoprotein (LDL) and high-density lipoprotein (HDL). Each are distinguished by a unique apolipoprotein content, hydrated density and different mobilities by agarose gel electrophoresis. Reproduced with permission from Wasan et al.
29
Characteristics of apolipoproteins found in humans. Modified with permission from Wasan et al. 29
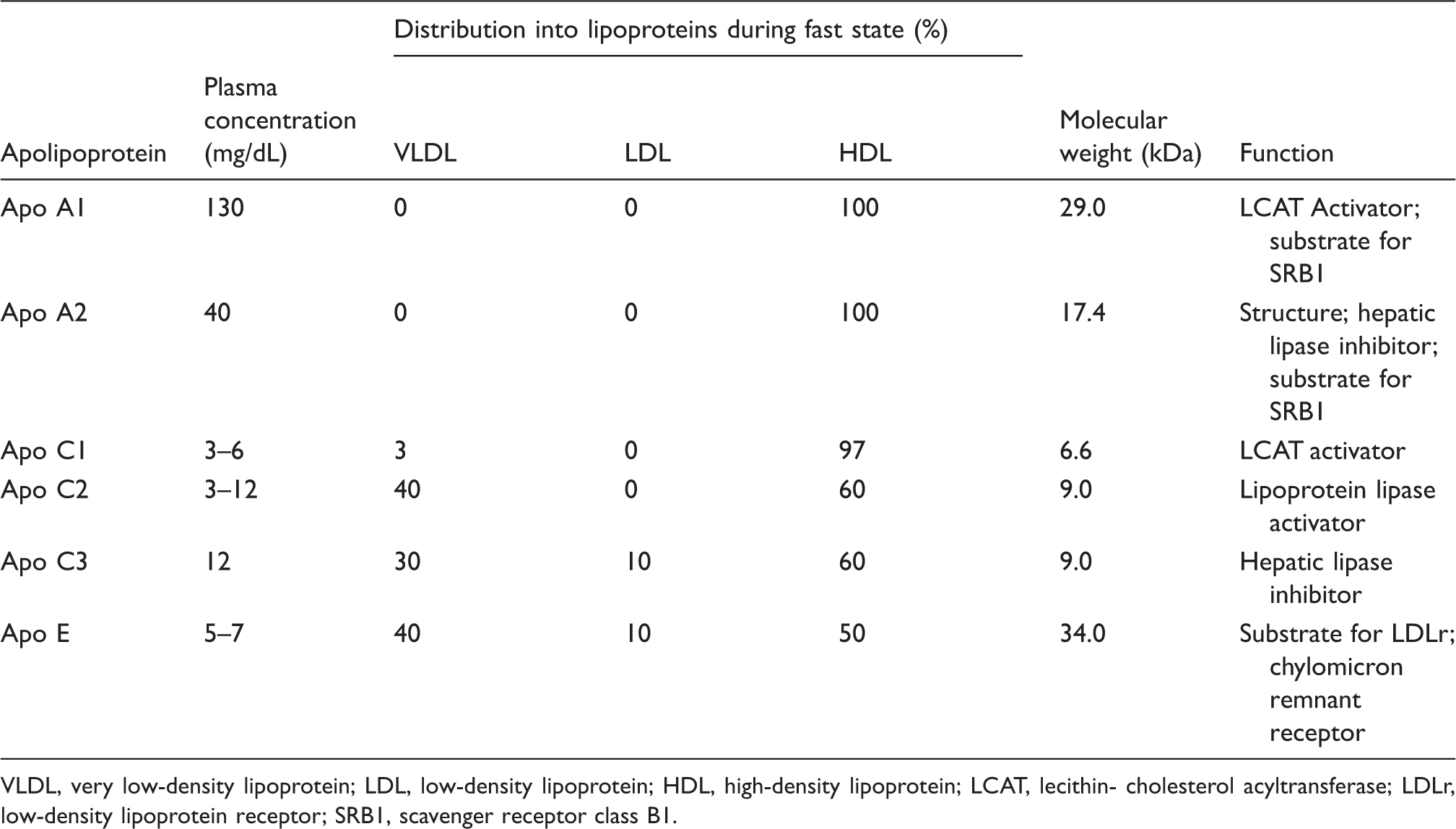
VLDL, very low-density lipoprotein; LDL, low-density lipoprotein; HDL, high-density lipoprotein; LCAT, lecithin- cholesterol acyltransferase; LDLr, low-density lipoprotein receptor; SRB1, scavenger receptor class B1.
