
Abstract
Erythrina variegata L., a member of the Fabaceae family, is a valuable medicinal plant with a rich history of traditional medicinal uses. However, its potential as an antidiabetic agent has remained largely unexplored. In this study, three isoflavonoid compounds, namely eryvarin M (
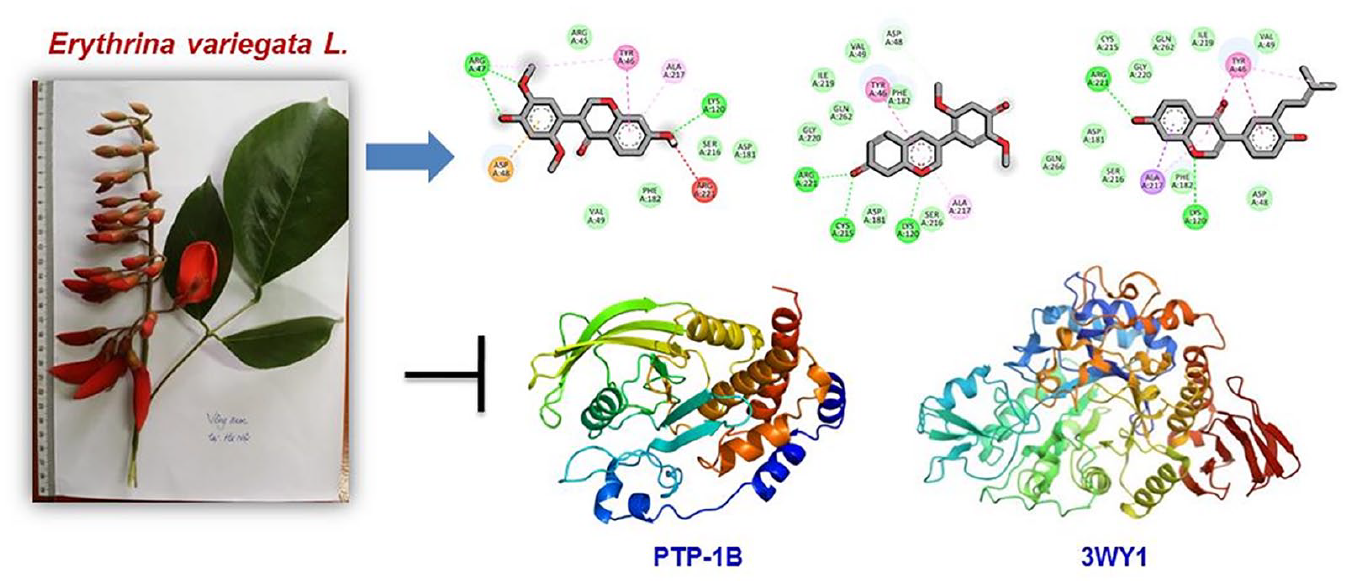
Introduction
Erythrina variegata L. var. orientalis (L.) Merr., also known by synonyms, such as Erythrina indica Lamk, Erythrina spathacea DC, and Erythrina orientalis (L.) Murr, belongs to the Fabaceae family.1,2 The name “Erythrina” is derived from the Greek word “erythros” which means “red” referring to the striking red color of this plant species flowers. 3 E. variegata is indigenous to the coastal regions of East Africa and is distributed across Africa, tropical regions of South Asia, Southeast Asia, Pacific Islands, Australia, and Southern China. In Vietnam, E. variegata grows naturally and can be found scattered throughout lowland mountainous provinces, the central region, and the delta. It is commonly cultivated as a hedge plant in fields and home gardens. E. variegata tree holds various medicinal uses in traditional medicine. It is regarded as a nerve sedative, an ingredient in ophthalmic eye drops, an anti-asthmatic agent, an anticonvulsant, an antiseptic, and an astringent.3–5 Traditional literature suggests that E. variegata leaves possess drying and calming properties and are associated with the heart and spleen meridians. They are known for their soothing and antiseptic effects. 6 In Vietnam, people frequently consume E. variegata leaves and apply heated leaves to treat hemorrhoids. The leaves are also used for treating insomnia and chronic colitis. A combination of E. variegata leaves and lotus leaves is employed to address nosebleeds and bloody stools, and to treat infantile diarrhea. 7
E. variegata has been found to contain more than 100 compounds, including alkaloids, flavonoids, pterocarpan, triterpenes, steroids, alkyl trans-ferulate, proteins, and lecithin. Alkaloids and flavonoids are the two major class of compounds present in different parts of the plant.6,8–14 Currently, many studies have isolated flavonoids from E. variegata with isoflavonoids reported as the primary constituents among prenyl isoflavones, flavan-3-ols, flavonols with sugar moieties, and triterpenoids.15–32 Given its rich chemical composition, several studies have investigated the plant’s biological activities, such as antibacterial effects,22,33–35 antioxidant activity,5,36 pain relief, anti-inflammatory effects,36–39 cardiovascular effects, 40 anticonvulsant effects, and anxiolytic effects.3,5,8,41–44 However, there is limited research on the antidiabetic effects of this plant. In a study by Santhiya et al. 45 in 2016, the E. variegata L. bark methanolic extract showed in vitro stimulatory effects on glucose uptake assay by isolated rat hemidiaphragm and glucose uptake by Yeast cells, and inhibitory effects on α-amylase and α-glucosidase enzymes as well. In addition, methanol extract of E. variegata leaf significantly and dose dependently reduced and normalized blood glucose levels in streptozotocin (STZ)-induced diabetic Wistar rats. 46
