AMP-activated protein kinase is a crucial regulator of cellular metabolism with potential therapeutic implications in various diseases. In this study, the benzimidazole scaffold was chosen for investigation with EX-229 as the lead compound. Modifications were made to the terminal carboxyl group, and the methylindole moiety was replaced with phenylpyrrolidinone to synthesize and assess the AMP-activated protein kinase-activating properties of 13 benzimidazole derivatives. In vitro studies have shown that most of the synthesized compounds have significant excitatory effects on AMP-activated protein kinase. Particularly, A11 demonstrated the most potent activity with an EC50 value of 39 nM. Combining activity data and molecular docking analysis revealed the necessity of end-molecule hydrogen bond donors for LYS29, and the importance of an appropriate torsion angle at the pyrrolidinone position to facilitate the formation of a crucial hydrogen bond with LYS31. These discoveries offer preliminary insights into the interaction of small-molecule drugs with the AMP-activated protein kinase protein and establish a foundation for the future development of AMP-activated protein kinase activators guided by structure–activity relationships.
A series of new benzimidazole analogues were synthesized and evaluated as AMPK agonists. Among the synthesized compounds, A11 exhibited a strong affinity for AMPK. The EC50 is 39 nM. These results suggest that compound A11 is a potent AMPK agonist.
AMP-activated protein kinase (AMPK) is an enzyme that plays a key role in regulating cellular energy metabolism. As a nutrient and energy sensor, it plays a key role in the whole body energy homeostasis.1 AMPK exhibits reduced activity in diseases with caloric excess, indicating dysregulation of cellular metabolism and energetics and decreased AMPK activity may play a causal role in disease etiology. For example, AMPK inhibits cell proliferation by activating catabolic processes and inhibiting anabolic processes under energy stress.2–5 Therefore, AMPK has great potential as a target in the treatment of diabetes, pulmonary inflammatory diseases, and cancer.6–8 As a result, activating AMPK by small molecules may be a viable method to restore energy metabolism and manage disease.9
It is still important to develop more AMPK agonists, here are some AMPK small-molecule agonists (Figure 1).
Some small-molecule agonists acting on AMPK receptor.
A-769662, a direct activator of AMPK discovered by Abbott,10,11 exerts its activation on AMPK allosterically and suppresses fatty acid synthesis in rat hepatocytes, demonstrating an EC50 value of 3.2 mM.10MT63-78 was selected by Zadra et al.,12 with an EC50 of 25 μM, which was 16 to 40 times higher than that of A-769662. Mirguet et al. from GSK conducted a series of structural modifications based on Compound A for similar structures, aiming to enhance oral bioavailability and selectivity. Their research revealed that the compound’s intrinsic acidity, in the presence of the cyanide group, resulted in low membrane permeability. Substituting the cyanide group with a carboxyl group was found to enhance AMPK activity.13 With the introduction of electron-absorbing groups (e.g. F and Cl atoms) into R1, oral availability is significantly improved. However, plasma clearance and cell membrane permeability remain suboptimal. When combined with P-protein blockers, these groups can effectively reduce plasma clearance.14 In the subsequent development of PF-06409577 by Pfizer, the substitution of carboxylic acid groups and the parent Cl led to an EC50 of 7 nM, resulting in improved selectivity. This implies that the presence of the carboxylic acid on the right side and the chlorine substitution on the core nucleus are essential.15,16
Since 2010, Merck and Metabasis Therapeutics have filed multiple patents related to the AMPK agonist of benzimidazole mother-core.17–20 They have preserved the 4-(2-hydroxyphenyl) phenyl fragment of A-769662. Various substituents such as CH3, OCH3, F, Cl, and CN were utilized to explore the positioning of Cl on the parent nucleus. The findings align closely with those of Mirguet et al., indicating that only Cl and F substitutions lead to a significant increase in EC50 value, with Cl substitution being the most effective. Furthermore, their analysis of carboxylic groups revealed that groups with high steric hindrance and carboxylic acid exhibited greater activity at the 2′ position. Mercaptan and phenol link were also tested, and based on the EC50 values, the O connection was found to be slightly more effective than the S connection. This could be attributed to the lower electronegativity of sulfur atoms compared with oxygen atoms, which hinders the formation of intermolecular forces with proteins. The resulting Compound B exhibited excellent performance with an EC50 of 3 nM, and EX-229 showed a value of 2 nM. In addition, the binding affinity of the EX-229 was found to be 10 times greater than that of A-769662.21PF-739 is obtained by Merck by introducing a large number of natural groups into the 2′ position of the pyridine-imidazole parent nucleus using the isosteric principle of electrons.22,23PF-739 is an orally active and non-selective AMPK activator EC50 of 9 nM.24
However, up to now, no small-molecule drugs that can directly stimulate AMPK have passed clinical trials, and the drugs targeting AMPK in clinical practice are mainly metformin and statins that can indirectly stimulate AMPK.25–27 Therefore, the development of small-molecule AMPK direct agonists remains critical. Based on EX-229, we used molecular docking software to design and synthesize 13 compounds to explore the structure–activity relationship (SAR) of AMPK targets, hoping to find AMPK agonists with better activity.
Results and discussion
Based on the reported activities of EX-229 and its homologs, the SAR was preliminarily inferred:28 the introduction of electron-withdrawing groups (e.g. F, Cl) at position-6 of the benzimidazole ring was beneficial to enhance the activity. When the position-5 group is aromatic ring or heterocyclic compound, AMPK activity is significant. The pharmacophore with large hindrance of electronegative group has excellent activity at position-2′.
