
Abstract
A purified complex of metabolically labeled [3H]lipooligosaccharide (LOS) and recombinant human myeloid differentiation factor 2 (MD-2), [3H]LOS·MD-2, has been used to demonstrate pM affinity binding interactions with soluble TLR4 ectodomain (TLR4ecd). For measurement of the binding parameters of membrane-bound TLR4, we took advantage of the stability of endotoxin·MD-2 and tyrosine(s) present on the surface of MD-2 to radioiodinate LOS·MD-2. Radioiodinated LOS·MD-2 generated a reagent with an estimated 1:1 molar ratio of [125I] to sMD-2 with 20-fold higher specific radioactivity and TLR4-activating properties comparable to metabolically-labeled LOS·MD-2. LOS·MD-2[125I] and [3H]LOS·MD-2 have similar affinities for soluble FLAG TLR4ecd and for membrane-bound TLR4 in HEK293T/TLR4 cells. In a similar dose-dependent manner, sMD-2 and LOS·MD-2 inhibit LOS·MD-2[125I] binding to TLR4 indicating the pM affinity binding of LOS·MD-2[125I] is agonist-independent. LOS·MD-2[125I] allowed measurement of low levels of cell-surface human or murine TLR4 expressed by stable cell lines (2000–3000 sites/cell) and quantitatively measures low levels of ‘MD-2-free’ TLR4 (est. 250 molecules/cell) in cells co-expressing TLR4 and MD-2. Occupation of 50–100 TLR4/cell by LOS·MD-2 is sufficient to trigger measurable TLR4-dependent cell activation. LOS·MD-2[125I] provides a powerful reagent to measure quantitatively functional human and murine cell-surface TLR4, including in cells where surface TLR4 is potentially functionally significant but not detectable by other methods.
Introduction
TLRs are essential innate immune sensors that couple molecular recognition of specifically conserved and structurally unique microbial molecules to rapid mobilization of innate immune effector systems and induction of adaptive immunity.1,2 Substantial progress has been made in deciphering the mechanism(s) of activation and subsequent signal transduction upon TLR engagement by an agonist. However, data are limited with regard to ligand recognition properties of TLRs, including the structural requirements of ligand recognition and the binding parameters of specific ligand:TLR interactions. As a consequence, measurement of receptor activation by various compounds, rather than direct binding, has often been utilized to infer TLR receptor recognition.
TLR4 confers exquisite sensitivity to many different species of invading Gram-negative bacteria (GNB) by virtue of its role in recognition and response to endotoxins (E), unique and abundant surface glycolipids of GNB. Endotoxins are the most potent natural regulators of TLR4 stimulating host inflammatory responses at fmol/ml concentrations of endotoxin, corresponding to as few as 100 invading GNB/ml. The ability of TLR4 to respond to pM concentrations of E does not reflect direct E–TLR4 interactions, but depends on the ordered action of three extracellular and cell surface host proteins: LPS-binding protein (LBP), soluble (s) and glycophosphatidylinositol (GPI)-linked membrane (m)-associated forms of CD14 and secreted or TLR4-associated MD-2.3–8 LBP, CD14 and MD-2 dramatically alter the physical presentation of E, resulting in amplification of host responsiveness to E. One GNB containing about 106 E molecules can be converted to 106 TLR4-activating monomeric E·protein complexes.7,9,10 Individual E monomers, extracted from GNB or purified E aggregates, form monomeric E·protein complexes that, at pM concentrations, engage TLR4 either indirectly (E·CD14 with MD-2·TLR4) or directly (E·MD-2 with TLR4), and cause receptor and cell activation. TLR4 activation by E requires simultaneous binding of E and TLR4 by MD-2. E·MD-2 is unique in its ability to directly engage and activate host cells expressing TLR4 without MD-2.4,7,9
The identification and characterization of monomeric E·MD-2 complexes as potent TLR4 agonists or antagonists, depending on E and/or MD-2 structure, have clearly demonstrated that it is the properties of the E·MD-2 complex, not E alone, that specifies TLR4 activation or receptor occupancy without activation.4,9–11 In stark contrast to the instability of sMD-2 monomers,12–16 the stability and solubility of monomeric E·MD-2 have permitted the purification of stable, monomeric and active E·MD-2 complexes.9–11 The use of purified and radiolabeled [3H] lipooligosaccharide (LOS)·MD-2 in combination with the predicted TLR4 ectodomain (TLR4ecd) has allowed detection and quantitative analysis of specific, high-affinity (pM) E·MD-2 and TLR4ecd interactions.7,17 In addition, the stability of E·MD-2 and TLR4ecd·E·MD-2 complexes has allowed the use of NMR to examine and compare structural properties of [13C]-labeled fatty acyl chains of E in both an agonistic wild type (wt) and an antagonistic mutant MD-2 complex alone and bound to TLR4ecd. 18
While solution studies using TLR4ecd have been informative regarding E·MD-2 interactions, the ability to extend binding studies to full-length TLR4 present on various cell types has remained elusive owing apparently to the low levels of cell-surface TLR4 on many E-responsive cells and the limits of detection of available reagents, including [3H]E·MD-2. The stability of E·MD-2 complexes 9 and the presence of tyrosines on the surface of MD-219,20 suggest that iodination of purified LOS·MD-2 could provide a reagent of enhanced specific radioactivity with sufficient sensitivity for binding studies to TLR4-containing cells. Both biochemical and X-ray crystal structural data suggest tyrosine residues that are exposed and not involved in sites of interactions with E or TLR4 necessary for E·MD-2 function.19–21 Data presented here indicate that iodination of purified LOS·MD-2 under gentle conditions generates LOS·MD-2[125I], a sensitive, high-affinity ligand for TLR4 that has the same functional characteristics as LOS·MD-2, but with a specific activity (∼500,000 cpm/pmol) approximately 20-fold greater than metabolically-labeled [3H]LOS·MD-2. LOS·MD-2[125I] provides a high-affinity TLR4 ligand capable of quantifying as few as 100 ‘free’ cell surface TLR4/cell in both human and murine cells, including in cells expressing both TLR4 and MD-2 where levels of free TLR4 have been too low for detection. The sensitivity of the binding assays described here reveal how few TLR4/cell need to be engaged for measurable cell activation, underscoring the remarkable potency of this innate immune sensor.
Materials and methods
Materials
Metabolically-labeled endotoxin ([14/13C] LOS, 15 cpm/pmol or [3H]LOS, 25,000 cpm/pmol) was isolated from Neisseria meningitidis serogroup B (NMB) grown in Morse medium supplemented with either 2 mM 1-[12C], 2-[13C]-acetate (Moravek Chemicals, Brea, CA, USA) and 1 uCi/ml of 1,2-[14C] acetate or 2 mM sodium acetate containing 5 mCi/ml [3H]acetate (Moravek Chemicals), as described previously.22,23 Human serum albumin (HSA) was an endotoxin-free, 25% stock solution from Baxter Health Care (Glendale, CA, USA). Chromatography matrices (Sephadex G10, Sephacryl HR S100 and 200 and Ni2+ FF-Sepharose) were purchased from GE Healthcare (Piscataway, NJ, USA). BSA, anti-FLAG M2 agarose, and other chemical reagents were from Sigma (St. Louis, MO, USA). Iodogen was purchased from Pierce (Rockford, IL, USA) and used according to the manufacturer’s instructions. The stable HEK293 cell line expressing human TLR4 was a generous gift from Dr Jesse Chow (Eisai Research Institute, Andover, MA, USA) and the stable HEK293 cell line expressing murine TLR4 was purchased from Invivogen (San Diego, CA, USA). Conditioned medium containing secreted FLAG-tagged TLR4 ectodomain-vlr-Fc protein used in capture assays was obtained from Freestyle HEK293F cells stably transfected with TLR4 (amino acids 27-527) in a CMV vector containing additional hagfish amino acid sequence at the C-terminal end followed by the Fc domain. The stable transfected cells were a generous gift from Dr Richard Tapping (University of Illinois, Urbana, IL, USA). Control conditioned medium (i.e. not containing FLAGTLR4ecd) obtained from HEK293 cells was used as a negative control. The pCMV-FLAG-TLR4 vectors encoding either TLR4 wt or mutant D299G.T399I were a generous gift from Dr Stefanie Vogel (University of Maryland, Baltimore, MD, USA). The IL-8 ELISA kit (BD OptEIA) was purchased from BD Biosciences (San Diego, CA, USA) and used according to manufacturer’s instructions. Binding data were analyzed using GraphPad Prism 5 Software (La Jolla, CA, USA).
Preparation of recombinant human MD-2
Recombinant human His6-MD-2 was generated as described previously, 9 except that cDNA encoding human MD-2 was optimized for transcription in High Five cells (Tricoplusia ni) by DNA 2.0 (Menlo Park, CA, USA) generating a designer gene for human MD-2. The cDNA encoding human MD-2 was inserted using SacII and XhoI restriction sites into the baculovirus transfection vector, pBAC3 (Novagen, Billerica, MA, USA) that contains a six-residue polyhistidine (His6) tag at the N-terminal end and a 5′ flanking signal sequence (gp64) to promote secretion of the expressed protein from the insect cells. Baculovirus stocks containing the gene for His6-MD-2 were generated by transfection of BacVec3000 and plasmid into Sf9 cells using the BacVec3000 kit from Novagen, according to the manufacturer’s instruction. The generated baculovirus was then amplified in Sf9 cells and used to infect High Five™ (Invitrogen, Grand Island, NY, USA) insect cells in serum-free medium for protein production. Large-scale (20 l) preparations of conditioned insect medium containing secreted His6-MD-2 were produced by BlueSky Biotech (Worcester, MA, USA).
Preparation of LOS·MD-2 complexes
We have recently described the preparation of endotoxin·albumin complexes (Mr ∼70,000) and conditions in which endotoxin·albumin complexes react with conditioned insect medium containing sMD-2 to generate endotoxin·MD-2.18,24 [14/13C]LOS·albumin complexes (15 cpm/pmol) were generated from [14/13C]LOS aggregates dispersed in 100 mM Tris·HCl/5 mM EDTA, pH 8.0, sonicated for 15 min, supplemented with 1.0% HSA and then incubated for 18 h (overnight) at 37℃. [14/13C]LOS·albumin complexes were isolated by size-exclusion chromatography (Sephacryl S200, 2.6 × 100 cm) on a column equilibrated in 100 mM Tris/5 mM EDTA, pH 8.0. The complexes were kept at 4℃ and used within 2–3 d to generate LOS·sMD-2.