Given that eritoran is a structural analogue of the bioactive lipid A moiety found in LPS, we hypothesized that HDL-mediated deactivation of the drug over time during sepsis is a consequence of its association with apolipoproteins found on HDL. The objective of this study was to elucidate whether, and which, apolipoproteins play a role in facilitating deactivation by HDL in the hope that this will lend insight into how drug deactivation can be avoided and, thus, improve the use of eritoran as a therapeutic for patients with sepsis.
Materials and methods
Reagents
Eritoran (E5564) was a obtained from the Eisai Research Institute (Andover, MA, USA). Initially, 10 mg of lyophilized drug was re-suspended in 10 ml of dH20 to make a 1 mg/ml solution. Fifty µl aliquots of the 1 mg/ml solution were created for storage at −20°C and fresh drug was used when required. Before use, the drug was further diluted to achieve a working concentration of 500 n
Plasma lipoprotein separation by ultracentrifugation
Human normolipidemic plasma (Bioreclamation, Hicksville, NY, USA) was separated into lipoprotein fractions by density gradient ultracentrifugation for the collection of HDL. Tubes containing plasma were placed into individual titanium buckets and capped. Buckets were loaded on a SW 41 Ti swing rotor (Beckman Coulter, High Wycombe, UK) and centrifuged at ∼200,000 g, 15°C for 18 h. The HDL fraction from each tube was pooled into a 50 ml BD Falcon™ tube and stored at 4°C, while other lipoprotein fractions were discarded. Subsequently, HDL was desalted using desalting columns (Econo-Pac desalting pre-packed gravity flow columns, Bio-Rad®, Hercules, CA, USA) as per the manufacturer’s instructions. Eluted fractions were measured for protein content using the DC-protein assay (Bio-Rad®) and bovine serum albumin as a standard (Thermo Pierce, Rockford, IL, USA). Total cholesterol (TC) was measured using the Wako Cholesterol E kit (Wako Chemicals, Richmond, VA, USA). Fractions with the highest concentration of protein and TC were pooled together and adjusted to a 0.9% NaCl (Sigma-Aldrich®) solution. Total cholesterol and protein was measured again to determine the stock concentration of HDL to be used in subsequent assays.
Creating reconstituted HDL (rHDL) with apolipoproteins
To create spherical particles that best mimic the physical structure of human HDL3, the whole lipid fraction from delipidated human HDL was combined with protein in a molar ratio of 80:1 phospholipid:protein when using individual apolipoproteins. 21 The whole lipid fractions were isolated from human HDL as previously described in Wang et al. 22 Briefly, HDL was mixed with 60 volumes of hexane/2-propanol (3:2), centrifuged at 365 g for 20 min and the lipid fractions collected and dried under nitrogen. The extracts were then resuspended in a chloroform/methanol/0.74% KCl (8:4:3 v/v/v) solution, centrifuged again at 365 g for 5 min and the aqueous phase discarded. The lipids were dried down under nitrogen and finally resuspended in chloroform containing 0.1% butylated hydroxytoluene to prevent oxidation and stored at −20°C.
When creating rHDL with a combination of different apolipoproteins, molar ratios of apolipoproteins used were based on the circulating plasma concentration of each protein and its association with HDL. Initially, the whole lipid fraction was added to a glass vial and evaporated completely under nitrogen gas, after which 1 ml of reconstitution buffer (NaCl 150 m
Tumour necrosis factor-α assay in human whole blood: testing eritoran inhibitory activity against LPS
The inhibitory activity of eritoran was assessed using the whole blood assay according to Rose et al.
23
Initially, to induce drug sequestration, 50 n
Eritoran was used at final concentration of 10 n
Statistics
Treatment and control groups on each plate were assayed in triplicate. Moreover, plates for each apolipoprotein construct were repeated by using blood from three separate human subjects (one female, two male). Raw data were processed in Excel 97 (Microsoft, Redmond, WA, USA). Paired t-tests were performed using SigmaStat 3.5 (Systat®, Chicago, IL, USA) to determine the effects of rHDL-containing apolipoproteins on eritoran inhibitory activity against LPS-induced TNF-α release. A one-way analysis of variance (ANOVA) was used to determine the effects of increasing concentrations of native HDL on eritoran inhibitory activity against LPS. Error was reported as standard error of the mean (SEM). A P-value of <0.05 was considered statistically significant.