Furthermore, it was noted that starting from the initial A-769662, the majority of compound 5 sites exhibited a preference for incorporating substituted biphenyls, resulting in consistently higher activity levels. It was hypothesized that this phenomenon could be attributed to the ability of biphenyls to generate the appropriate torsion angle in this context. Currently, the termini of the compounds fabricated by predecessors are predominantly acids or hydroxyl groups, with limited exploration on the terminal carboxylic group of the molecule. So we replace the terminal carboxylic acid with esters, amides, and hydroxamic acid derivatives, thereby investigating the SAR in this context. The relationship between the torsion angle and the activity was studied by linking the substituted phenyl group with pyrrolidone.
Thus, it was attempted to replace the indole ring of EX-229 with (2-oxopyrrolidin-1-yl)phenyl, or 4-(2-oxopiperidin-1-yl) phenyl to get target compounds in order to mimic similar effects as PF-06409577via bioisosterism, while the necessary electronegative 6-F or Cl was retained. It was also tried to synthesize the 4-membered 4-(2-oxoazetidin-1-yl)phenyl analogs, but failed. Substituents were introduced into the phenyl ring connected to the 1H-benzimidazole-5-yl core. The carboxylic compounds were also converted to their esters, amides, or hydroxamic acid (right part) for the sake of compound diversity. Hence, 13 target compounds were designed, synthesized, and AMPK activity evaluated in vitro in this paper. Among them, A11 exhibits excellent AMPK activity with EC50 of 39 nM. It was also investigated the docking pattern to verify our hypothesis.
At present, there is only one synthetic route of EX-229 reported in the literature.29 In the route described in US201113086563, a similar approach was adopted (Scheme 1). Starting from 1 (R1=F or Cl), it was converted to the bromo 2, which was brought into the diamino 3; 3 was treated with carbon disulfide for cyclization to construct the benzimidazole ring. The afforded 4 was methylated with iodomethane to produce 5, which was further converted into its sulfonyl 6; 6 was protected with SEM-Cl to give 7; 7 was substituted with methyl 5-hydroxy-2-methylbenzoate or methyl 3-hydroxybenzoate to obtain 8; 8 is then subjected into the Suzuki coupling with corresponding borate ester to yield 9. After the removal of the protecting group, 9 was further hydrolyzed to yield target compounds A1 and A2.
In the process of synthesis, it is noteworthy that the synthesis involves starting materials substituted with F and Cl (where F exhibits greater electron absorption strength than Cl). This variance impacts the yield in the synthesis process. Generally, the yield in the synthesis process is slightly lower for F substitution compared with Cl substitution. Moreover, the yield in the nitro reduction process is notably lower for F substitution than for Cl substitution (3a-3b). The yield of the eighth step Suzuki coupling reaction is also significantly influenced by these factors (9a-9b).
After α1β1γ1 AMPK assay (Table 1), the EC50 of A1 was 258 nM. However, the activity was lost when the chlorine at R1 was changed to fluorine (A2: EC50 > 1000 nM). As reported in the previous literature, the substitution effect of Cl at R1 was significantly better than that of F (Scheme 1). We attempted to replace pyrrolidone with piperidone to obtain A3 (Scheme 2), resulting in a threefold decrease in EC50, followed by an attempt to synthesize azocyclic butanone to get 13, but its instability resulted in ring-opening during subsequent Suzuki couplings. Then, A1 was selected for further optimization, and its carboxyl group was replaced.
EC50 values are the mean of duplicate measurements.
Reagents and conditions: (a) arylborates, Pd(dppf)Cl2, X-Phos, K2CO3, DMF, 120℃, 4 h; (b) a. 1 M Bu4NF, THF, 80℃, 45 min; b. 2.5 N NaOH, MeOH, 45℃, 2 h; (c) 1 M Bu4NF, THF, 80℃, 45 min; (d) 2.5 N NaOH, MeOH, 45℃, 2 h; (e) for A5: O-(tetrahydro-2H-pyran-2-yl)hydroxylamine, HATU, Et3N, DMF, rt, 14 h; for A6℃A9: Corresponding amines, THF, HATU, DIPEA, rt, 12 h.
As shown in Scheme 2, A4 was obtained without the final ester hydrolysis during the synthesis of A1. A5 was obtained by condensation with hydroxylamine with A1. Similarly, the amide analogs A6‒A9 were obtained by the condensation of A1 with the corresponding amines.
According to the activity test (Table 1), it could be found that the acid compound showed moderate activating effects. The hydroxamic acid showed very high activity, while its esters or amides were all inactive. These indicated that acid or hydroxamic acid is essential for the action.