In a typical purification, isolated LOS·albumin complexes (1 mg LOS, 200 nmol) were incubated with conditioned insect medium containing secreted human His6-sMD-2 (2 l, 90 nmol, ∼1 ug/ml sMD-2) for 3 h at 37℃ with gentle shaking. The reaction mixture was dialyzed (Spectrapor 1, MMCO 3500) against 20 mM phosphate, 0.5 M NaCl, 5 mM imidazole, pH 7.6, ‘binding buffer’. The dialyzed material was loaded onto Ni FF Sepharose (5 cm × 25 cm) using GEHealthcare Explorer fast protein liquid chromatography at 4℃ at a flow-rate of 3 ml/min. After washing the column with 20 mM imidazole in binding buffer, the adsorbed material was eluted by an imidazole gradient in ‘binding buffer’ (pH 7.6). The peak of [14/13C]LOS·MD-2 eluted toward the end of the 0.5 M imidazole gradient. The radioactive fractions were combined and concentrated using a Centriplus-70 (Millipore, Billerica, MA, USA) to a final volume ∼2 ml and then injected onto a Sephacryl S100 column (1.6 × 100 cm) equilibrated in 20 mM phosphate, 150 mM NaCl, pH 7.1. The peak fractions containing radioactivity were combined and concentrated using a Millipore Centricon MMCO 10 K to a final volume 0.5–1 ml. Coomassie blue stain of SDS-PAGE PhastGel, 10–15% gradient gel (GEHealthcare), confirmed the purity of the sample. 18 LOS·MD-2 F126A complexes were prepared and purified using an identical procedure. 18
Complexes of [3H]LOS·MD-2 (25,000 cpm/pmol) used in analytical binding and cell activation were generated as described previously.7,9,22
Radioiodination of LOS·MD-2
Iodination was done at room temperature in borosilicate tubes coated with 10 ug IODOGEN that had been delivered in chloroform followed by removal of the solvent under a stream of nitrogen. To an iodogen-containing tube, 70 μl 20 mM phosphate, 150 mM NaCl, pH 7.1 (buffer A) was added, followed by 9.2 µg MD-2 in the form of LOS·MD-2 (20 μl containing 100 ng LOS and 460 ng MD-2 per μl). Iodine stock solution {0.5 mCi of Na125I (1000 Ci/mmol [125I], Perkin Elmer, Waltham, MA, USA} (10 μl in buffer A) was added to the tube and the sample was mixed occasionally for 7–9 min. Sodium metabisulfite (10 mg/ml buffer A, 10 μl) was added to stop the reaction and the sample was applied to a Sephadex G10 column (1 cm × 16 cm) equilibrated in buffer A, 0.1% BSA. Fractions (1 ml) were collected and an aliquot counted to determine the peak of radioactivity that eluted near the void volume containing LOS·MD-2. Application of [3H]LOS·MD-2 to G10 Sephadex under the same conditions was used to establish the elution pattern and recovery (100%) of [3H]LOS·MD-2. Radioactivity determinations for radioiodinated samples were done in the Packard Cobra (Meridan, CT, USA) II autogamma counter. The void volume radioactive peak fraction had the greatest % of trichloroacetic acid (TCA) precipitable counts (80–85%) and was further purified from free [125I] by dilution in buffer A, followed by concentration using Millipore Ultracel MMCO 10 K to the original volume. This process of dilution and concentration was repeated three times. The recovered retentate was diluted with buffer A to a final volume of 1 ml. This recovered iodinated product was re-evaluated by TCA precipitation; 98% of the [125I] counts were TCA-precipitable, i.e. protein-bound. Iodinated LOS·MD-2 was aliquoted and stored at –80℃; material was used within 2–3 months of production. Specific radioactivity of the final product was approximately 500,000 cpm/pmol LOS·MD-2[125I]. Iodination of LOS·MD-2 was repeated three times with virtually identical results in recovery, specific radioactivity and functional properties.
To verify that the purified iodinated product corresponded to LOS·MD-2[125I], an aliquot of this sample (0.04 pmol, 18,000 cpm), as well as [3H]LOS·MD-2 (0.2 pmol, 5,500 cpm), were applied to Sephacryl S200 (1.6 cm × 60 cm) and eluted in PBS, 0.1% HSA, pH 7.4, on the AKTA Purifier10 at a flow rate 0.5 ml/min. The Sephacryl S200 column was calibrated by application of HSA and BioRad gel filtration standards (BioRad, Hercules, CA, USA) containing thyroglobulin, IgG, ovalbumin, myoglobin and vitamin B12. Fractions (1 ml) were evaluated for radioactivity— LOS·MD-2[125I] using Packard Cobra II autogamma counter and [3H]LOS·MD-2 by liquid scintillation spectroscopy using the Beckman LS6500 scintillation counter (Beckman Coulter, Schaumberg, IL, USA). Recovery of total cpm applied to the column for both reagents was ∼100%. LOS·MD-2[125I] was also examined by 4–25% PhastGel (GEHealthcare) SDS-PAGE/image analysis to confirm radioiodination of MD-2 (Mr ∼ 23,000) in comparison to [14C]LOS·MD-2 (LOS Mr ∼5000) and covalently cross-linked LOS·MD-2[125I] (Mr ∼28,000) run in neighboring lanes as references.
Anti-FLAG agarose co-capture of the product of LOS·MD-2 reaction with FLAGTLR4ecd-vlr
Co-capture assay of radiolabeled LOS·MD-2 binding to FLAG-tagged TLR4 ectodomain was performed as described previously.7,17 Increasing concentrations of [3H]LOS·MD-2 (25,000 cpm/pmol) or LOS·MD-2[125I] (500,000 cpm/pmol) were incubated with control conditioned medium or conditioned medium containing FLAGTLR4ecd-vlr (10 μl) for 30 min at 37℃ (total volume 0.4 ml in PBS, 0.1% HSA, pH 7.4). After incubation, the samples were mixed 1 h at 25℃ (room temperature) on a rotating wheel with anti-FLAG-agarose (30 μl 1:1 slurry) equilibrated in the same buffer. The samples were spun at 2000 g for 5 min at 4℃, supernatant was removed and the beads were then washed twice with 1 ml cold PBS, 0.1% HSA, pH 7.4. The supernatants, washes and radioactivity associated with the affinity gel (‘capture’) were evaluated by liquid scintillation spectroscopy using a Beckman LS6500 scintillation counter for tritiated samples and by direct counting in a Packard Cobra II autogamma counter for iodinated samples. In analysis of the data, incubations containing control conditioned medium was used to determine non-specific binding (capture) in the dose curve for both ligands.
TLR4-dependent binding of LOS·MD-2 to cells
For experiments with transfected cells, T75 flasks of HEK293T cells were grown to confluence in DMEM, 10% FCS and transfected with 10 µg control empty vector or pCMV-FLAG containing either wt TLR4 or TLR4 D299G. T399I using either PolyFect (Qiagen, Valencia, CA, USA) or Lipofectamine2000 (Invitrogen) in OptiMEM I (Gibco, Grand Island, NY, USA) medium followed by the addition of DMEM, 0.1% HSA. After 12 h, the medium was changed to DMEM, 10% FCS; after an additional 24 h, the cells were harvested, spun and re-suspended (1 ml) in PBS, 0.1% HSA, pH 7.4. For each preparation, the number of viable cells was determined in the re-suspended pellet by trypan blue staining and cell counting with a hemacytometer. Cell count was also determined using the Accuri flow cytometer. The cells were distributed in 1.5 ml siliconized microfuge tubes (approximately 1 or 2 × 106 cells/tube as indicated in figure legends) and brought to 1 ml with PBS, 0.1% HSA.
For experiments with stable HEK293 cell lines, control parent cells (not expressing TLR4), and transformants expressing either human or murine TLR4, cells were grown to confluence in T75 flasks, dislodged, spun and re-suspended in PBS, 0.1% HSA, pH 7.4, for distribution (approximately 2 × 106 cells/tube) into 1.5 ml siliconized microtubes and brought to a final volume of 1 ml in the same buffer.
To determine binding parameters on membrane-bound TLR4, increasing concentrations of [3H]LOS·MD-2 or LOS·MD-2[125I] were added to control and TLR4-containing cells in duplicate, and the samples were incubated for 30 min at 37℃ on an orbital rocker. Parent or mock transfected cells provided a control of non-specific cell binding for each ligand at each dose. After 30 min, the samples were placed on ice and spun at 2000 g for 5 min at 4℃. The supernatants were removed and the cell pellets were washed twice with ice cold PBS, 0.5% HSA, pH 7.4 (1 ml). For cells bound with LOS·MD-2[125I], radioactivity associated with the cells, in the supernatants and washes was determined by direct counting in the gamma counter (Packard Cobra II autogamma counter). For cells bound with [3H]LOS·MD-2, the pellet was re-suspended and radioactivity in the re-suspended pellet, supernatant and washes was determined by liquid scintillation spectroscopy (Beckman LS 6500 scintillation counter). Binding of [3H]LOS·MD-2 or LOS·MD-2[125I] to control cells was subtracted from the total [3H]LOS·MD-2 or LOS·MD-2[125I] bound to the TLR4 expressing cells at each dose of LOS·MD-2 to calculate specific TLR4-dependent binding. Based on 1:1 mol/mol binding of LOS·MD-2 to TLR47,18,21 and the known specific radioactivity of LOS·MD-2[125I], the number of molecules of TLR4 on the cell surface/cell could be derived from the Bmax value acquired by Scatchard analysis of binding data on 2 × 106 cells/sample.
For experiments examining inhibition of LOS·MD-2[125I] binding to TLR4 in transiently transfected cells, increasing doses of sMD-2 or LOS·MD-2 were added to the cells (2 × 106 /sample) before addition of LOS·MD-2[125I] (60 pM). Binding experiments were then performed as described earlier for 30 min at 37℃. A similar binding assay was performed with LOS·MD-2[125I] using bone marrow derived macrophages (BMDM) from wt and TLR4-/- C57BL/6 mice that were immortalized by transformation with a J2 transforming retrovirus expressing myc and raf. 25 These immortalized BMDM (iBMDM) were developed and generously provided by Dr Katherine Fitzgerald (University of Massachussetts Medical School, Worcester, MA, USA). The cells were maintained in DMEM, high Glc, 10% heat inactivated FBS supplemented with ciprofloxin (10 µg/ml), dislodged for binding assays with Versene (Gibco), and cell binding was performed in solution as described earlier. At each dose of LOS·MD-2[125I], samples were incubated ± sMD-2 (5 nM), a dose that saturated TLR4, to determine sMD-2-sensitive cell binding of LOS·MD-2[125I].
For all cell-binding experiments, the number of cells/sample was normalized by cell counting and gating using the Accuri flow cytometry apparatus. Total protein/aliquot of each cell preparation was measured using the BCA protein assay (Pierce) to confirm similar cell mass/sample.
Cell activation by LOS·MD-2
Cell activation by [3H]LOS·MD-2, [13/14C]LOS·MD-2 or LOS·MD-2[125I] in control (no TLR4) and TLR4-expressing cells was determined on a portion of the cells described above that had been dislodged, re-suspended, and counted before use in binding experiments. This permitted a direct correlation between TLR4-dependent cell binding and cell activation by LOS·MD-2 in the same cell population. Cells were transferred to 96-well plates—approximately 105 cells/well in 0.2 ml. Cells were stimulated with increasing concentrations of [3H]LOS·MD-2 or LOS·MD-2[125I] for 6 h before the supernatants were collected and secreted IL-8 was measured in the medium using the BD OptEIA human IL-8 ELISA assay (BD Biosciences). In the case of iBMDM cells (wt, TLR4-/-, and CD14-/-), cells were washed twice with warm (37℃) PBS, pH 7.4, and incubated with the indicated concentrations of LOS·MD-2 wt or LOS·MD-2 F126A for 18 h at 37℃ in DMEM/0.1% HSA. Cell activation was assessed by measuring accumulation of extracellular TNF-α by ELISA (BD Biosciences).