Results
Concentration-dependent effects of high-density lipoprotein on eritoran activity
As rHDL constructs containing individual apolipoproteins have never been explored as a means of reducing eritoran activity, a concentration range to test rHDL was established by creating a concentration-response curve using native HDL. At 0.1 mg/ml of native HDL by protein, there was approximately a 30% reduction in activity of eritoran (deactivation) after LPS stimulation (30.9%, P < 0.05) (Figure 2). Lower levels of HDL, below 0.8 mg/ml (which is equivalent to a plasma concentration of 40 mg/dl or 1.03 mmol HDL cholesterol), were shown to deactivate similar to 0.1 mg/ml. As septic patients are hypocholesterolaemic and typically have very low circulating levels of HDL, we chose to make rHDL constructs at 0.1 mg/ml (equivalent to a plasma concentration of 10 mg/dl or 0.23 mmol HDL cholesterol) as this would best mimic levels found in a septic patient.
Concentration-dependent effects of high-density lipoprotein (HDL) on eritoran activity. TNF-α release from human whole blood after stimulation with 10 ng/ml LPS in the presence of the eritoran (10 n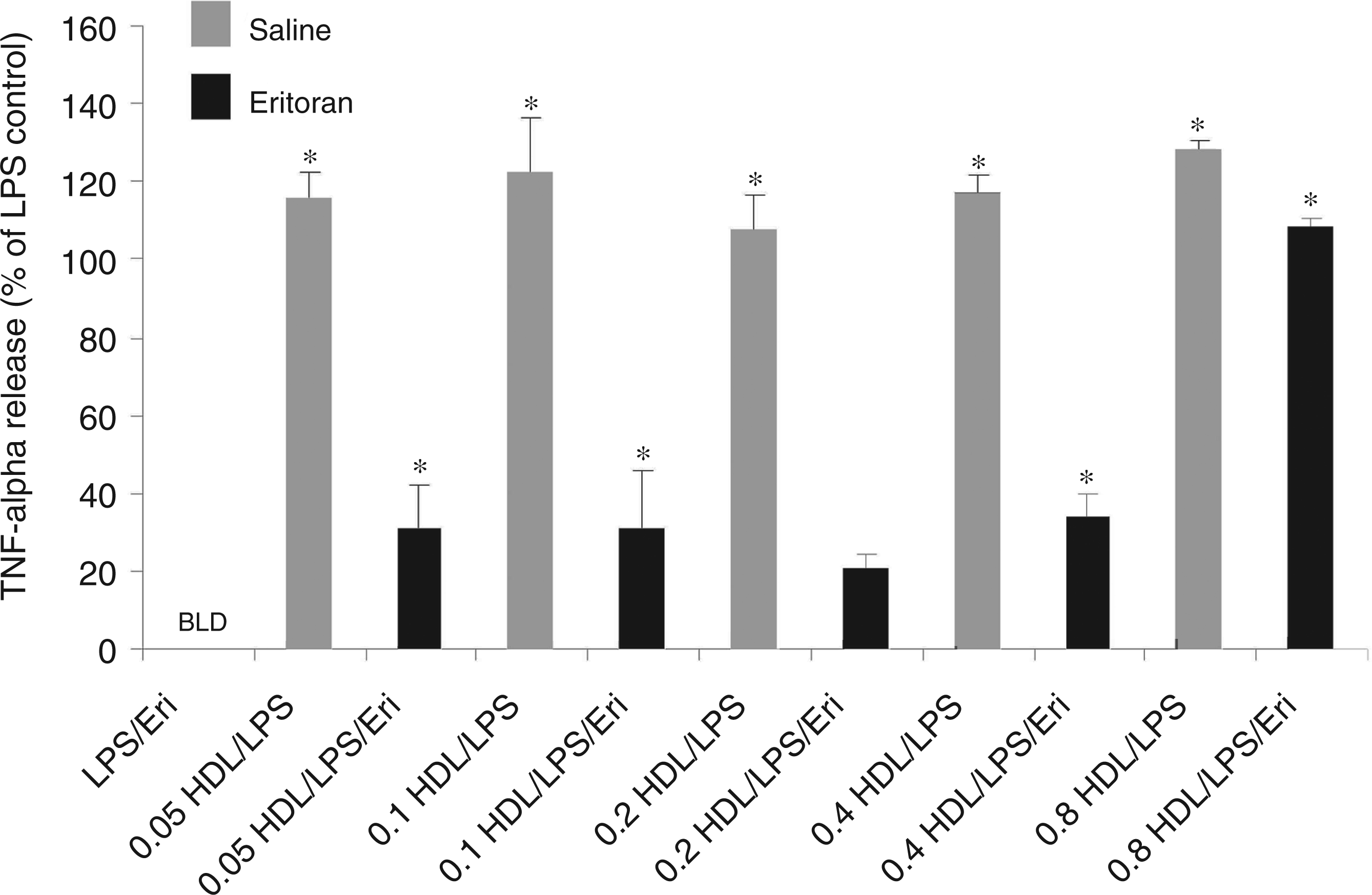