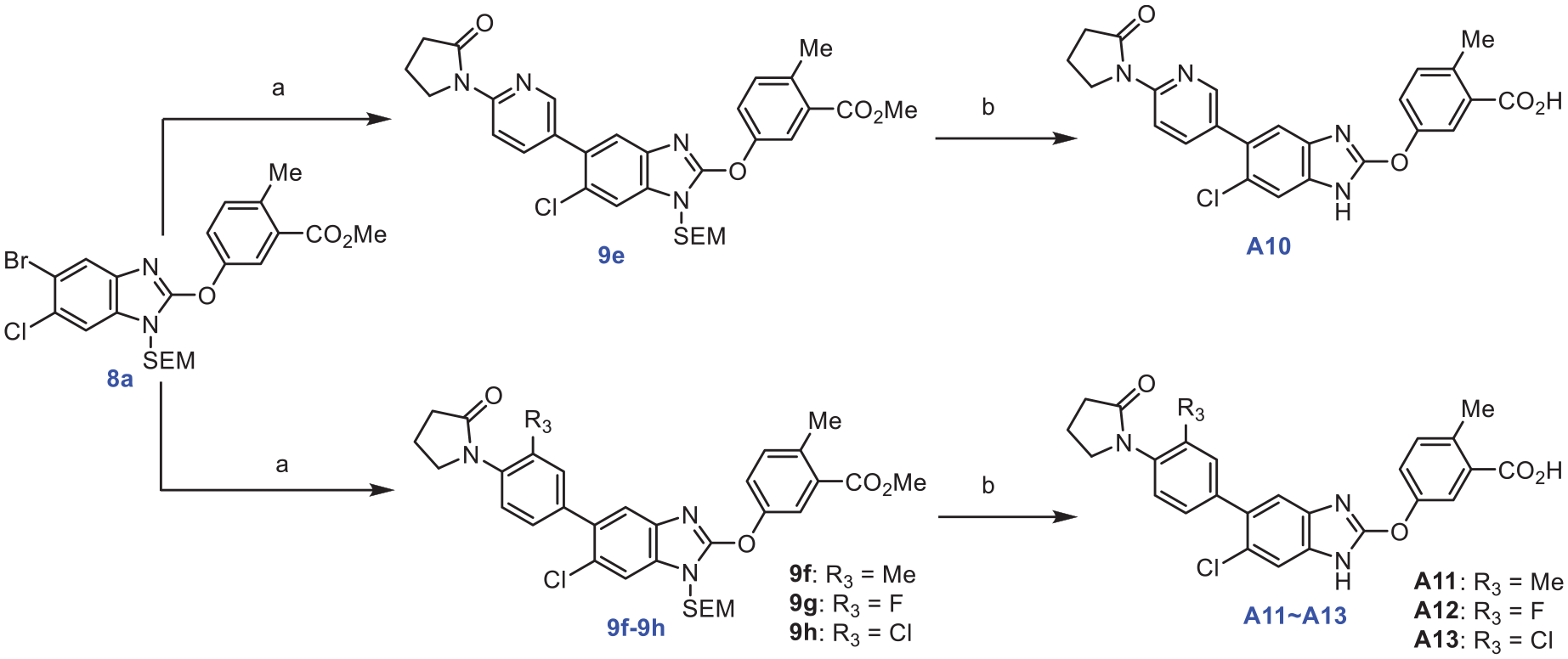
Further modification was carried on. A10, a hetero analog of A1, was similarly obtained, and A11‒A13 were synthesized from the corresponding fluoro, chloro, methyl intermediates as shown in Scheme 3.
Reagents and conditions: (a) arylborates, Pd(dppf)Cl2, X-Phos, K2CO3, DMF, 120℃, 4 h; (b) a. 1M Bu4NF, THF, 80℃, 45 min; b. 2.5N NaOH, MeOH, 45℃, 2 h.
To induce torsion between the pyrrolidone ring and the adjacent benzene ring plane, Compounds A10-A14 were synthesized by incorporating fluorine, chlorine, or a methyl group at the R3 position of the benzene ring based on Compound A1, or by substituting the benzene ring with pyridine. The activity test revealed that the twisted compound exhibited significantly higher efficacy compared with Compound A1 (EC50: 39–165 nM), with the A11 derivative containing a methyl group showing the most potent activity with an EC50 of 39 nM. It was suggested here that the introduction of R3 groups facilitated a suitable torsion angle with the pyrrolidone to give an improved activity.
Docking analysis
In addition, the interaction mode between A11 and AMPK (PDB: 4CFE) was explored via molecular docking simulation. As expected, the introduction of methyl at R3 resulted in a specific twist with pyrrolidone (Figure 2). Studies have shown that A11 can strongly interact with 4CFE (Figure 3) through hydrophobic interactions, electrostatic interactions, and hydrogen bonding. The most important ones are LYS31, ASP88, ARG83, and LYS29. The pyrrolidone carbonyl group of A11 forms hydrogen bonds with LYS31 and also interacts electrostatically with its neighboring phenyl. The hydrogen on the benzimidazole N forms hydrogen binding with ASP88, and ARG83 has some electrostatic interactions with the imidazole ring. In addition, LYS29 was found to interact electrostatically with the hydrogen on the carboxyl group of the terminal benzoic acid and to form hydrogen bond with the N atom on imidazole.
Binding mode of A11 within the active pocket of AMPK (PDB ID: 4CFE).
Specific interaction of A11 (green) with AMPK Key amino acid (PDB: 4CFE). Hydrogen bonds are represented by lines (green), electrostatic interactions with orange dashed lines.
To clarify the reasons for the difference in activity between A11 and its analogs, the torsion angle was analyzed by SeeSAR. After A10 linked a fluorine atom at position 3 of phenyl (Figure 4(a)), the torsion angle reached 102.1° due to the existence of steric hindrance. When A11 replaced fluorine with the methyl group (Figure 4(b)), the torsion angle reached 93.4°. After compound A12 substituted pyridine ring for benzene ring (Figure 4(c)), the torsion angle changed to 100.4°. The torsion angle of A12 is 98.0° after replacing F atom with chlorine atom (Figure 4(d)). In order to explore the relationship between the twist angle and structure activity, the distribution of hydrogen bond acceptor and hydrogen bond donor around the cavity was analyzed by Discovery Studio 2019. It can be seen from Figure 4(e) that it is difficult to form a suitable twist angle when in A1 there is no substituent in the benzene ring, which makes it difficult for the carbonyl group of pyrrolidone to form hydrogen bond with LYS31, which is one of the important amino acids in the kinase domain. When the substituent of small molecule is introduced into the benzene ring, the pyrrolidone is twisted properly due to the existence of steric hindrance. A11 successfully turns the carbonyl group of pyrrolidone to LYS31 due to the presence of methyl group (Figure 4(f)), in which methyl plays a filling and supporting role, and the sterically hindered carbonyl group of pyrrolidone can only be twisted to LYS31. A10 has a similar effect with A12, but its large torsion angle causes the pyrrolidone carbonyl to be counterclockwise slightly away from LYS31, so the EC50 value of A11 is the best. A13 is substituted with chlorine and it is difficult to form support at the bottom, but it is in the opposite position of methyl group, which leads to a change in spatial conformation. In short, the twist angle of A11 is the most suitable for the formation of hydrogen bond and thus its best activity.