Results
Radioiodination of LOS·MD-2
We have previously described the generation of complexes of LOS·MD-2 on a preparative scale using recombinant human sMD-2 generated by baculoviral infection of insect cells and metabolically labeled ([14/13C]) endotoxin, LOS, from N. meningitidis.
18
Purified complexes of LOS·MD-2 were isolated by a combination of metal chelation and size exclusion chromatographies. While metabolically-labeled [3H]LOS·MD-2 has proven useful in quantifying the high-affinity binding of endotoxin·MD-2 with the TLR4 ectodomain in solution,7,17 the apparently low levels of membrane-bound TLR4 receptors/cell in many endotoxin-responsive cells necessitated development of a ligand that could be detected and measured more sensitively when levels of TLR4/cell and/or the number of TLR4-expressing cells available for study were limited. To generate such a reagent, we have taken advantage of the stability of LOS·MD-2 and the presence of several tyrosines (Figure 1A) on the surface of human MD-2, including several residues (shown in blue in Figure 1A) that are not required for interactions with endotoxin and/or TLR4.11,26–29 Tyrosines that have been shown to be essential for activity based on mutagenesis studies for both murine and human sMD-2 are shown in red (Y34, 36, 76, 102, and 131) in Figure 1A.11,26–29 A purified complex of human LOS·MD-2 was iodinated under mild reaction conditions using the reagent Iodogen. A monomeric LOS·MD-2 complex in which 98% of the [125I] counts are protein-bound, as judged by precipitation by ice-cold 10% TCA eluted co-incident with [3H]LOS·MD-2 on Sephacryl S200 exclusion chromatography (Figure 1B), confirming that LOS·MD-2[125I] remains a monomeric complex. LOS·MD-2 is dissociated by SDS treatment. The peak of [125I] recovered from application of LOS·MD-2[125I] to, and elution from, Sephacryl S200 co-migrated on SDS-PAGE with MD-2 (data not shown), consistent with radioiodination of MD-2 in the LOS·MD-2 complex. LOS·MD-2[125I] recovered after removal of free iodine had a specific radioactivity ∼500,000 cpm/pmol. Based on the specific radioactivity of the [125I], the amount of MD-2 used in the reaction, and the amount of [125I]product recovered in the peak fraction, the calculated molar ratio of [125I] to MD-2 in LOS·MD-2[125I] is approx. 1:1.
Iodination of MD-2 in a purified, monomeric LOS·MD-2 complex. (A) Two views of the tyrosine residues in human MD-2 are represented in ribbon models of MD-2 derived from the X-ray crystal structure by Park et al.
21
of LPS/MD-2/TLR4ecd. Based on mutagenesis studies of human and murine MD-2,11,26–29 tyrosines essential for MD-2 function are shown in red; non-essential tyrosines are shown in blue. (B) [3H]LOS·MD-2 and LOS·MD-2[125I] co-elute on Sephacryl S200 size exclusion chromatography (column 1.6 cm × 70 cm) in PBS, 0.1% HSA, pH 7.4. Recovery of the applied cpm for both reagents was ∼100%. The void volume and albumin (HSA) peak absorbances are indicated for reference. The internal volume of the column as judged by elution of vitamin B12 was 67 ml.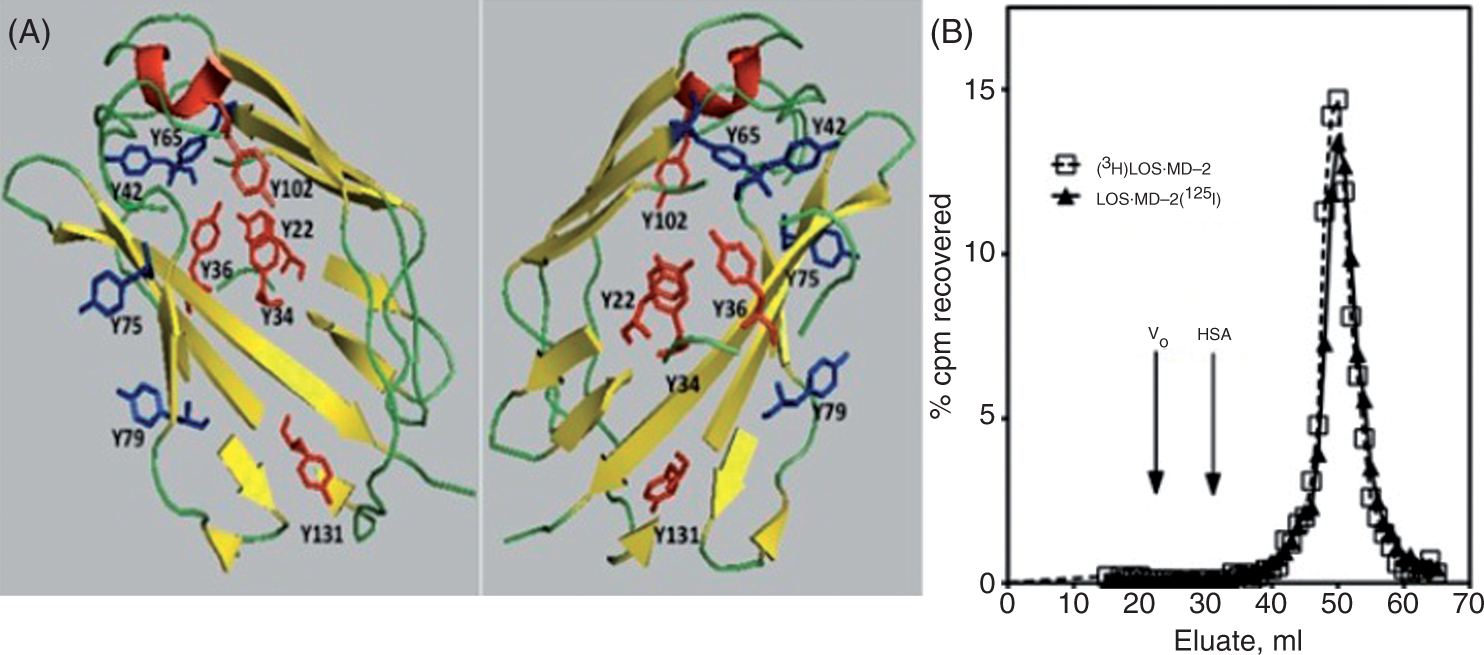
Comparison of the TLR4-binding properties of metabolically-labeled [3H]LOS·MD-2 and radioiodinated LOS·MD-2[125I]
The TLR4-binding properties of LOS·MD-2[125I] were evaluated in assays using TLR4ecd in order to directly compare LOS·MD-2[125I] with metabolically-labeled [3H]LOS·MD-2. Dose-dependent interaction of LOS·MD-2[125I] and [3H]LOS·MD-2 with soluble N-terminal FLAG-tagged TLR4ecd (27–527 a.a.) was measured using a previously described co-capture assay.
17
Increasing doses of LOS·MD-2[125I] or [3H]LOS·MD-2 were incubated with conditioned medium ± approximately 100 pM FLAGTLR4ecd. Treatment with anti-FLAG Ab agarose beads resulted in co-capture of [3H]LOS·MD-2 or LOS·MD-2[125I] bound to FLAGTLR4ecd. At each dose of [3H]LOS·MD-2 and LOS·MD-2[125I], specific FLAGTLR4ecd-dependent capture was calculated by subtraction of cpm adsorbed to the beads in the absence of FLAGTLR4ecd (‘non-specific’ capture) from cpm captured after incubation of [3H]LOS·MD-2 or LOS·MD-2[125I] with FLAGTLR4ecd (‘total’ capture). As seen in Figure 2A and B, Scatchard analyses of these data show specific, saturable and high-affinity binding of LOS·MD-2[125I] to TLR4ecd that is similar to binding of [3H]LOS·MD-2 to FLAGTLR4ecd: Kd = 117 ± 8 pM for [3H]LOS·MD-2 and 153 ± 92 pM for LOS·MD-2[125I].