Loss of eritoran antagonistic activity by rHDL containing apo A1 and serum amyloid A
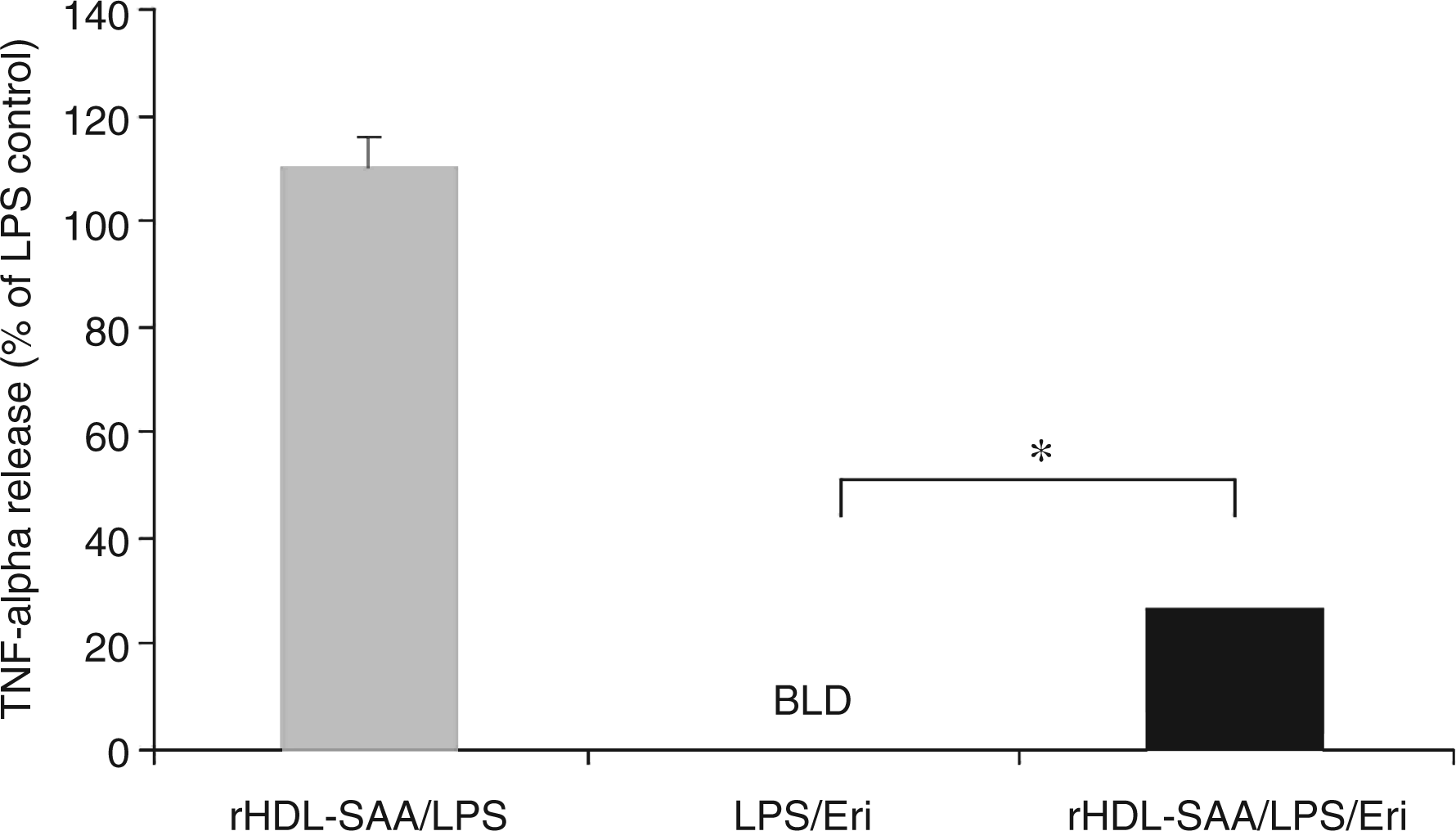
To assess the effects of different apolipoproteins on eritoran drug activity in a physiologically relevant form (lipid-bound), reconstituted HDLs were made with individual apolipoproteins. By making these constructs, we were able to maintain the lipid composition of rHDLs while varying the protein type and, therefore, investigate changes in drug activity based on the presence of a single apolipoprotein. At 0.1 mg/ml, apo A1 caused a significant loss in drug activity in an rHDL construct (47.4% deactivation, P < 0.02) as shown by the inability of eritoran to fully suppress TNF-α release after LPS stimulation (Figure 3). Similarly, eritoran antagonistic activity in whole blood was considerably inhibited in the presence of rHDL-containing SAA at 0.1 mg/ml by 26.4% (P < 0.001) as compared to untreated controls (Figure 4).