Torsion angle between pyrrolidone and phenyl or pyridinyl ring: (a) A10; (b) A11; (c) A12; (d) A13; (e) distribution of hydrogen-bonded donors and receptors around A1; and (f) distribution of hydrogen-bonded donors and receptors around A11.
Conclusion
In this paper, the AMPK agonist EX-229 was utilized as the lead, and its core structure of benzimidazole was retained. A series of compounds were designed and synthesized based on traditional drug design theories, in combination with computer-aided drug design and other modifications. Most of the compounds were excellent agonists of AMPK. The EC50 of A11 was as low as 39 nM, and the SAR was preliminarily discussed based on activity and molecular docking. Our investigation revealed the essential role of the terminal carboxyl group in forming a crucial hydrogen bond with LYS29, necessitating an electron donor to serve as a hydrogen bond donor. We observed that a series of amides are unsuitable for this interaction, with increased substitution on the amide NH correlating with decreased activity. Our findings suggest that a directly exposed donor is more favorable. We have illustrated the correlation between activity and LYS31 by introducing a torsionable group at position 5 of the parent nucleus. This group must act as a hydrogen bond donor and be positioned in proximity to LYS31 to facilitate the formation of hydrogen bonds. These insights into SARs offer valuable guidance for the future design of AMPK agonists.
Experimental
Materials and reagents
All chemical reagents are purchased from commercial sources and are either chemically or analytically pure. For nuclear magnetic resonance (NMR) analysis, 400 and 600 Bruker NMR spectrometer with tetramethylsilane (TMS) as an internal standard was used. The solvent employed was DMSO-d6. The mass spectra used were Waters QAB-1837 tandem quadrupole liquid mass spectrometry and Agilent 1100 quadrupole liquid chromatography mass spectrometry. The type of silica gel used for column chromatography is 200‒300 mesh coarse-pore silica gel, specifically the GF254 model produced by Qingdao Ocean Chemical Industry. Thin layer chromatography uses a 254 thin layer plate (Merck silica gel 60 F254).
General procedure for the synthesis of compounds 2a‒8b.
4-bromo-5-chloro-2-nitroaniline (2a)
To a 500 mL flask containing 5-chloro-2-nitroaniline (23.40 g, 134.03 mmol) and methanol (300 mL) was portionwisely added NBS (23.86 g, 134.03 mmol) at room temperature. The mixture was stirred at 65°C for 5 h until completion of the reaction detected by TLC. It was rotary evaporated, and treated with ethyl acetate 400 mL and water 200 mall The crude product was purified by column chromatography, with an elution gradient of 0%–10% EtOAc in PE. The separated organic was concentrated to obtain the crude 30.55 g as a light brown solid in a yield of 93.90%. 1H NMR (400 MHz, DMSO-d6) δ 8.24 (s, 1H), 7.63 (s, 2H), 7.29 (s, 1H). ESI-MS:(m/z) 249.6 [M-H]-.
2b were synthesized according to the procedure described in 2a; 27.54 g light brown solid is obtained with a yield of 91.47%.
4-bromo-5-chlorobenzene-1,2-diamine (3a)
To a 1 L flask containing 2a (30.05 g, 119.30 mmol) and ethanol (500 mL) was portionwisely added SnCl2 (113.10 g, 596.51 mmol) at room temperature. The mixture was stirred at 60°C for 5 h until the completion of the reaction detected by TLC. It was cooled to room temperature, filtered, and the filtrate was vacuum concentrated. The residue was diluted with ethyl acetate (3 × 500 mL), and treated with NaHCO3 solution. The organic was washed with brine (500 mL), and dried over anhydrous Na2SO4. Finally, the crude product was purified by column chromatography, with an elution gradient of 0-20% EtOAc in PE. It was concentrated under vacuum to obtain 21.84 g yellow solid in a yield of 82.65%. 1H NMR (400 MHz, DMSO-d6) δ6.74 (s, 1H), 6.64 (s, 1H), 4.89-4.85 (m, 4H). ESI-MS:(m/z) 220.7 [M-H]-.
3b were synthesized according to the procedure described in 3a; 14.21 g yellow solid is obtained with a yield of 65.15%.
To a 1 L flask containing 3a (20.02 g, 90.30 mmol) and ethanol (250 mL) was added in an ice water bath. Then were added potassium hydroxide (7.60 g, 135.45 mmol) and CS2 (8.25 g, 108.36 mmol). The mixture was stirred at 50°C for 3 h until completion of the reaction detected by TLC. Cooling and filtering the mixture, the obtained filtrate was combined with water (300 mL) and AcOH (50 mL) to produce a precipitate. The suspension liquid was then filtered using a pump, and the filter cake was washed with water and a small amount of ethanol. The crude product was purified by column chromatography, with an elution gradient of 0%–20% EtOAc in PE. It was concentrated under vacuum to obtain 18.45 g brown powder in a yield of 77.45%. ESI-MS:(m/z) 262.0 [M+H]+.
4b were synthesized according to the procedure described in 4a; 23.24 g dark yellow solid is obtained with a yield of 77.14%.
To a 1 L flask containing 4a (14.03 g, 53.12 mmol) and ethanol (300 mL) was added in an ice water bath. Then added K2CO3 (3.67 g, 26.56 mmol), iodomethane (3.77 g, 26.56 mmol), and after stirring at room temperature for 1 h, K2CO3 (3.67 g, 26.56 mmol) and iodomethane (3.77 g, 26.56 mmol) were added to the mixture. The mixture was stirred at room temperature overnight until completion of the reaction detected by TLC. The obtained filtrate was combined with water (200 mL) and diluted with ethyl acetate (3 × 500 mL). The crude product was purified by column chromatography, with an elution gradient of 10%–20% EtOAc in PE. It was concentrated under vacuum to obtain 13.57 g brown powder in a yield of 92.03%. ESI-MS:(m/z) 277.0 [M+H]+.