Comparison of dose-dependent binding of [3H]LOS·MD-2 (A) and LOS·MD-2[125I] (B) to soluble FLAGTLR4ecd by anti-FLAG agarose co-capture. Increasing concentrations of [3H]LOS·MD-2 (A) or LOS·MD-2[125I] (B) were incubated for 30 min at 37℃ in PBS, 0.1% HSA supplemented with conditioned medium expressing FLAGTLR4ecd or from control medium without FLAGTLR4ecd. Co-capture of [3H] or [125I] labeled LOS·MD-2 by anti-FLAG affinity gel was measured as described in Materials and methods. Inserts in (A) and (B) show dose-dependent capture of [3H]LOS·MD-2 (A) and LOS·MD-2[125I] (B) after incubation and subsequent capture with conditioned medium containing FLAGTLR4ecd (total) and medium from control cells (non-specific). Specific capture shown as a saturation curve in (A) and (B) was calculated as the difference between total and non-specific capture. Scatchard analyses of these data by GraphPad Prism were used to determine Kd and Bmax. Binding to FLAGTLR4ecd indicated Kd = 117 ± 8 pM for [3H]LOS·MD-2 and 153 ± 92 pM for LOS·MD-2[125I]. Data shown are from one experiment with duplicate samples for each dose and are representative of three closely similar experiments.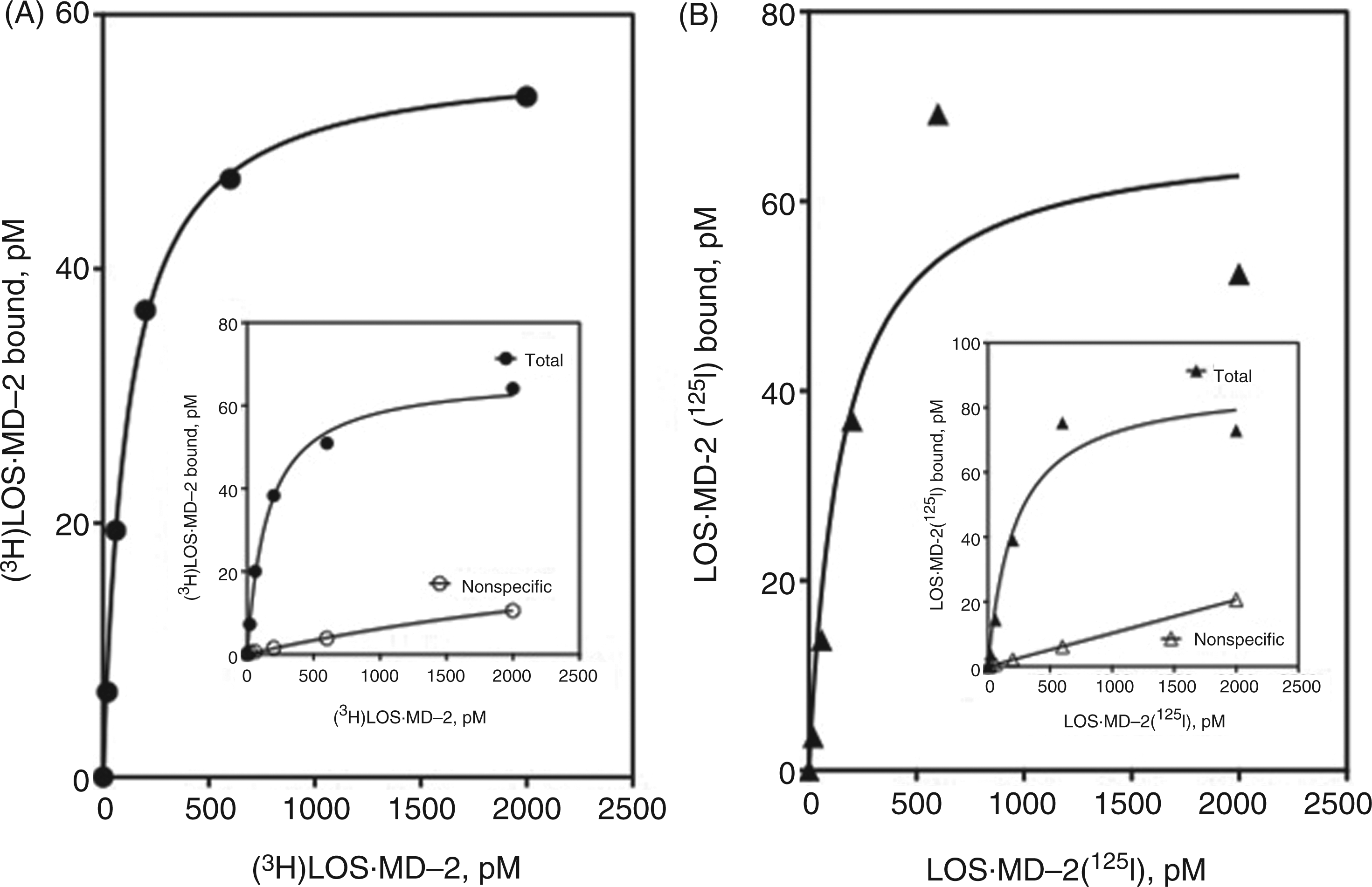
To extend the studies to interactions with full-length TLR4 so that binding of LOS·MD-2 and subsequent TLR4-dependent cell activation could be compared, HEK293T cells were transiently transfected with TLR4 or an empty control vector, and the cell binding parameters and endotoxin-dependent TLR4 cell activation by LOS·MD-2[125I] and [3H]LOS·MD-2 were evaluated. Transfection using Lipofectamine2000 resulted in levels of TLR4 expression that facilitated comparisons of TLR4-dependent cell binding of LOS·MD-2[125I] and [3H]LOS·MD-2. Non-specific binding of increasing doses of the radioactive LOS·MD-2 was determined after incubation with the radioactive ligands and cells that had been transiently transfected with control empty vector. LOS·MD-2[125I] and [3H]LOS·MD-2 complexes bound cell surface TLR4 with similar high affinity: Kd = 367 ± 136 pM and 694 ± 101 pM for [3H]LOS·MD-2 and LOS·MD-2[125I] respectively (Figure 3A, B). Non-specific binding to control transfected cells accounted for <10% of the total binding (see Figure 3A and B inserts) at LOS·MD-2 concentrations ≤Kd. Specific TLR4 binding was similar at 4℃, indicating that binding was to TLR4 at the cell surface (data not shown). The derived Bmax for TLR4-dependent LOS·MD-2 binding to the transiently transfected cells indicates an average of approximately 10,000 TLR4s/cell on the cell surface. Comparison of dose-dependent TLR4 binding (Figure 3A, B) and TLR4-dependent cell activation (Figure 3C and D) by both [3H]LOS·MD-2 and LOS·MD-2[125I] revealed maximal cell activation at doses of LOS·MD-2 (50–100 pM) corresponding to approximately 10% occupation of available TLR4 (i.e. approximately 1000 TLR4/cell) and appreciable cell activation (i.e. ∼10–20% of maximum) at LOS·MD-2 doses (2–6 pM) occupying ≤2% of cell surface TLR4 (≤200 TLR4/cell).
Comparison of [3H]LOS·MD-2 (A, C) and LOS·MD-2[125I] (B, D) dose-dependent binding to, and activation of, membrane-bound TLR4 in transiently transfected HEK293T cells. Binding of increasing concentrations of [3H]LOS·MD-2 (A) and LOS·MD-2[125I] (B) to HEK293T cells (2 × 106 cells/dose) transiently transfected (Lipofectamine2000) with TLR4 or control vector during 30 min incubation at 37℃. Inserts in (A) and (B) show dose-dependent [3H]LOS·MD-2 (A) and LOS·MD-2[125I] (B) bound after incubation with TLR4-transfected cells (total) and mock-transfected cells (non-specific). Specific binding shown in (A) and (B) as a saturation curve was calculated as the difference between total and non-specific cpm bound. Scatchard analyses of these data by GraphPad Prism were used to determine Kd and Bmax. Binding to transiently transfected TLR4 cells indicated Kd = 367 ± 287 pM for [3H]LOS·MD-2 and Kd = 694 ± 101 pM for LOS·MD-2[125I]. Binding experiments were performed as described in Materials and methods. Data shown represent a composite of more than three experiments, each dose done in duplicate, using aliquots of the same cell preparation for both [3H]LOS·MD-2 (A) and LOS·MD-2[125I] (B) for binding. Bmax ranged from 26 to 51 pM, corresponding to an average of 8000–15,000 cell surface TLR4s/cell. Dose-dependent cell activation by [3H]LOS·MD-2 (C) and LOS·MD-2[125I] (D) on aliquots of the transiently transfected TLR4 or mock-transfected cells used in binding experiments in (A) and (B) was measured as extracellular accumulation of secreted IL-8 by ELISA, as described in Materials and methods. Data shown are from one experiment with duplicate samples for each dose and are representative of more than three similar experiments.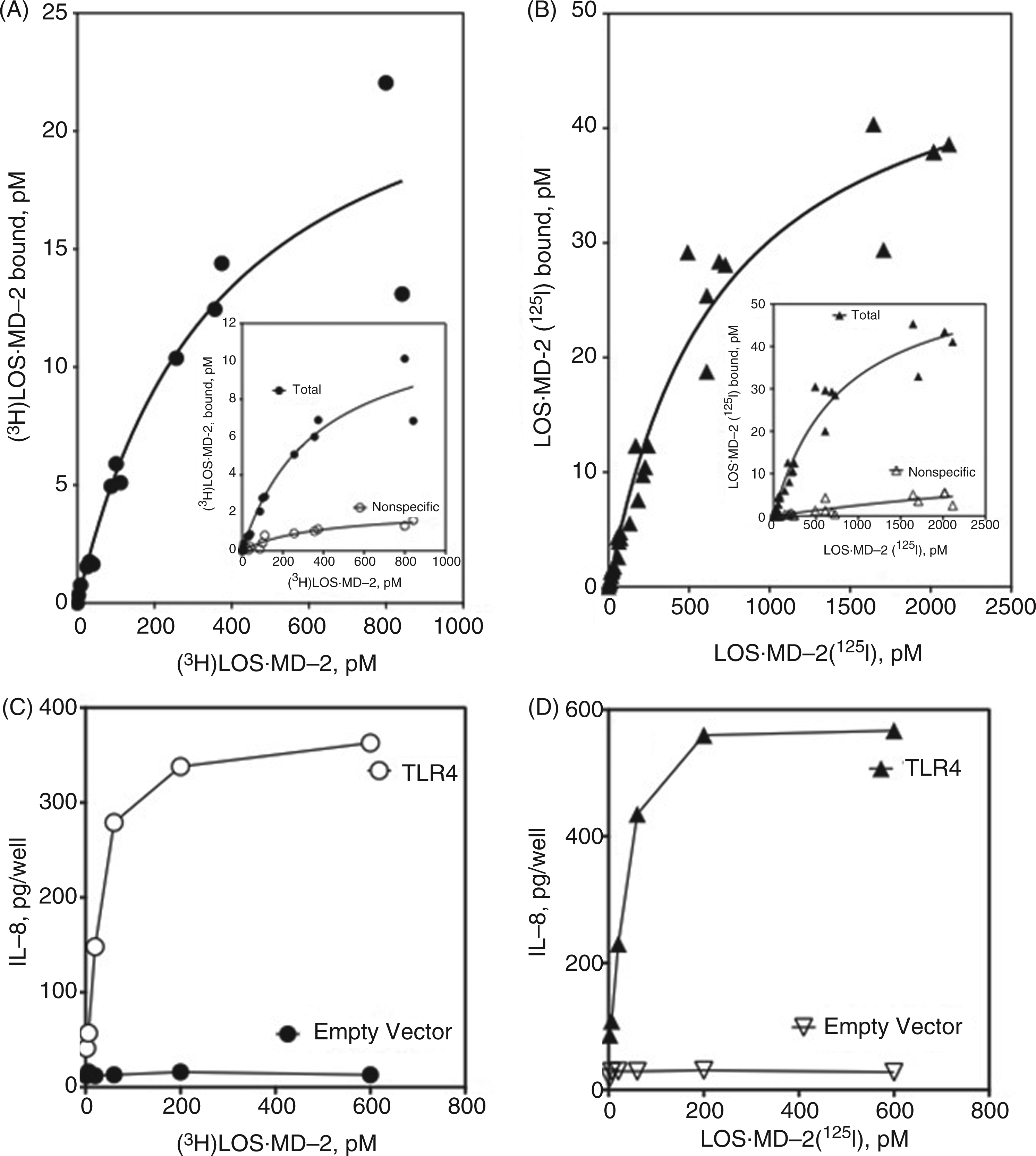
High-affinity binding of LOS·MD-2 to cellular TLR4 corresponds to agonist-independent binding of MD-2 to TLR4
Two functionally and topologically distinct binding sites within the TLR4 ectodomain for ligand·MD-2 have been defined: (i) sites within N- and central-leucine-rich repeat (LRR) regions of the TLR4ecd mediating agonist-independent interactions of MD-2 and of endotoxin·MD-2 with TLR4 within a binary (MD-2·TLR4) and ternary (endotoxin·MD-2·TLR4) complex, respectively, and (ii) sites within the C-terminal LRR region of TLR4 in one ternary complex mediating interactions with TLR4-activating endotoxin·MD-2 of a second neighboring ternary complex.19,21 As shown in Figure 4, sMD-2 alone was as potent as TLR4-activating LOS·MD-2 in causing dose-dependent inhibition of LOS·MD-2[1251] binding to cell surface TLR4. These findings indicate that agonist-independent interactions of LOS·MD-2 with cell surface TLR4, i.e. interactions within a single ternary complex, are sufficient to confer the high-affinity pM interactions observed.