Binding of insulin to its receptors leads to the phosphorylation of insulin-sensing substrates, which triggers the activation of a series of intracellular signaling pathways that lead to a cascade of biochemical reactions including glucose transport into cells and glycogen synthesis. 47 Protein tyrosine phosphatase (PTP) together with protein tyrosine kinases are involved in the regulation of a variety of cellular functions, including proliferation, differentiation, and apoptosis. PTPs dephosphorylate tyrosine residues of proteins and are considered as negative regulators (back correctors) of insulin signaling. Among the various PTPs, protein tyrosine phosphatase 1B (PTP1B) plays an important role in the regulation of body weight and glucose homeostasis by acting as a negative regulator of the insulin and leptin signaling pathway, thus has attracted considerable attention from scientists as a potential target for research in the treatment of diabetes and metabolic disorders. 48 Overexpression of PTP1B enzyme leads to the inhibition of the insulin receptors signaling cascade, and increased expression of this protein leads to insulin resistance, 49 while in a PTP1B, knockdown mice showed increased insulin sensitivity and enhanced resistance to obesity. 50 Therefore, active substances capable of inhibiting PTP1B enzyme activity not only have great potential in researching and finding drugs to treat type 2 diabetes but also effective agents in the treatment of obesity.51–55
In our study, the extraction and isolation of isoflavonoids (
Results and discussion
Chemistry
The 1H nuclear magnetic resonance (NMR) spectrum of compounds
In-depth analysis of the 1H NMR and 13C NMR spectra of compound
Compound

Chemical structure of compounds (
Biology
Compounds (
In vitro inhibitory activity of compounds (

Results are expressed as IC50 values (µM), determined by regression analysis and expressed as the means ± SD of three replicates.
Positive control.
A study by Bae et al.
59
in 2006 reported that the both prenylated isoflavanones, orientanol E and 2,3-Dihydroauriculatin, exhibited strong PTP1B inhibitory activities with IC50 values of 10.1 ± 0.3 and 2.6 ± 0.5 μM, respectively. In contrast, eryvarin M (
Molecular docking study
PTP1B contains two different sections, the catalytic site and the allosteric site, both of which are crucial to the enzyme’s operation and control. The core domain of the enzyme occupies PTP1B’s catalytic site, which dephosphorylates tyrosine residues on the target protein. The allosteric site is situated on the C-terminal domain, about 20 residues from the catalytic site, and is surrounded by helices 3, 6, and 7 (Figure 2(a)). The allosteric site is significantly less polar and poorly conserved among PTPs, in contrast to the active site of PTP1B.