5b was synthesized according to the procedure described in 5a; 20.56 g brown powder solid is obtained with a yield of 88.43%.
To a 500 mL flask containing 5a (12.01 g, 43.23 mmol) and DCM (250 mL) was portionwisely added m-CPB(19.75 g, 129.70 mmol) at room temperature. The mixture was stirred at room temperature for 10 min until completion of the reaction detected by TLC. The reaction liquid was washed with 10% NaHCO3 to separate and concentrate the organic phase. Then 30 mL of MeOH was added. The suspension liquid was then filtered using a pump, and the crude product was purified by column chromatography, with an elution gradient of 5%–20% EtOAc in PE. It was concentrated under vacuum to obtain 14.20 g white powder in a yield of 70.73%. ESI-MS:(m/z) 307.8 [M+H]+.
6b was synthesized according to the procedure described in 6a; 14.21 g white powder is obtained with a yield of 63.29%.
To a 500 mL flask containing 6a (12.00 g, 38.76 mmol) and THF (250 mL) was portionwisely added Et3N (7.85 g, 77.53 mmol) at room temperature. Then SEM-Cl (8.40 g, 50.39 mmol) was added to the reaction bottle. The mixture was stirred at 25°C for 1 h until completion of the reaction detected by TLC. It was rotary evaporated, combined with water (200 mL), and treated with ethyl acetate (3 × 300 mL). The organic was washed with 2N HCl aqueous solution and brine. After drying the organic phase with anhydrous sodium sulfate and vacuum concentration, the crude product was purified by column chromatography, with an elution gradient of 10%–20% EtOAc in PE. It was concentrated under vacuum to obtain 14.80 g white solid in a yield of 87.01%.
7b were synthesized according to the procedure described in 7a; 17.95 g white solid is obtained with a yield of 88.77%.
To a 500 mL flask containing 7a (13.00 g, 30.71 mmol) and DMF (250 mL) was portionwisely added K2CO3 (10.61 g, 76.76 mmol) at room temperature, then 5-hydroxy-2-methylbenzoate (7.01 g, 46.06 mmol) was added to the reaction bottle. The mixture was stirred at 25°C for 24 h until completion of the reaction detected by TLC. It was rotary evaporated, combined with water (200 mL), and treated with ethyl acetate (3×300 mL). The organic was washed with brine. After drying the organic phase with anhydrous sodium sulfate and vacuum concentration, the crude product was purified by column chromatography, with an elution gradient of 25% EtOAc in PE. Pure fractions were evaporated to dryness to afford 8a (11.32 g, 74.41%) as a yellow solid. ESI-MS:(m/z) 511.0 [M+H]+.
8b were synthesized according to the procedure described in 8a; 13.58 g yellow solid is obtained with a yield of 77.37%.
General procedure for the synthesis of A1‒A13
Synthesis of A1
To a 50 mL flask containing 8a (0.20 g, 0.38 mmol) and DMF (25 mL) was added arylborates (0.163 g, 0.57 mmol) at room temperature, then Pd(dppf)Cl2 (0.013 g, 0.019 mmol), X-Phos (0.009 g, 0.019mmol), and 1M K2CO3 solution (2.4 mL) were added to the reaction bottle. Under the protection of nitrogen, the mixture was stirred at 120°C for 4 h. It was rotary evaporated, combined with water (60 mL), and treated with ethyl acetate (3 × 100 mL). The organic was washed with brine and dried with anhydrous sodium sulfate and vacuum concentration, and the crude product was purified by column chromatography, with an elution gradient of 10%–30% EtOAc in PE. Pure fractions were evaporated to dryness to afford a yellow white solid (0.1 g). Add Bu4NF (1.0 M of THF, 5 mL, 2.08 mmol) into the THF (20 mL) solution of the product of the previous step (0.1 g, 0.164 mmol) through a syringe into a 50 mL flask. The mixture was stirred at 85°C for 45 min, then Bu4NF (3 mL) was added to the reaction solution. The mixture was stirred at 85°C for 4 h until completion of the reaction detected by TLC. The reaction liquid was treated with ethyl acetate (200 mL) and KHSO4 (~pH 3) saturated water, and then extracted with of ethyl acetate (100 mL). The organic was washed with brine and dried with anhydrous sodium sulfate and vacuum concentration which contains 9a. Dissolve the concentrated solution in MeOH (50 mL) and NaOH solution (10 mL 2.5 N) was added to mixture. The mixture was stirred at 45°C for 2 h until completion of the reaction detected by TLC. Then it was combined with water (50 mL) and washed with ethyl acetate (2 × 100 mL). The water phase is acidified with 2 N HCl to pH~1 and treated with ethyl acetate (3 × 300 mL). The organic was washed with brine. After drying the organic phase with anhydrous sodium sulfate and vacuum concentration, the crude product was purified by column chromatography, with an elution gradient of 30% MeOH in H2O. Pure fractions were evaporated to dryness to afford A1 (0.05 g, 61.20%).1H NMR (400 MHz, DMSO-d6) δ 7.81 (d, J=2.0 Hz, 1H), 7.80 (d, J = 2.8 Hz, 1H), 7.79 (s, 1H), 7.61 (s, 1H), 7.52 (d, J=2.5 Hz, 1H), 7.51 (d, J=4.2 Hz, 1H), 7.50 (s, 1H), 7.43 (d, J=8.5 Hz, 1H), 7.41 (s, 1H), 3.96 (t, J=7.0 Hz, 2H), 2.62 (s, 3H), 2.60 (t, 2H), 2.17 (p, J=7.4 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 174.38, 169.01, 158.41, 151.50, 139.13, 136.09, 135.65, 133.08, 133.02 (2C), 130.34 (3C), 124.56, 123.28 (2C), 121.87 (2C), 119.32 (3C), 48.52, 32.83, 21.02, 17.93. HRMS (ESI-) m/z calcd for C25H20ClN3O4 [M-H]-, 460.1070; found, 460.1115.