sMD-2 and LOS·MD-2 inhibit binding of LOS·MD-2[125I] to TLR4. Binding of LOS·MD-2[125I] (60 pM) to cells transiently transfected (Lipofectamine2000) with TLR4 was inhibited in a dose-dependent manner by increasing concentrations of sMD-2 and [14/13C]LOS·MD-2 (15 cpm/pmol). Conditions for binding were as described in Materials and methods. Data are plotted as MD-2 concentration versus % maximal LOS·MD-2[125I] bound per 2 × 106 cells. Data shown are from one experiment with triplicate samples for each dose and are representative of three closely similar experiments.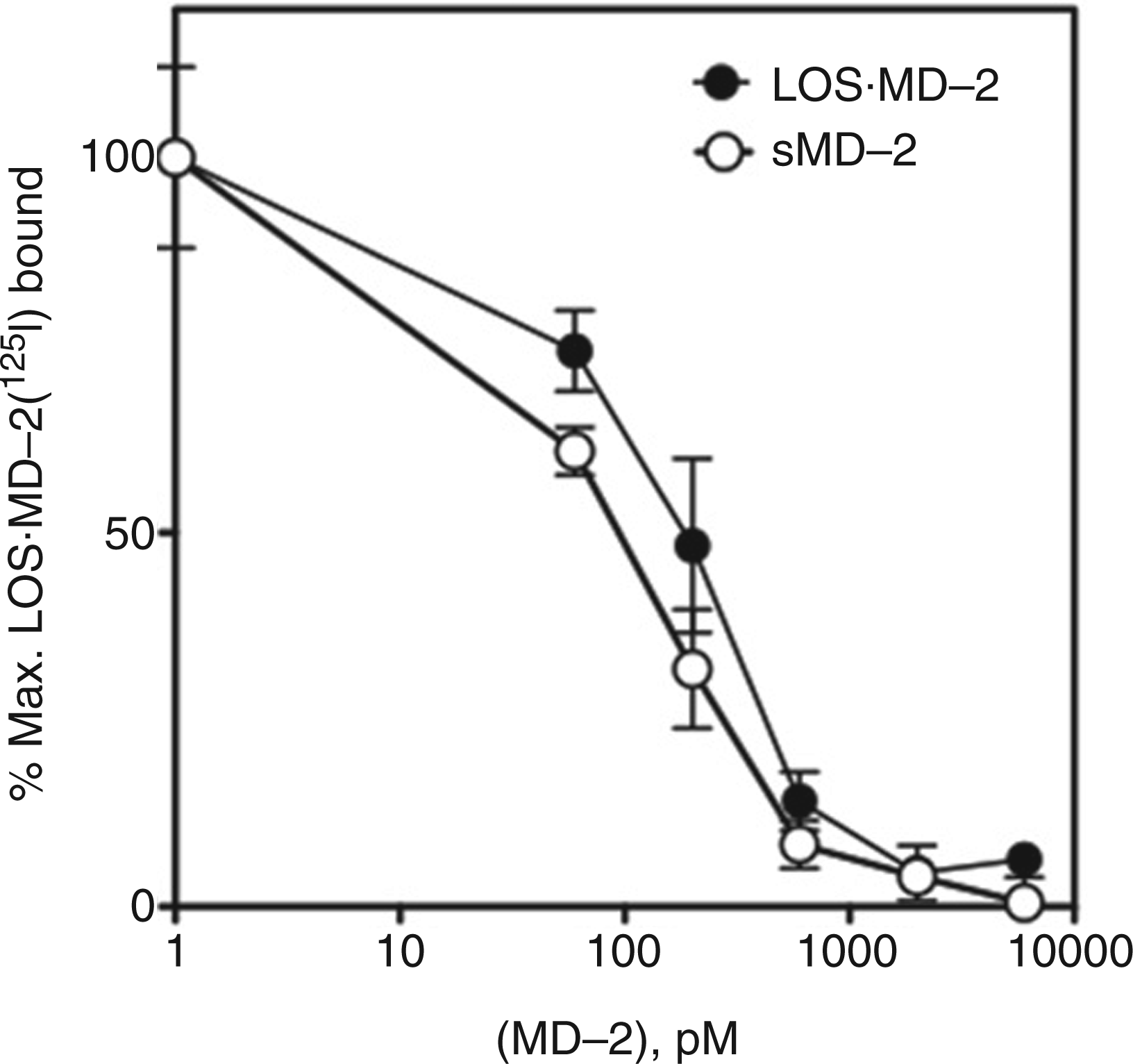
Use of LOS·MD-2[125I] to measure low levels of functional cell surface TLR4
An important advantage of LOS·MD-2[125I] versus [3H]LOS·MD-2 should be its ability to measure levels of functional surface TLR4 in endotoxin-responsive cells that express very low levels of surface TLR4. To test this feature more directly we compared TLR4-dependent cell binding of [3H]LOS·MD-2 and of LOS·MD-2[125I] to stable transformants of HEK293 cells (HEK/TLR4 cells) that we anticipated expressed lower levels of TLR4/cell than the transiently-transfected cells. Scatchard analysis of dose-dependent binding of LOS·MD-2[125I] to these cells, as well as parental HEK293 cells as a negative control, confirmed this prediction. TLR4-dependent high-affinity binding of LOS·MD-2[125I] to the stable HEK/TLR4 cells (Kd = 734 ± 40 pM) was similar to the transiently transfected cells with, on average, 4–5-fold fewer LOS·MD-2-reactive TLR4 receptors/cell, i.e. Bmax = 12.7 ± 0.3 pM/2 million HEK/TLR4 cells ml-1 corresponding to approximately 2500 surface TLR4/cell (Figure 5B, C). In contrast, the lower signal/noise (i.e. TLR4-dependent/non-specific LOS·MD-2 binding) in the stably transformed HEK/TLR4 cells precluded reliable Scatchard analysis with the metabolically labeled [3H]LOS·MD-2 (Figure 5A). In these cells, TLR4-dependent cell activation was readily detected when cells were incubated with 6–20 pM LOS·MD-2 (Figure 5D), corresponding to approximately 50–100 surface TLR4 occupied by TLR4-activating LOS·MD-2.
Ability of LOS·MD-2[125I] to detect and analyze limited human TLR4 binding sites in a stable HEK293 cell line. Binding of increasing concentrations of [3H]LOS·MD-2 (A) or LOS·MD-2[125I] (B) to HEK293 cells stably expressing TLR4 or HEK parent cells (2 × 106 cells/sample) during 30 min incubation at 37℃. Specific TLR4-dependent binding of LOS·MD-2[125I] was calculated as the difference between total and non-specific cpm bound (C). Scatchard analysis of the data by GraphPad Prism was used to determine Kd and Bmax: Kd = 734 ± 40 pM and Bmax = 12.7 ± 0.3 pM for binding of LOS·MD-2[125I] to HEK/TLR4. Data are a composite of more than three experiments, each dose done in duplicate. (D) Dose-dependent cell activation by LOS·MD-2[125I] of HEK/TLR4 or parent HEK cells was measured as extracellular accumulation of secreted IL-8 by ELISA, as described in Materials and methods. Data shown are representative of one experiment with duplicate samples for each dose and are representative of more than three similar experiments. The dose curve of only low concentrations of LOS·MD-2 is shown to highlight the sensitivity of TLR4 activation by LOS·MD-2.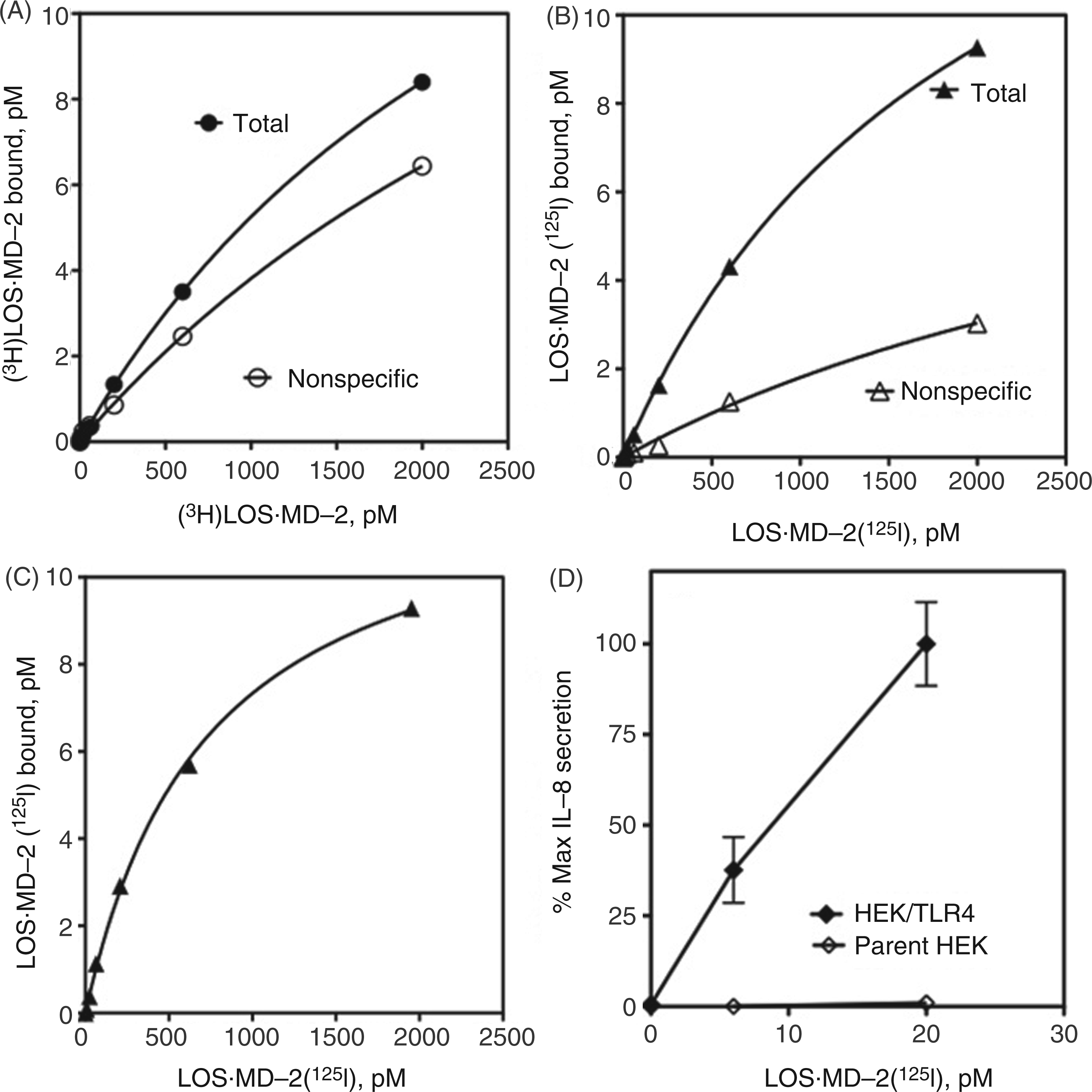
Comparison of LOS·MD-2 binding to wt and polymorphic variant TLR4
Two missense TLR4 variants, D299G and T399I, have been described that can result in reduced responsiveness to endotoxin.30–35 We have reported previously that wt TLR4ecd and D299G.T399I TLR4ecd display similar high affinity for LOS·MD-2 (Kd = ∼200 pM), but the steady-state accumulation of functional secreted D299G.T399I TLR4ecd was reduced, particularly in the absence of co-expression of MD-2.