65
That means, to potentially achieve PTP specificity, targeting PTP1B through its allosteric site, which is distinct from other PTPs, is a viable strategy. Therefore, focusing on the allosteric site may present a viable strategy for creating PTP1B inhibitors that have higher bioavailability and selectivity. In this study, three isoflavonoids (
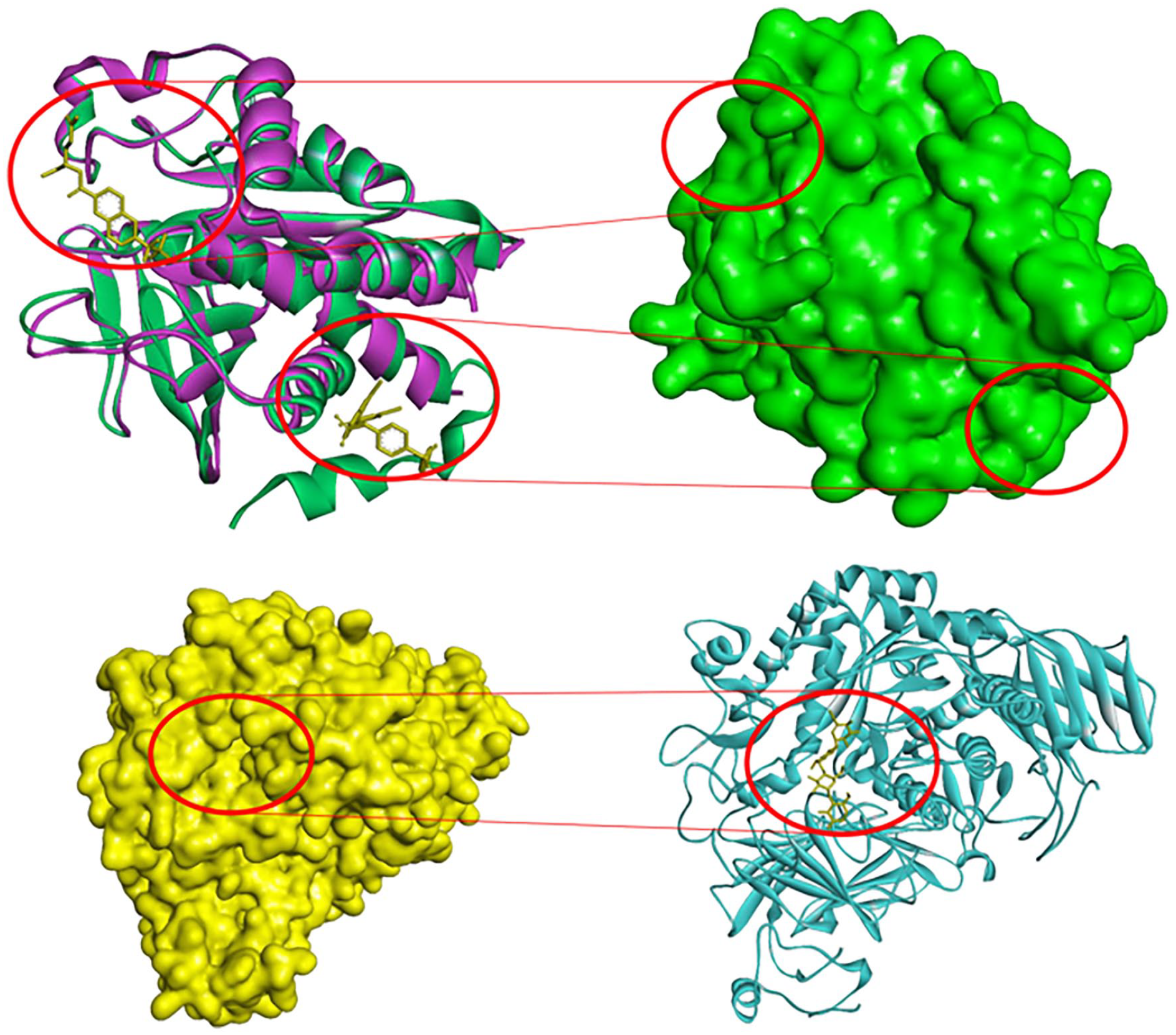
Docking site of protein. (a = upper) Two crystal proteins of PTP1B (1BZC and 1T4J) was aligned with two crystal ligands at catalytic site and allosteric site. (b = under) α-glucosidase with crystal ligand.