Eritoran activity as measured by TNF-α release in the presence of reconstituted high-density lipoprotein (rHDL)-A1. Values shown here are the means calculated from all three subjects (one female, two male). Tumour necrosis factor-α release is shown as % of LPS. Final concentrations of rHDL-A1 are 0.1 mg/ml by protein. The rHDL control (without eritoran) is shown in grey. Error bars represent + SEM. Statistical analysis was performed using a paired t-test. *Denotes a significant difference from LPS/Eri (P < 0.05). N = 3 BLD, below the limit of detection; LPS, lipopolysaccharide; TNF-α, tumour necrosis factor-α; Eri, eritoran; rHDL, reconstituted high-density lipoprotein. Eritoran activity as measured by TNF-α release in the presence of reconstituted high-density lipoprotein (rHDL)-SAA. Values shown here are the means calculated from all three subjects (one female, two male). Tumour necrosis factor-α release is shown as % of LPS. Final concentrations of rHDL-SAA are 0.1 mg/ml by protein. The rHDL control (without eritoran) is shown in grey. Error bars represent + SEM. Statistical analysis was performed using a paired t-test. *Denotes a significant difference from LPS/Eri (P < 0.05). N = 3 BLD, below the limit of detection; LPS, lipopolysaccharide; TNF-α, tumour necrosis factor-α; Eri, eritoran; rHDL, reconstituted high-density lipoprotein.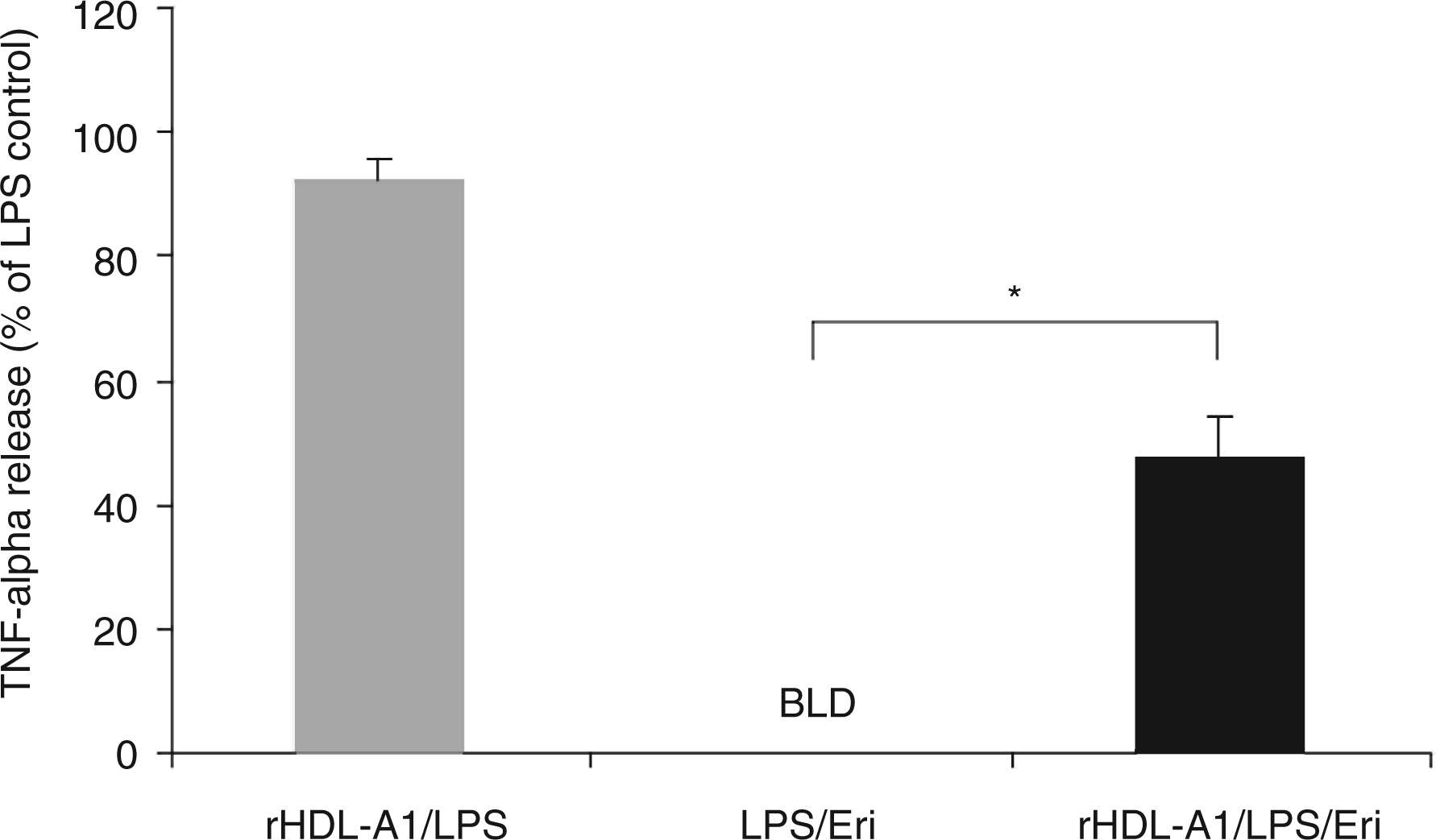
Loss of eritoran antagonistic activity by rHDL containing apo A2 and C1
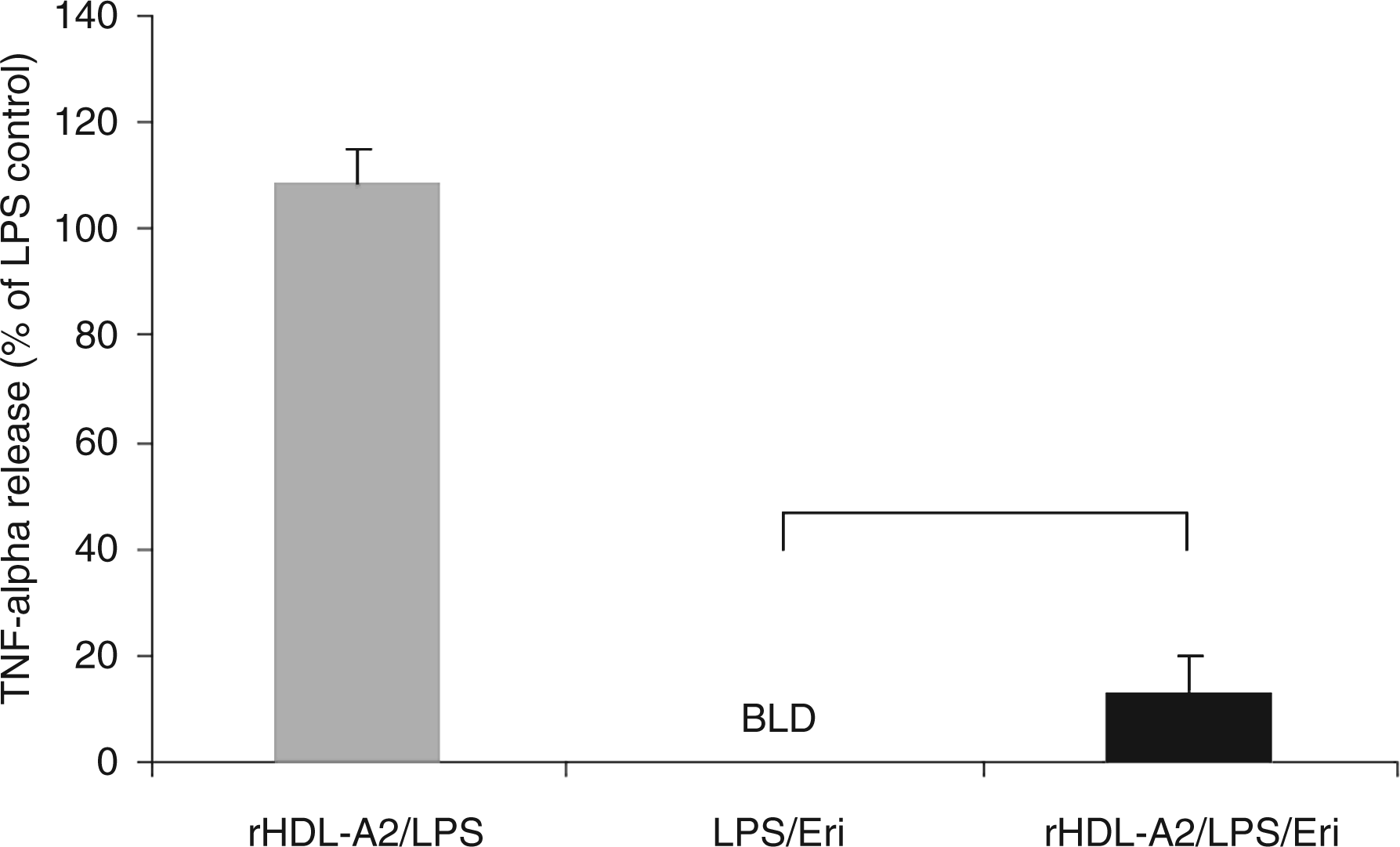
Minor HDL apolipoproteins, A2 and C1, which were found to deactivate eritoran based on earlier free protein studies (data not shown), were tested in rHDL constructs to determine their effects on the drug in a lipid-bound form. The reductions in drug effect seen with apo A1 were not observed when eritoran activity was tested after pre-incubation with rHDL-C1 (13.0% deactivation, P > 0.05) at 0.1 mg/ml (Figure 5). Likewise, a less than 15% decrease in drug activity was observed when eritoran was tested for its ability to block TNF-α release in the presence of 0.1 mg/ml rHDL-A2 (13.1% deactivation, P > 0.05) (Figure 6).