17
Similar findings were observed when [3H]LOS·MD-2 binding to cell surface wt and D299G.T399I TLR4 were compared, but the lower levels of functional surface variant TLR4 precluded quantitative analysis by Scatchard analysis.
17
We took advantage of the more sensitive measurement of cell surface TLR4 possible with LOS·MD-2[125I] to re-examine the functional levels of cell surface wt and D299G.T399I TLR4 using the same transient transfection conditions in HEK293T cells used previously.
17
Under these conditions, there was similar transfection efficiency and transcription of expression vectors encoding wt and D299G.T399I TLR4, as confirmed by qRT-PCR.
17
Scatchard analysis of LOS·MD-2[125I] binding, as described earlier, confirmed similar high affinity of LOS·MD-2 for wt and D299G.T399I TLR4 (Kd = 528 ± 100 for wt TLR4 and 531 ± 74 pM for D299G.T399I TLR4) and reduced steady-state surface expression of the variant TLR4 (with 2 × 106 cells/ml Bmax = 12.7 ± 0.9 pM vs. 4.0 ± 0.2 pM for cells expressing wt vs. D299G.T399I TLR4; Figure 6A, B). Reduced surface expression of D2996.T399I TLR4 was paralleled by reduced cell activation by LOS·MD-2, especially at very low concentrations of added LOS·MD-2 (Figure 6C). When dose-dependent LOS·MD-2-induced cell activation was expressed as a function of the amount of TLR4-dependent LOS·MD-2 binding (Figure 6D), there was no diminution in the ability of LOS·MD-2-occupied D299G.T399I versus wt TLR4 to trigger cell activation.
Comparison of LOS·MD-2[125I] dose-dependent binding to, and cell activation of, HEK293T cells transiently transfected with full-length wt (A) or D299G.T399I (B) TLR4. Binding of increasing concentrations of LOS·MD-2[125I] to HEK293T cells transiently transfected (PolyFect) with either wt or D299G.T399I TLR4 or control vector (106 cells/sample) during 30 min incubation at 37℃ was performed as described in Materials and methods and in Prohinar et al.
17
Inserts in (A) and (B) show dose-dependent LOS·MD-2[125I] bound after incubation with TLR4-transfected cells (total) and mock-transfected cells (non-specific). Specific binding, shown as a saturation curve in A and B, was calculated as the difference between total and non-specific cpm bound. Scatchard analyses of these data by GraphPad Prism were used to determine Kd and Bmax: Kd = 528 ± 100 pM and Bmax = 12.7 ± 0.9 pM with wt TLR4 cells and Kd = 531 ± 74 pM and Bmax = 4.0 ± 0.2 pM with D299G.T399I TLR4 cells. (C) Dose-dependent cell activation by LOS·MD-2[125I] of transiently transfected wt, D299G.399I TLR4 or mock-transfected cells was measured as extracellular accumulation of secreted IL-8 by ELISA, as described in Materials and methods. Results shown are from one experiment representative of at least three experiments with each dose in triplicate. In (D) the amount of TLR4-dependent cell activation as measured by IL-8 secretion is plotted as a function of the amount of LOS·MD-2[125I] bound to wt or D299G.T399I TLR4 cells. Data are compiled from dose curves of binding and cell activation by LOS·MD-2[125I] to wt and D299G.T399I TLR4 as represented in (A), (B) and (C).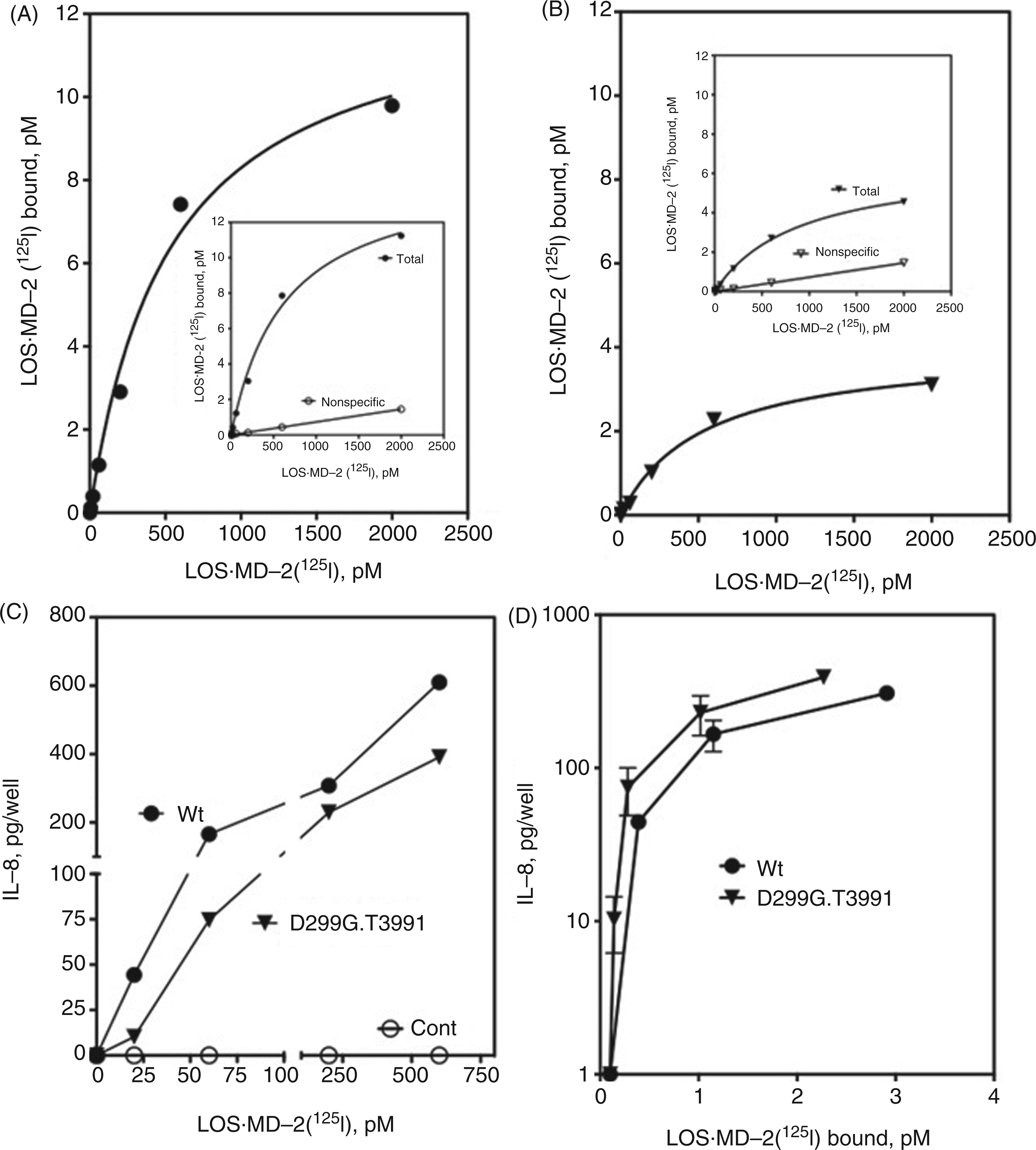
High-affinity binding of LOS·human MD-2 to human and murine TLR4
Our studies have used endotoxin·MD-2 complexes containing human MD-2 exclusively, in part because of the much greater difficulty to recover functional secreted murine MD-2.
36
Both agonist-independent and agonist-dependent interactions of murine MD-2 with TLR4 exhibit pronounced species selectivity, whereas these interactions are similar between endotoxin·human MD-2 and murine, as well as human, TLR4.27,36–41 Therefore, we expected that LOS·MD-2[125I] would display similar high-affinity binding to cells expressing murine TLR4. A stable transformant line of HEK293 cells expressing murine TLR4 was examined to test this prediction. Comparison by Scatchard analysis of TLR4-dependent cell binding of LOS·MD-2[125I] to cells expressing human TLR4 (Figures 3B, 5A and 6A) versus cells expressing murine TLR4 (Figure 7) demonstrated similar high-affinity binding of LOS·MD-2[125I] to human (Kd range from 528–730 pM) and murine (Kd = 572 ± 15 pM) TLR4.