Molecular interaction of the protein PTP1B.

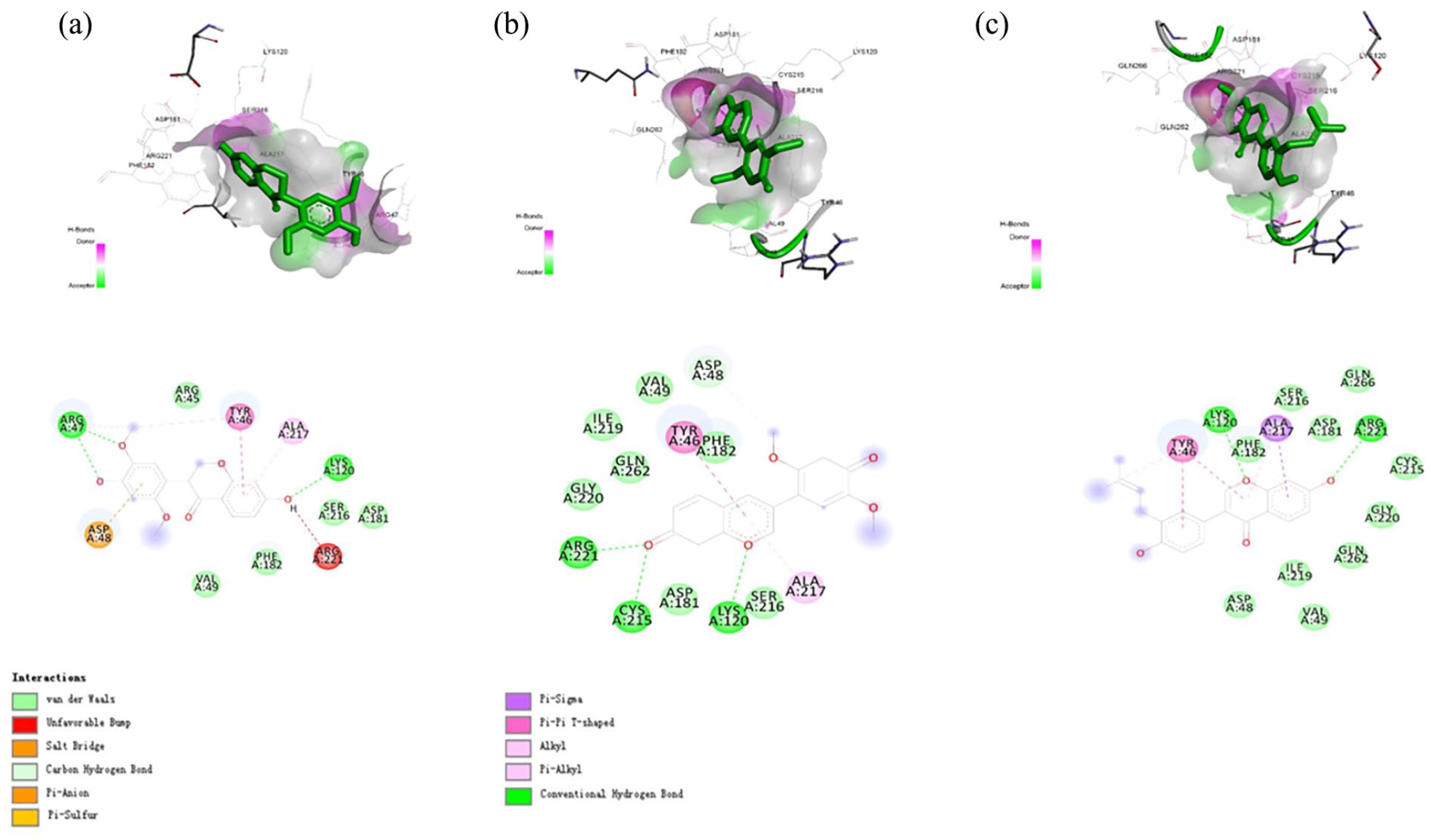
2D ligand interaction diagram and binding modes for the PTP1B at catalytic site. (a) Eryvarin M. (b) Eryvarin H. (c) Neobavaisoflavone.