Eritoran activity as measured by TNF-α release in the presence of reconstituted high-density lipoprotein (rHDL)-C1. Values shown here are the means calculated from all three subjects (one female, two male). Tumour necrosis factor-α release is shown as % of LPS. Final concentrations of rHDL-C1 are 0.1 mg/ml by protein. The rHDL control (without eritoran) is shown in grey. Error bars represent + SEM. Statistical analysis was performed using a paired t-test. *Denotes a significant difference from LPS/Eri (P < 0.05). N = 3 BLD, below the limit of detection; LPS, lipopolysaccharide; TNF-α, tumour necrosis factor-α; Eri, eritoran; rHDL, reconstituted high-density lipoprotein. Eritoran activity as measured by TNF-α release in the presence of reconstituted high-density lipoprotein (rHDL)-A2. Values shown here are the mean calculated for all three subjects (one female, two male). Tumour necrosis factor-α release is shown as % of LPS. Final concentrations of rHDL-A2 are 0.1 mg/ml by protein. The rHDL control is shown in grey. Error bars represent + SEM. Statistical analysis was performed using a paired t-test. *Denotes a significant difference from LPS/Eri (P < 0.05). N = 3 BLD, below the limit of detection; LPS, lipopolysaccharide; TNF-α, tumour necrosis factor-α; Eri, eritoran; rHDL, reconstituted high-density lipoprotein.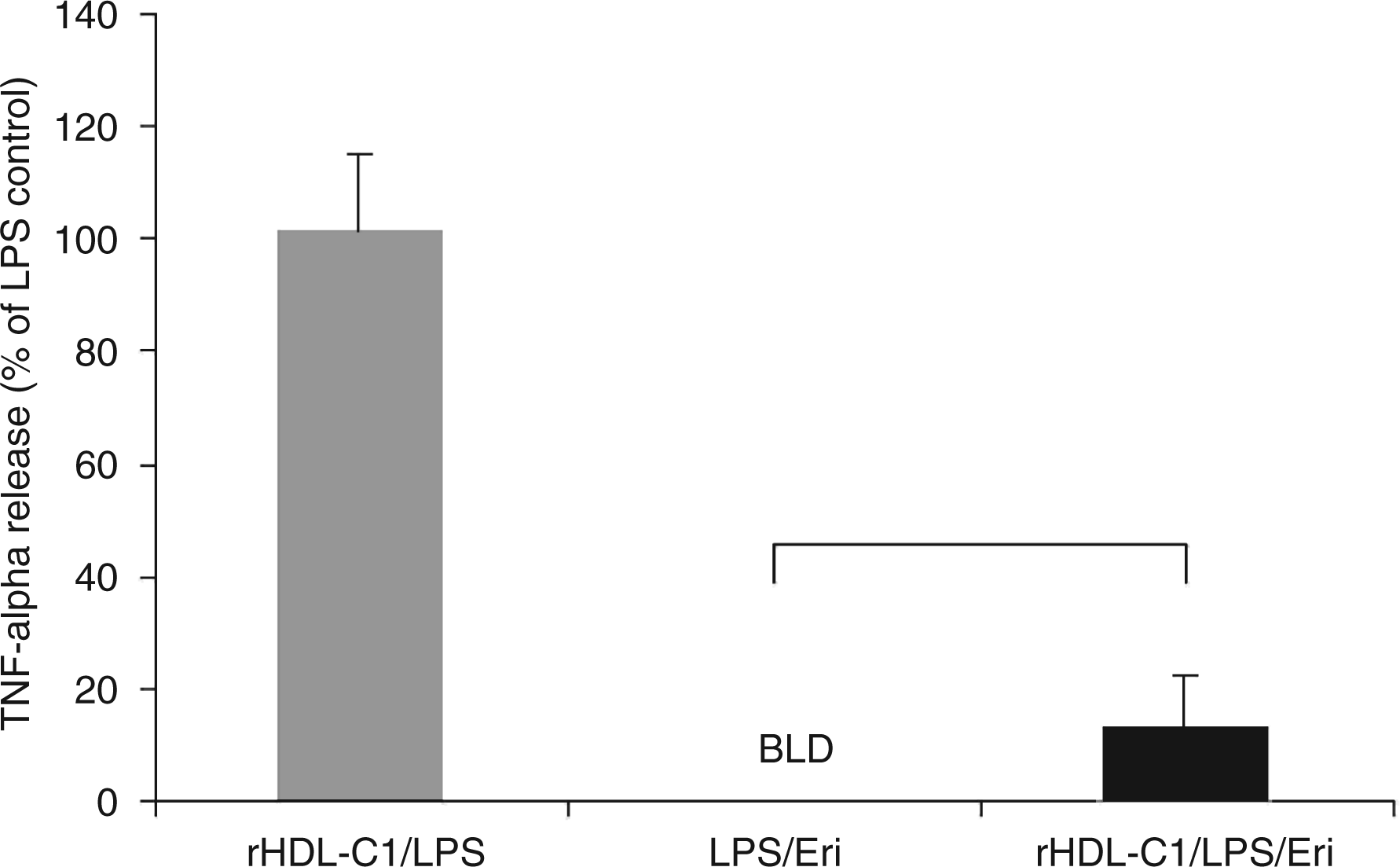
Discussion
Time-dependent deactivation of eritoran, an antagonist of LPS at the TLR4 receptor, by HDL in clinical subjects has become a point of interest and also concern regarding drug efficacy and dosing regimens in critically ill, septic patients. The results of this study suggest that exchangeable apolipoproteins are potential factors in eritoran deactivation by HDL. Their ability to reduce drug action as part of a reconstituted HDL has confirmed that changes made to the protein components of rHDL will produce changes in drug activity (i.e. different apolipoproteins cause varying degrees of deactivation). This supports the concept that apolipoproteins and not other lipid constituents (e.g. phospholipids or cholesterol) are the reason for eritoran association and deactivation by HDL.