Saturable, high-affinity binding of LOS·MD-2[125I] to murine TLR4. Binding of increasing concentrations of LOS·MD-2[125I] to HEK293 cells stably expressing murine TLR4 or parent cells during 30 min incubation at 37℃ was performed as described in Materials and methods on 2 × 106 cells/sample. Insert shows dose-dependent LOS·MD-2[125I] bound after incubation with HEK/ murine TLR4 cells (total) and parent cells (non-specific). Specific binding shown as a saturation curve was calculated as the difference between total and non-specific cpm bound. Scatchard analyses of the data by GraphPad Prism were used to determine Kd and Bmax: Kd = 572 ± 15 pM and Bmax = 10.0 ± 0.1 pM. Results shown are from one experiment representative of at least three experiments, with duplicate samples for each dose.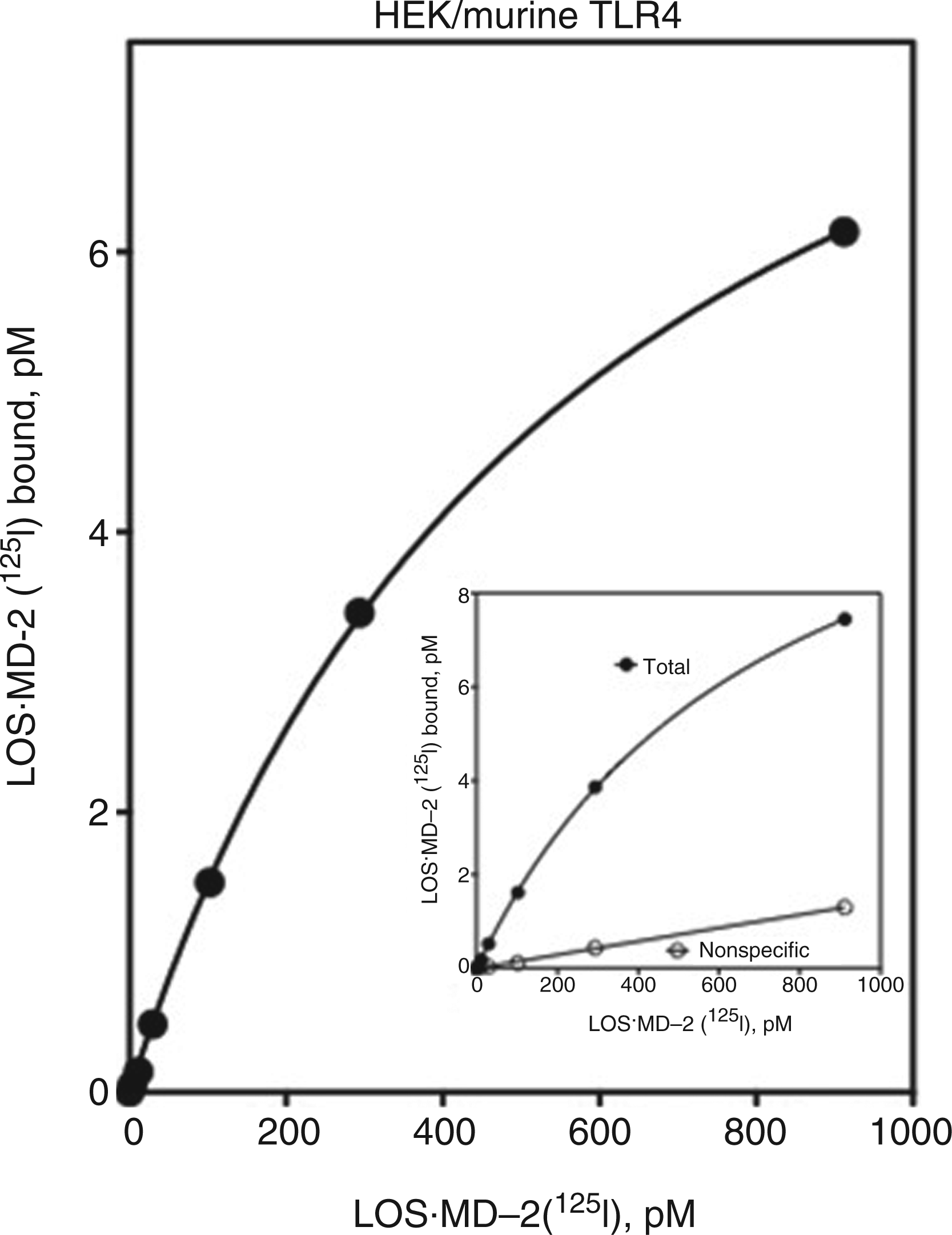
Detection of LOS·MD-2-reactive TLR4 in cells expressing both TLR4 and MD-2
The ability of sMD-2 to fully inhibit LOS·MD-2[125I] binding to TLR4 (Figure 4) strongly suggests that LOS·MD-2[125I] binding to cell surface TLR4, as previously shown for TLR4ecd,
7
is specific for TLR4 molecules that are not already occupied by MD-2. This property implies that LOS·MD-2[125I] binding could be used to measure levels of ‘MD-2-free’ TLR4 in cells expressing both TLR4 and MD-2. Crucial to this assay is that inhibition of cell binding of LOS·MD-2[125I] by sMD-2 is TLR4-dependent, i.e. sMD-2 does not inhibit non-specific cell binding of LOS·MD-2[125I]. We therefore compared cell binding of LOS·MD-2[125I] to murine BMDM cell lines (iBMDM) derived from wt and TLR4-/- mice. The wt cell line provided a test case for probing for the presence of minute numbers of membrane-bound TLR4 ‘free’ of pre-bound MD-2, as indicated by LOS·MD-2[125I] cell binding that is inhibited by sMD-2. The TLR4-/- cells were used to test that sMD-2-sensitive LOS·MD-2[125I] cell binding was TLR4-dependent. There were low levels of non-specific (TLR4-independent) binding of LOS·MD-2[125I] to the TLR4-null cells (<0.5% of total LOS·MD-2[125I] added) that was unaffected by added excess sMD-2. Saturable sMD-2-sensitive binding of LOS·MD-2[125I] was seen only in the wt murine iBMDM (Figure 8A) where similar sMD-2-insensitive, non-specific (TLR4-independent) binding of LOS·MD-2[125I] as in TLR4-null cells was also seen. Scatchard analysis of the sMD-2-sensitive binding of LOS·MD-2[125I] to the wt cells revealed a Kd = 448 ± 194 pM (Figure 8A). The high affinity and TLR4-dependence of the sMD-2-sensitive binding of LOS·MD-2[125I] measured are consistent with the presence of a subpopulation of macrophage cell surface TLR4 that is reactive with LOS·MD-2[125I], i.e. corresponds to ‘MD-2-free’ TLR4. Scatchard analysis indicated 0.9 ± 0.1 pM sites per 2 million iBMDM ml-1 (Bmax), i.e. approximately 250 ‘MD-2-free’ TLR4/cell.
LOS·MD-2[125I] detects ‘free’ TLR4 receptors devoid of pre-associated MD-2 in a murine iBMDM cell line. (A) Binding of increasing concentrations of LOS·MD-2[125I] to wt (TLR4+/+) or TLR4-/- iBMDM cells (2 × 106 cells/sample) during 30 min incubation at 37℃ was performed as described in Materials and methods. Cell binding of LOS·MD-2[125I] was done in the absence and presence of saturating amounts of sMD-2 (100 μl, 5 nM) added to saturate ‘MD-2-free’ TLR4 binding sites. Specific binding to’ ‘MD-2-free’ TLR4 in iBMDMs was determined as the difference between the total LOS·MD-2[125I] bound in wt and TLR4-/- cells minus the amount of LOS·MD-2[125I] bound in the presence of sMD-2. Scatchard analyses of the data by GraphPad Prism were used to determine Kd and Bmax. Kd = 448 ± 194 pM and Bmax = 0.9 ± 0.1 pM for specific binding of LOS·MD-2[125I] to ‘free’ TLR4 in iBMDM cells (2 × 106 cells/sample). Results shown are from one experiment representative of at least three experiments, with duplicate samples for each dose. (B, C) Dose-dependent cell activation of wt (B) or (C) CD14-/- iBMDM cells (both TLR4+/+, 105 cells/sample) by LOS·MD-2 wt or LOS·MD-2 F126A was measured after 18 h incubation at 37℃. Cell activation was assessed by measuring accumulation of extracellular TNF-α by ELISA (BD Biosciences). Results shown are from one representative experiment, with each dose in triplicate.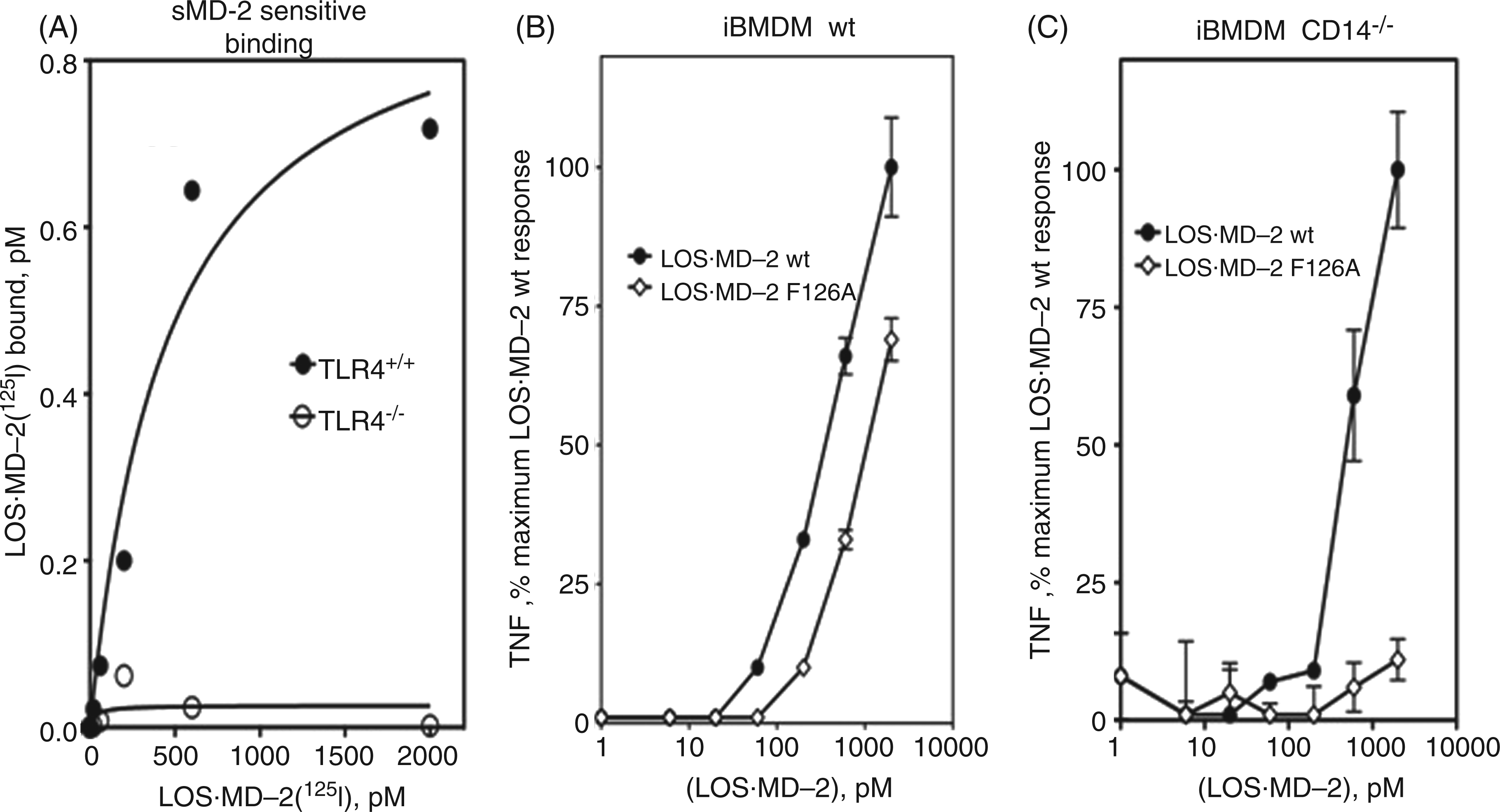
To confirm the presence of functional ‘MD-2-free’ TLR4 in wt mouse iBMDM, we compared activation as measured by extracellular accumulation of TNF-α of these cells by LOS·MD-2 wt versus by LOS·MD-2 F126A. In wt iBMDM, LOS·MD-2 wt can activate TLR4 both by direct engagement of ‘MD-2-free’ TLR47,9 and by transfer of bound endotoxin (LOS) from MD-2 to cellular mCD14 followed by transfer of LOS to and activation of cellular MD-2·TLR4 heterodimer. 42 LOS·MD-2 F126A can also transfer LOS to mCD14,11,42 but binding of this mutant complex to TLR4 directly induces little or no activation of TLR4.11,18 Thus, the presence of ‘MD-2-free’ TLR4 in wt mouse iBMDM should result in more cell activation by LOS·MD-2 wt than by LOS·MD-2 F126A. The data in Figure 8B support this prediction by showing more cell activation by LOS·MD-2 wt at each dose of wt and mutant LOS·MD-2 tested. This difference is consistent with the added contribution of activation of ‘MD-2-free’ TLR4 by LOS·MD-2 wt to the activation achieved by both wt and mutant complexes via transfer of LOS to cellular MD-2·TLR4 through mCD14. Similar cell activation assays using iBMDM derived from CD14-/- mice showed cell activation by LOS·MD-2 wt, but little or no activation by LOS·MD-2 F126A (Figure 8C), confirming the ability of LOS·MD-2 wt to induce CD14-independent activation of TLR4 by direct engagement and activation of ‘MD-2-free’ TLR4 in these cells. Note that the greater difference in cell activation by LOS·MD-2 wt versus LOS·MD-2 F126A in wt murine iBMDM was most apparent at the lower doses of LOS·MD-2 tested (≤200 pM), consistent with the high-affinity engagement and activation of ‘MD-2-free’ TLR4 by LOS·MD-2.