2D ligand interaction diagram and binding modes for the PTP1B at allosteric site. (a) Eryvarin M. (b) Eryvarin H. (c) Neobavaisoflavone.
Herein, the co-ligand in PTP1B (1BZC), 4-carbamoyl-4-{[6-(difluoro-phosphono-methyl)-naphthalene-2-carbonyl]-amino}-butyric acid, showed eight hydrogen bonds at Gly218, Gly220, Ser216, Ala217, Arg47, Asp48, Cys215, and Ile219 and three hydrophobic interactions with Arg221, Phe182, and Tyr46. Figure 3 shows the bidding pose and dynamics of co-ligand 1BZC. The phosphate group in this compound was found to form hydrogen bond with Cys215, the key residues in the activity of PTP1B.66,67 Compared to our three compounds, eryvarin M (
To compare the interaction between compounds and PTP1B at allosteric site, co-ligand in PTP1B (1T4J), 3-(3,5-dibromo-4-hydroxy-benzoyl)-2-ethyl-benzofuran-6-sulfonic acid [4-(thiazol-2-ylsulfamoyl)-phenyl]-amide, was docked. This reference compound showed three hydrogen bonds at Arg79, Ser285, and Ile281 and four hydrophobic interactions at Glu200, Asp236, Ile281, and Arg199. Figure 4 shows that eryvarin M (
The deep gap that separates the two subunits where α-glucosidase’s active site is located has numerous conserved residues that are involved in substrate binding and catalysis. In this study, the crystal ligand, acarbose, was selected as reference compound. Acarbose showed four hydrogen bonds between residues Ser676, Ser679, Asp282, and Asp616 and sugar moieties of acarbose (Table 3). Compared to the crystal ligand, eryvarin M (
Molecular interaction of the protein α-glucosidase.


2D ligand interaction diagram and binding modes for the α-glucosidase. (a) Eryvarin M. (b) Eryvarin H. (c) Neobavaisoflavone.
Structurally, the hydroxyl groups and heterocyclic oxygen in flavonoid framework play an important role in strong interactions with protein. In this case study, compared to eryvarin M (
For accessing the druggability of potential medicinal compounds, the “Lipinski’s rule of five” has long been used. According to this rule, there are factors taken into account, such as molecular weight, the number of hydrogen bond donors and acceptors, and log P. In this study, the three isolated isoflavonoids including eryvarin M (
Physicochemical properties of isoflavonoids (

MW: molecular weight; log P: log of octanol/water partition coefficient; nHBD: number of hydrogen bond donor(s); nHBA: number of hydrogen bond acceptor(s); TPSA: total polar surface area; MR: molar refractivity; log S: log of solubility; nRotB: number of rotatable bond(s).
In silico ADME predictions of the three studied compounds are exhibited in Table 5. The log of skin permeability (log Kp) values ranges from −5.14 to −6.60. These isoflavonoids (
ADME prediction.