As the principal and most abundant apolipoproteins on HDL in both the healthy and septic state, apolipoproteins A1 and SAA account for up to 70% and 80% of HDL protein content during normal and inflammatory conditions, respectively.25,26 Thus, from a quantitative standpoint the availability of these proteins on HDL particles make them more likely to physically associate with the drug as opposed to other apolipoproteins and, therefore, it was not unexpected that apolipoprotiens A1 and SAA had a notable impact on drug activity in the physiological, lipid-bound form. Nevertheless, different from HDL found in vivo, in this particular study these apolipoproteins were tested individually at a defined, abnormally low protein concentration of 0.1 mg/ml, which suggests that there is something specific or exclusive about apolipoproteins A1 and SAA that confer a greater deactivating effect on drug activity than the minor apolipoproteins, A2 and C1.
All apolipoproteins have a conserved structure containing a signature feature that connects them: the presence of several 11-residue long (11-mer) amino acid repeats. 27 The resulting physical feature of these 11-mer repeats is their ability to form amphipathic α-helices, the majority of which are those belonging to class A, which are defined by a unique clustering of positively charged residues at the polar/non-polar interface and negatively charged residues at the centre of the polar face. 28
These helices bestow apolipoproteins the ability to avidly bind lipid and maintain the structural integrity of lipoproteins as carriers of hydrophobic molecules throughout the blood while interacting with other blood components such as LPS, likely by electrostatic interactions via their polar/charged surface
Footnotes
Acknowledgements
This work was financially supported by Eisai Inc. and the Canadian Institutes of Health Research (CIHR) operating grant MOP-12660 to G.A.F and operating grant MOP-9905 to K.M.W. Special thanks to Nature and the Nature Publishing Group (NPG) for permission to reproduce Figure 1 and ![]() .
.