Synthesis of A2
A2 0.31 g was synthesized according to the procedure described in A1, as a white powder, and the yield was 63.41%. 1H NMR (400 MHz, DMSO-d6) δ 12.71 (s, 1H), 7.88 (d, J=2.7 Hz, 1H), 7.85 (d, J=2.0 Hz, 1H), 7.83 (d, J=2.0 Hz, 1H), 7.65 (d, J=1.8 Hz, 1H), 7.63 (d, J=1.8 Hz, 1H), 7.60 (dd, J=8.4, 2.7 Hz, 1H), 7.50 (d, J=8.4 Hz, 2H), 7.40 (d, J=11.2 Hz, 1H), 3.97 (t, J=7.0 Hz, 2H), 2.64 (s, 3H), 2.62–2.60 (m, 2H), 2.2 –2.15 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 174.39, 168.31, 151.60, 139.12, 136.68, 133.36 (2C), 132.18, 132.06, 129.64 (2C), 129.61 (2C), 124.17 (2C), 122.38, 122.22, 122.15, 119.77 (3C), 48.51, 32.81, 21.07, 17.89. HRMS (ESI-) m/z calcd for C25H20FN3O4 [M-H]-, 444.1365; found, 444.1395.
Synthesis of A3
A3 0.31 g was synthesized according to the procedure described in A1, as a buff powder, and the yield was 43.23%. 1H NMR (400 MHz, DMSO-d6) δ 12.74 (s, 1H), 7.83 (d, J=2.7 Hz, 1H), 7.59 (s, 1H), 7.55 (dd, J=8.3, 2.6 Hz, 1H), 7.49 (s, 1H), 7.47 (s, 2H), 7.40 (s, 2H), 7.38 (s, 1H), 3.70 (t, J=5.6 Hz, 2H), 2.59 (s, 3H), 2.46 (t, 2H), 1.95–1.87 (m, 4H). 13C NMR (100MHz, DMSO-d6) δ 169.42, 168.27, 162.80, 158.36, 151.51, 143.15, 137.79, 136.83, 133.40 (2C), 133.09, 132.14, 130.41 (2C), 126.19 (2C), 124.56, 124.27 (2C), 122.24 (2C), 51.25, 33.10, 23.50, 21.36, 21.07. HRMS (ESI)-m/z calcd for C26H22ClN3O4 [M-H-], 474.1226; found, 474.1333.
Synthesis of A4
A4 was synthesized according to the procedure described in A1; A4 was obtained after the SEM protection group was removed with tetrabutylammonium fluoride, and 0.089 g yellow solid is obtained with a yield of 71.54%.
To a 50 mL flask containing A1 and DCM was portionwisely added tetrahydropyranyl hydroxylamine (1.5 eq) and HATU (1.5 eq) at room temperature. The mixture was stirred at room temperature for 14 h until completion of the reaction detected by TLC. The reaction liquid was combined with water and treated with ethyl acetate. After drying the organic phase with anhydrous sodium sulfate and vacuum concentration, the crude product was dissolved in methanol and toluene sulfonic acid monohydrate (1.5 eq). The mixture was stirred at room temperature for 2 h until completion of the reaction detected by TLC. The reaction liquid was combined with brine and treated with ethyl acetate. After drying the organic phase with anhydrous sodium sulfate and vacuum concentration, the crude product was purified by column chromatography, with an elution gradient of 0%–10% MeOH in CH2Cl2. Pure fractions were evaporated to dryness to afford A5; 0.076 g yellow solid is obtained with a yield of 63.42%. 1H NMR (600 MHz, DMSO-d6) δ 11.00 (s, 1H), 8.29 (s, 1H), 8.00 (s, 1H), 7.78 (s, 1H), 7.77 (s, 1H), 7.59 (s, 1H), 7.48 (s, 1H), 7.47 (s, 1H), 7.44 (dd, J=8.2, 2.7 Hz, 1H), 7.40 (d, J=8.5 Hz, 1H), 7.38 (s, 1H), 7.36 (d, J=2.5 Hz, 1H), 3.93 (t, J=6.9 Hz, 2H), 2.58 (d, J=7.9 Hz, 2H), 2.41 (s, 3H), 2.16–2.12 (m, 2H). 13C NMR (150MHz, DMSO-d6) δ 174.38, 165.52, 162.78, 158.27, 151.31, 139.13, 136.19, 135.62, 133.44, 133.11, 132.24 (2C), 130.34 (2C), 124.58, 121.94 (2C), 119.61 (2C), 119.32 (2C), 48.52, 32.83, 19.11, 17.93. HRMS (ESI-) m/z calcd for C25H21ClN4O4 [M-H]-, 475.1179; found, 475.1096.