ADME: Absorption, Distribution, Metabolism, and Excretion; Log Kp: log of skin permeability; GI Abs: Gastrointestinal absorption; BBB Per: blood–brain barrier permeability; P-gp: P-glycoprotein; CYP: cytochrome-P.
Toxicity of isoflavonoids (

Conclusion
In conclusion, isoflavanone (
Moreover, the results from docking simulations align well with the experimental findings, confirming the strong binding affinities of isoflavanone (
Experimental
Chemistry
The stem bark of E. variegata was gathered in Hanoi, Vietnam. Dr Nguyen Quoc Binh conducted the botanical authentication of the sample, and a voucher specimen (VN01HN) was formally deposited at the Department of Chemical Analysis, Institute of Natural Products Chemistry, Vietnam Academy of Science and Technology (VAST).
General procedure for the isolation of compounds
The isolation process begins with the stems of E. variegata. Impurities are meticulously removed, and the stems are finely chopped, thoroughly washed, and then dried at a temperature of approximately 50 °C until the moisture content reaches around 10%. Subsequently, the dried sample is ground into a powder with a particle size of approximately 0.2 mm, yielding 2 kg of E. variegata stem powder. This powder is then subjected to three consecutive extractions using n-hexane solvent at a ratio of 1/3 (weight/volume) with the assistance of an ultrasonic apparatus, conducted at 35 °C for a duration of 90 min. Following this process, the solvent is separated, and the remaining powder residue is obtained. The powder residue undergoes further extraction with ethyl acetate (EtOAc) solvent at a ratio of 1/5 (weight/volume) using the same ultrasonic apparatus, operated at 35 °C for a duration of 120 min. The resulting extract is then filtered, and the solvent is removed via vacuum distillation, ultimately yielding a total EtOAc extract (115.6 g). This total EtOAc extract is subsequently loaded onto a normal-phase silica gel chromatography column, featuring particle sizes ranging from 63 to 200 μm. Elution is carried out using a solvent system consisting of n-hexane/acetone. Initially, elution is performed with a polarity ratio of 10/1, which is gradually increased to 1/10 (volume/volume). This multi-step process yields 15 fractions, which are denoted as EVF.1–EVF.15. Fraction EVF.12 is further subjected to chromatography on an RP-C18 column (4.0 × 60 cm; 150 μm particle size). A stepwise gradient of MeOH–H2O (ranging from 1:4 to 4:1) is employed, resulting in 10 subfractions (F.12-1–F.12-10). Subfraction F.12-3 is then subjected to further purification using semi-preparative Agilent HPLC. An isocratic solvent system of 30% MeOH in H2O is used over a period of 42 min. This process yields compounds
Eryvarin M (
Eryvarin H (
Neobavaisoflavone (
Biology
PTP1B assay
To determine the inhibitory activity of compounds isolated from the stems and branches of the E. variegata against the enzyme PTP1B, an in vitro enzymatic assay was conducted following the method developed by To and colleagues in 2021. 70 The principle of this method involves the hydrolysis of p-nitrophenyl phosphate (p-NPP) by the enzyme PTP1B, resulting in the production of p-nitrophenol. The p-nitrophenol product generated after the phosphatase reaction has a yellow color. The amount of this color compound formed is directly proportional to the activity of the enzyme PTP1B. The enzyme activity is assessed by measuring the absorbance of the sample at 405 nm. Each well (final volume 110 μL) is loaded with 2 mM p-nitrophenyl phosphate (p-NPP) and 0.05–0.1 µg of PTP1B enzyme in a buffer solution containing 50 mM citrate (pH 6.0), 0.1M NaCl, 1 mM EDTA, and 1 mM dithiothreitol (DTT), with or without the test compound. The mixture is then incubated at 37 °C for 10 min, followed by the addition of 50 μL of p-NPP in a buffer solution. After incubating at 20 °C for 20 min, the reaction is stopped by adding 10M NaOH. The amount of p-nitrophenol produced by the enzyme through the phosphatase reaction is determined by measuring the absorbance at a wavelength of 405 nm using a spectrophotometer. The hydrolysis of p-NPP without the presence of PTP1B enzyme is corrected by measuring the increase in absorbance at 405 nm in the absence of PTP1B. All experiments are repeated three times.