Discussion
The studies presented herein describe the preparation and characterization of an iodinated endotoxin·MD-2 complex, LOS·MD-2[125I], as a highly sensitive and specific TLR4 ligand that provides, for the first time, a reagent that can be used directly to detect and characterize the number of TLR4 receptors on cells that contain TLR4, but lack MD-2. Using 1–2 × 106 cells/ml in incubations with LOS·MD-2[125I], we could detect as few as ∼100 cell surface TLR4/cell and demonstrate that this is a sufficient number of TLR4 receptors to produce measurable TLR4-dependent cell activation when engaged by activating endotoxin·MD-2 complexes. It was possible to detect and quantify, for the first time, ‘free’ TLR4 (i.e. TLR4 without pre-associated MD-2) on the surface of cells expressing both TLR4 and MD-2 by utilizing the ability of added sMD-2, in substantial molar excess, to selectively inhibit TLR4-dependent cell binding of LOS·MD-2[125I] (Figure 8). The closely similar binding of LOS·MD-2[125I] to human and murine TLR4 (Figures 3, 5, 7 and 8) is consistent with earlier evidence,27,36–41 suggesting similar functional interactions of human MD-2 with human and murine TLR4, and extends the applicability of this new reagent to an array of human and murine cells and tissues. Essential to the utility of LOS·MD-2[125I] is its very high specific radioactivity (approximately 500,000 cpm/pmol) and retention of the TLR4-binding and TLR4-activating properties of metabolically-labeled LOS·MD-2 (Figures 2 and 3). These features suggest that iodination under the conditions presented herein do not affect essential tyrosines or features of MD-2 that are necessary for stability of endotoxin·MD-2, or key to TLR4 recognition and activation. LOS·MD-2[125I] binds to both TLR4ecd in solution, as well as cell surface, transmembrane TLR4 in transiently transfected cells with the same high affinity as [3H]LOS·MD-2. An improved method of transient transfection of HEK293T cells with TLR4 resulted in sufficient presentation of TLR4 on the membrane-surface to allow for a saturation curve and Scatchard analysis of bound [3H]LOS·MD-2 so a direct comparison of its binding features could be made with LOS·MD-2[125I]. Comparison of the binding affinity of either [3H]LOS·MD-2 or LOS·MD-2[125I] to membrane-bound TLR4 versus TLR4ecd in solution indicates a slightly lower affinity of LOS·MD-2 for transmembrane TLR4 (Kd ∼200 pM for TLR4ecd versus ∼500 pM for membrane-bound TLR4). These findings are consistent with the view that all binding interactions between LOS·MD-2 and TLR4 occur within the ectodomain of TLR4, presumably explaining the efficient binding we observe at 4℃. The basis of the small difference in the Kd of interactions of LOS·MD-2 with TLR4ecd versus full-length transmembrane TLR4 is not known, but could reflect constraints to molecular motion of TLR4 imposed by insertion of full-length TLR4 into the membrane in comparison to more freely ‘tumbling’ TLR4ecd in solution.
The ability of pM endotoxin concentrations to activate TLR4 is consistent with the role of TLR4 as a sentinel for early detection of even small numbers of invading GNB. However, these data alone do not indicate how many TLR4 receptors on the surface of endotoxin-responsive cells need to be occupied to trigger measurable TLR4-dependent cell activation. The unique ability of LOS·MD-2 to directly engage and activate TLR4 together with the very high specific radioactivity of LOS·MD-2[125I] made it possible to quantify the number of cell surface TLR4s needed to be occupied by an activating ligand, LOS·MD-2, to trigger cell activation. Comparison of dose-dependent TLR4-dependent cell binding and activation by LOS·MD-2[125I] revealed that, in transiently transfected HEK293 cells, no more than 10–20% maximal occupancy of cell surface TLR4 is needed for maximal cell activation and that occupancy of as little as 1–2% of surface TLR4, corresponding to around 100–200 TLR4/cell, is sufficient for appreciable cell activation, as measured by extracellular accumulation of IL-8 (Figure 3). Data in stable transformants of HEK293 cells expressing TLR4 indicate that, in these cells, occupancy of as few as 50–100 TLR4/cell is sufficient for measurable TLR4 activation (Figure 5). These findings demonstrate dramatically both the sensitive—low Kd—and robust—small number of activated receptors sufficient for graded cellular responses—properties of TLR4, fully consistent with its sentinel role in innate immune recognition and response to invading GNB. The small number of engaged TLR4 sufficient for cell activation underscores the importance of sensitive and quantitative detection of TLR4 that LOS·MD-2[125I] provides. If dimerization of TLR4-activating ternary complexes (e.g. LOS·MD-2·TLR4) is required for TLR4 activation by endotoxin, our data strongly suggest that dimerization occurs remarkably efficiently, even when there are only 50–100 TLR4-activating ternary complexes within an entire cell surface.
An example in which changes in steady-state levels of functional surface TLR4 expression affect cellular response to endotoxin is illustrated by our comparative studies of wt and variant D299G.T399I TLR4 expression in transfected HEK293T cells. 17 The high specific radioactivity of LOS·MD-2[125I] made possible full saturation curves and Scatchard analysis of LOS·MD-2 binding to these cells, and confirmed and extended our earlier evidence that the principal effect of the combined D299G and T399I substitutions within the TLR4 ectodomain is on the steady-state levels of functional surface TLR4 and not the intrinsic functional properties of the expressed TLR4 molecule (Figure 6A, B, D). 17 The reduced responsiveness of transfected cells expressing D299G.T399I rather than wt TLR4 was most apparent at low doses of LOS·MD-2 (Figure 6C), i.e. conditions in which the reduced surface expression of variant TLR4 resulted in levels of LOS·MD-2-occupied variant TLR4 below the threshold needed for maximal TLR4-dependent cell activation. However, as seen in Figure 6D, when data were normalized for the amount of LOS·MD-2 bound to available wt or D299G.T399I TLR4, a similar degree of cell activation was observed, demonstrating that LOS·MD-2-occupied variant TLR4 had an undiminished ability to trigger cell activation.
The absence of measurable exchange between MD-2 bound to LOS in the monomeric LOS·MD-2 complex with even 100-fold molar excess sMD-2 or LOS·MD-2, within the 30 min time frame of our experiments (data not shown), 42 made possible unambiguous comparison of the effects of excess sMD-2 and LOS·MD-2 on TLR4-dependent cell binding of LOS·MD-2[125I]. The demonstrated ability of sMD-2 to inhibit, as potently as LOS·MD-2, TLR4-dependent cell binding of LOS·MD-2[125I] (Figure 4) strongly suggests that ‘agonist-independent’ interactions of MD-2 with the TLR4 ectodomain principally drive the pM binding of LOS·MD-2[125I] to cell surface TLR4. Recent X-ray crystallographic analyses of MD-2·TLR4ecd and ligand·MD-2·TLR4ecd complexes19–21 have helped to define the sites within the TLR4 ectodomain, apparently mediating these agonist-independent interactions. Our findings suggest that assays of TLR4-dependent LOS·MD-2[125I] binding could be used to further define the structural determinants within TLR4, important not only for interactions with TLR4-agonist and TLR4-antagonist ligand·MD-2, but also with ligand-free MD-2, as needed for biogenesis of MD-2·TLR4 heterodimers. Competition assays of LOS·MD-2[125I] binding to TLR4 with other putative agonists of TLR4, such as aero-allergens, for example Der p 2, that are structurally and functionally related to MD-2,43–46 should also help to define the molecular and structural bases of the TLR4-dependent actions of these compounds.
Most cells expressing TLR4 also express MD-2. Given the pM interactions between ligand-free MD-2 and TLR4 that we have described, it has been generally presumed that in these cells surface TLR4 is expressed as a pre-formed heterodimer with MD-2. Thus, actions of exogenously generated ligand·MD-2-like complexes, such as Der p 2, would be largely restricted to those few cell types (e.g. airway epithelial and human corneal cells15,47,48) that express TLR4 with little or no MD-2 under resting conditions. Our ability to measure specifically sMD-2-sensitive LOS·MD-2[125I] binding to ‘free’ TLR4 in murine iBMDM suggests a broader range of cell types expressing cell surface functional TLR4 without MD-2. Our findings indicate that a potentially broader range of host cells may be responsive to MD-2-related aero-allergens, as well as extracellular endotoxin·MD-2 complexes that could be formed under inflammatory conditions when expression and secretion of sMD-2 are up-regulated.47,49–52 Our ability to produce ligand·MD-2[125I] that are either TLR4 agonists or antagonists, and to specifically measure ‘free’ TLR4 in both human and murine cells by assay of sMD-2-sensitive ligand·MD-2[125I] cell binding, should greatly facilitate future efforts to better define the possible sites and roles of ‘MD-2-free’ TLR4 in the actions of both endogenous ligand·MD-2 and other putative agonists of TLR4 that may need to engage TLR4 similarly to produce their immune effects.
Increasing evidence suggests even wider physiologic and pathophysiologic roles of TLR4 mediated, at least in part, by interactions of exogenous and/or endogenous host compounds—structurally unrelated to E—with MD-2·TLR4 or perhaps TLR4 alone.53–57 The compounds include a diverse range of molecules [damage associated molecular pattern molecules (DAMPS)], including viral proteins, parasite glycolipids, host proteins, glycosaminoglycans, hemin and lipids.17,53–58 In contrast to the insights that have been gained concerning the mechanism of E-induced MD-2·TLR4 activation, little is known about how these other putative TLR4 agonists affect TLR4 function and if their action is MD-2 dependent. Competition in binding assays with LOS·MD-2[125I] can be used to test if these DAMPS interact with TLR4 ectodomain in a manner analogous to ligand·MD-2.
Footnotes
Acknowledgements
We thank DeSheng Zhang for preparation, isolation and characterization of [14/13C]LOS. We also thank Dr Richard Tapping (University of Illinois, Urbana, Il, USA) for the generous gift of Freestyle HEK293F cells stably transfected with TLR4 (amino acids 27–527), Dr Stefanie Vogel (University of Maryland, Baltimore, MD, USA) for the pCMV-FLAG-TLR4 vector containing TLR4 wt and mutant D299G.T399I, and Dr Katherine Fitzgerald (University of Massachusetts Medical School, Worcester, MA, USA) for wt and TLR4-/- immortalized BMDM cell lines.
Funding
The work reported herein was supported by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development grant [1I01BX0000949-01A1 to T.L.G.) and grants to J.P.W. from the National Institute of Allergy and Infectious Disease, National Institutes of Health [grant numbers AI059372 and AI088372]. The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the granting agencies.
Conflict of interest
The authors have no conflicts of interest to report.