Synthesis of A6
To a 50 mL flask containing A1 and THF was portionwisely added 2N THF solution of NH3 room temperature, then HATU (1.5 eq) and DIPEA (3 eq) was added to the reaction bottle. The mixture was stirred at room temperature for 2 h until the completion of the reaction detected by TLC. After vacuum concentration of the mixture, the crude product was purified by column chromatography, with an elution gradient of 0%–10% MeOH in CH2Cl2. Pure fractions were evaporated to dryness to afford A6; 0.086 g buff solid is obtained with a yield of 50.41%. 1H NMR (600 MHz, DMSO-d6) δ 12.85 (s, 1H), 8.01 (s, 1H), 7.89 (s, 1H), 7.78 (dd, J=13.9, 8.1 Hz, 2H), 7.59 (d, J=36.9 Hz, 1H), 7.52 (s, 1H), 7.49 (d, J=8.4 Hz, 1H), 7.44 (d, J=2.5 Hz, 1H), 7.41 (d, J=2.6 Hz, 1H), 7.39 (s, 1H), 7.37 (d, J=9.9 Hz, 1H), 3.94 (t, J=6.5 Hz, 2H), 2.58 (d, J=8.0 Hz, 2H), 2.45 (s, 3H), 2.14 (t, J=7.8 Hz, 2H). 13C NMR (150MHz, DMSO-d6) δ 170.43, 162.79, 151.30, 139.16, 138.54, 132.93, 132.27 (2C), 130.34 (2C), 124.58, 121.65 (2C), 120.08, 119.38 (2C), 119.31 (2C), 118.52, 112.96, 111.62, 48.52, 31.24, 19.48, 18.41. HRMS (ESI-) m/z calcd for C25H21ClN4O3 [M-H]-, 459.1229; found, 459.1157.
Synthesis of A7
A7 0.31 g was synthesized according to the procedure described in A6, as a yellow powder, and the yield was 68.74%. 1H NMR (400 MHz, DMSO-d6) δ 12.67 (s, 1H), 8.30 (q, J=4.6 Hz, 1H), 7.77 (d, J=8.4 Hz, 2H), 7.59 (s, 1H), 7.50–7.45 (m, 2H), 7.41 (d, J=2.5 Hz, 1H), 7.39 (s, 3H), 3.93 (t, J=7.0 Hz, 2H), 2.81–2.78 (m, 3H), 2.58 (d, J=8.0 Hz, 2H), 2.40 (s, 3H), 2.15 (q, J=7.4 Hz, 2H). 13C NMR (100MHz, DMSO-d6) δ 174.41, 168.91, 158.42, 151.35, 139.17, 138.68, 135.56, 132.24 (2C), 130.34 (2C), 124.62, 121.65 (2C), 119.36 (2C), 119.33 (3C), 118.56, 112.92, 48.52, 32.82, 26.42, 19.31, 17.93. HRMS (ESI-) m/z calcd for C26H23ClN4O3 [M-H]-, 473.1386; found, 473.1299.
Synthesis of A8
A8 0.22 g was synthesized according to the procedure described in A6, as a buff powder, and the yield was 56.19%. 1H NMR (400 MHz, DMSO-d6) δ 12.71 (s, 1H), 7.80 (d, J=7.8 Hz, 2H), 7.62 (d, J=10.0 Hz, 1H), 7.57 (d, J=14.0 Hz, 1H), 7.50 (d, J=8.6 Hz, 2H), 7.42–7.40 (m, 2H), 7.30 (d, J=2.3 Hz, 1H), 3.96 (d, J=6.9 Hz, 2H), 3.07 (s, 4H), 2.86 (s, 3H), 2.60 (d, J=8.1 Hz, 2H), 2.17–2.13 (m, 2H). 13C NMR (100MHz, DMSO-d6) δ 173.94, 168.37, 151.50, 139.19, 137.63, 136.75, 135.42, 133.36, 133.08, 132.54, 132.52, 132.34, 132.00, 131.91, 129.30, 129.18, 128.35, 126.67, 124.53, 124.16, 122.20, 50.52, 31.24, 21.08, 19.18, 18.20. HRMS (ESI-) m/z calcd for C27H25ClN4O3 [M-H]-, 487.1542; found, 487.1554.
A10 0.18 g was synthesized according to the procedure described in A1, as a white powder, and the yield was 61.69%. 1H NMR (400 MHz, DMSO-d6) δ 12.68 (s, 1H), 7.78 (d, J=2.7 Hz, 1H), 7.56 (s, 1H), 7.51 (dd, J=8.3, 2.8 Hz, 1H), 7.41 (d, J=8.5 Hz, 1H), 7.33 (s, 2H), 7.27 (s, 2H), 3.72 (t, J=6.9 Hz, 2H), 2.55 (s, 3H), 2.44 (t, J=8.0 Hz, 2H), 2.20 (s, 3H), 2.14 (q, J=7.5 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 174.30, 168.74, 158.55, 157.55, 151.48, 140.25, 136.58, 133.25 (2C), 131.99, 131.77, 129.14, 127.61, 126.49, 126.46, 125.98, 124.37, 123.89, 122.12, 118.17, 117.96, 49.85, 30.98, 21.07, 19.07. HRMS (ESI-) m/z calcd for C26H22ClN3O4 [M-H]-, 474.1226; found, 474.1145.
Synthesis of A11
A11 0.25 g was synthesized according to the procedure described in A1, as a white powder, and the yield was 50.08%. 1H NMR (400 MHz, DMSO-d6) δ 12.68 (s, 1H), 7.78 (d, J=2.7 Hz, 1H), 7.56 (s, 1H), 7.51 (dd, J=8.3, 2.8 Hz, 1H), 7.41 (d, J=8.5 Hz, 1H), 7.33 (s, 2H), 7.27 (s, 2H), 3.72 (t, J=6.9 Hz, 2H), 2.55 (s, 3H), 2.44 (t, J=8.0 Hz, 2H), 2.20 (s, 3H), 2.14 (q, J=7.5 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 173.92, 168.26, 151.51, 139.18, 137.65, 136.82, 135.42, 133.40 (2C), 133.10, 132.34 (2C), 132.11, 128.35 (2C), 126.67 (2C), 124.55, 124.27, 122.24 (2C), 50.52, 31.24, 21.23, 19.18, 18.20. HRMS (ESI-) m/z calcd for C26H23ClN4O3 [M-H]-, 473.1386; found, 473.1299.