α-glucosidase assay
The method for determining the inhibitory activity of α-glucosidase enzyme is performed using a 96-well microplate, with a reaction volume of 200 µL per well. The reaction components include 2.5 mM p-nitrophenyl α-D-glucopyranoside, 100 mM phosphate buffer (pH 6.8), 0.2 U mL−1 α-glucosidase, and test samples (10 µL in a 200 µL reaction mixture) at different concentrations. In negative control samples, the test sample is replaced with 100 mM phosphate buffer, and acarbose is used as a positive control. The enzyme reaction mixture is incubated at 37 °C. After 30 min, the reaction is stopped with 0.1M Na2CO3. The absorbance of the reaction is determined at a wavelength of 405 nm using an Infinite F50 spectrophotometer (Tecan, Switzerland). The percentage inhibition of α-glucosidase activity (%I) is calculated using the following formula:

where %I is the percentage of α-glucosidase enzyme activity inhibited; AC is the absorbance of the control sample (without test sample); AT is the absorbance of the test sample; and A0 is the absorbance of the blank sample (containing only buffer, no α-glucosidase enzyme).
All tests are repeated three times. The IC50 values, representing the concentration of the test samples that inhibits 50% of α-glucosidase enzyme activity, are calculated based on the log (concentration of test sample) and percentage inhibition curve.
Molecular docking study
Protein preparation
In this study, protein structures were retrieved from the Protein Data Bank (RCSB). The structure of human PTP1B with a catalytic inhibitor (PDB ID: 1BZC) was downloaded for the molecular docking process at the catalytic site. However, since the crystal protein (PDB ID: 1BZC) lacked an allosteric inhibitor, we also selected the co-crystal ligand at human PTP1B (PDB ID: 1T4J) to validate the docking process at the allosteric site. To assess similarity and identify both the catalytic and allosteric sites, the two crystal structures of PTP1B were aligned. Subsequently, PDB ID: 1BZC was chosen for further processing based on factors, such as resolution and the presence of an α-helix chain (residues Gly283–Asp298) compared to PDB ID: 1T4J. For human α-glucosidase, the protein crystal structure was also obtained from the RCSB database (PDB ID: 5NN8). The protein preparation followed established protocols as described in previous publications (please provide the appropriate citations). The selected crystals for the simulation process underwent the removal of water molecules and co-ligands. Subsequently, energy minimization was performed using Swiss PDB Viewer with standard optimization parameters (please provide the relevant citation). To prepare for molecular docking, polar hydrogen molecules were added to both protein structures using AutoDock Tool 1.5.6, and the structures were exported in a dockable pdbqt format.
Ligand preparation
The 3D structures of the compounds were prepared using MarvinSketch software (ChemAxon, USA). Subsequently, the compounds were optimized and set up with the MMFF94 force field using Avogadro (please provide the relevant citation). The co-crystal ligands of the proteins (PDB ID: 1BZC, 1T4J, and 5NN8) were also extracted and subjected to the same protocol for reference. Finally, AutoDock Tool 1.5.6 was employed to automatically establish torsion bonds for all ligands and convert them to the pdbqt format, which is suitable for screening.
Molecular docking simulation
The software AutoDock Vina 1.1.2 was used to conduct the molecular docking process. The positions of the crystal ligands were employed to define the grid box encompassing the active site of the protein. Docking results were expressed in kcal mol−1. The Discovery Studio Visualizer 2020 program was employed to visualize the interactions between chemical compounds and protein targets.
ADMET predictions
Three isoflavonoids (
Footnotes
Acknowledgements
The authors thank Dr Nguyen Quoc Binh for his help in the identification of the plant materials.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was financially supported by the Vietnam Academy of Science and Technology under the grant no. UDPTCN 03/21-23.