Synthesis of A12
A12 0.35 g was synthesized according to the procedure described in A1, as a buff powder, and the yield was 58.74%. 1H NMR (400 MHz, DMSO-d6) δ 12.79 (s, 1H), 8.47 (d, J=2.5 Hz, 1H), 8.41 (d, J=8.6 Hz, 1H), 7.94 (dd, J=8.7, 2.5 Hz, 1H), 7.84 (d, J=2.7 Hz, 1H), 7.62 (s, 1H), 7.55 (dd, J=8.3, 2.7 Hz, 1H), 7.46 (d, J=3.4 Hz, 1H), 7.44 (s, 1H), 4.09 (t, J=7.1 Hz, 2H), 2.65 (t, J=8.0 Hz, 2H), 2.59 (s, 3H), 2.12 (p, J=7.7 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 175.18, 168.29, 151.48, 151.00, 139.35 (2C), 136.83, 133.39 (2C), 132.20, 131.39, 129.87, 129.13, 124.85, 124.23 (2C), 122.23 (2C), 113.16 (2C), 47.45, 33.52, 21.07, 17.68. HRMS (ESI-) m/z calcd for C24H19ClN4O4 [M-H]-, 461.1022; found, 461.0932.
Synthesis of A13
A13 0.24 g was synthesized according to the procedure described in A1, as a white powder, and the yield was 65.61%. 1H NMR (400 MHz, DMSO-d6) δ 7.73 (s, 1H), 7.64 (s, 1H), 7.61 (s, 1H), 7.51 (s, 2H), 7.44 (s, 1H), 7.41 (s, 1H), 7.37 (s, 1H), 3.79 (t, J=6.9 Hz, 2H), 2.55–2.54 (m, 3H), 2.49 (s, 2H), 2.21 (t, J=7.4 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 174.38, 168.78, 158.38, 151.51, 139.13, 136.27, 135.65, 133.72, 133.11, 133.09, 130.34 (3C), 124.58, 123.54 (2C), 121.96 (2C), 119.31 (3C), 48.52, 32.82, 21.02, 17.93. HRMS (ESI-) m/z calcd for C25H19Cl2N3O4 [M-H]-, 494.0680; found, 494.0608.
Docking simulations
Molecular docking of compound A11 into the three-dimensional X-ray structure of AMPK (PDB: 4CFE) was carried out using the CDOCKER protocol of Discovery Studio 2019.
The twist angle is calculated by using SeeSAR13.0.5. The docking results in Discovery Studio 2019 are imported into SeeSAR and analyzed by measure torsion function.
AMPK(α1β1γ1) assay
Materials.
Name
Vendor
Cat. No.
Lot. No.
AMPKα1/β1/γ1
Carna
02-113
07CBS-2720
ULight-CREBtide
PE
TRF0107-M
2961457
Eu-anti-P-CREB (Ser133)
PE
TRF0200-M
2462907
Triton X-100
Sigma
T9284-100ML
SLBS6421
HEPES, pH7.5
Gibco
11344-041
1653630
MgCl2
Sigma
M2670-500g
BCBM7703V
DTT
Sigma
D0632-10G
SLBK4951V
EDTA
Sigma
E5134
60-00-4
DMSO
Sigma
34869-4L
WXBD5538V
ATP
Sigma
A7699-1G
987-65-5
384-well plate
PerkinElmer
OptiPlate-384
8210-22271
384 well Echo plate
Labcyte
PP-0200
6386422
Testing instrument.
Name
Vendor
Model No.
Instrument No.
Acoustic Liquid Handler
LABCYTE
Echo650
E6XX-21146
Envision
Perkin Elmer
2104 Multilabel Reader
1041048
Biochemical incubator
Shanhai Boxun
SPX-100B-Z
_
Centrifuge
BECKMN COULTRE
Avanti J-15R
JBR20A055
Prepare compound
The compound is diluted with DMSO to a concentration of <100 times. The 40 μL compound diluent was then distributed into a 384-well echo plate. After echo, 200 nL of each hole is transferred to a 384-hole measuring plate for measurement.
Kinase reaction and detection
First, 10 μL kinase solution was added to each well of the measuring plate. Then the substrate and ATP were added to the kinase base buffer to prepare the substrate solution. 10 μL substrate solution was added to each hole of the measuring plate to start the reaction, incubate, and react at room temperature. The detection solution with twice the final concentration was prepared in the antibody dilution buffer; 20 μL detection solution was added to each hole of the measuring plate to stop the reaction. Then incubate at room temperature for 60 min.
Curve fitting
Copy values of Lance signal ratio from Envision program (665/615 nm). Convert ratio values to percent activation values:
a. Percent activation = (sample Lance signal ratio—max)/(max–min)*100.
b. “min” means the Ratio of no enzyme control and “max” means the Ratio of DMSO control.
Data were presented in MS Excel and the curves fitted by XLFit excel add-in version 5.4.0.8, and the EC50 calculation equation is
Supplemental Material
sj-docx-1-chl-10.1177_17475198241290882 – Supplemental material for Design, synthesis, and evaluation of some benzimidazole analogs as AMPK agonists
Supplemental material, sj-docx-1-chl-10.1177_17475198241290882 for Design, synthesis, and evaluation of some benzimidazole analogs as AMPK agonists by Runtao Tian, Jiajia Zhang, Yujia Zhang and Huaiwei Ding in Journal of Chemical Research
Footnotes
Acknowledgements
The authors thank Yujia Zhang, Shurui Yang, Xiaoyan Liao, Lin Li, and Bingbing Xie, who participated in the scientific research ability training course in their school and were involved in the synthesis of compounds.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the college students’ innovative and entrepreneurial training program (Grant No. S202110163040).
ORCID iD
Huaiwei Ding
Supplemental material
Supplemental material for this article is available online.
LaiYCKviklyteSVertommenD, et alBiochem J. 2014; 460:363–375.
29.
EdmondsonSDFisherMHKimD, et alPatent US201113086563, USA, 2011.
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.