
Abstract
Sphingosine kinase is a lipid kinase that catalyzes the phosphorylation of sphingosine to sphingosine-1-phosphate. Sphingosine-1-phosphate is a bioactive lipid that regulates biological processes. The overexpression of sphingosine kinases is related to a variety of pathophysiological conditions. For example, SphK1 has been shown to be highly expressed in various cancer cells including ovarian, cervical, colon, stomach, lung, and brain cancer. Inhibition of sphingosine kinases is a promising way to treat diseases such as cancer. Through computer-aided drug design, we have discovered a new SphK1 inhibitor named Amb30572637 (SAMS10). In this report, we describe the discovery process and biological characteristics. In biochemical experiments, SAMS10 shows a prominent inhibitory effect on SphK1, with an IC50 value of 9.8 μM. Subsequent MTT experiments show that SAMS10 has anticancer effects toward A549, SKVO3, A375, and LOVO cell lines and has essentially no cytotoxicity against the healthy cell L929. SAMS10 has significant inhibitory activity against the A549 and LOVO cell lines, with IC50 values of 14.64 and 14.48 μM, respectively. It belongs to a moderately active SphK1 inhibitor with lower anticancer activity than the control compound cisplatin, but the effect of SAMS10 toward SphK1 and its anticancer activity indicate that it is a promising lead compound for the development of effective SphK1 anticancer inhibitors.
Introduction
Targeting the sphingosine-1-phosphate (S1P) signaling pathway is promising for the treatment of many diseases because elevated S1P levels is related to cardiovascular diseases, autoimmune/inflammatory diseases, cancers, and other pathophysiological conditions. 1 Sphingosine kinases (SphKs) are key kinases in the S1P signaling pathway, which can regulate the balance between sphingosine (Sph), ceramide (Cer), and S1P, and regulate overall S1P levels.2,3 Inhibiting the activity of SphKs can inhibit Sph from being transforming into S1P that promotes cell proliferation, migration, differentiation, and survival.4,5 The SphK family includes two isoforms: SphK1 and SphK2. 6 The SphK1 crystal structure and co-crystal structures of SphK1 with three inhibitors (SK-II, PF-543, Amgen 23; Figure 1) have been resolved, but the crystal structure of SphK2 has not been resolved yet.7–9 SphK1 contains two domains: N-terminal domain (NTD) and C-terminal domain (CTD). 10 The ATP binding site is located in NTD, the Sph binding site is located in CTD, and the catalytic center of SphK1 is located between NTD and CTD. 10 To date, the development of SphK1 inhibitors has mainly focused on compounds that bind to the Sph binding pocket and compete with Sph.2,11 In this work, we also target the Sph binding site to develop novel sphingosine kinase 1 inhibitors. The shape of the Sph binding site resembles the letter J. 10 The end near the NTD is called the polar head, there are several polar amino acids in the polar head, Asp264, and Asp167 are the most critical amino acids. 10 The end away from the NTD is called the hydrophobic tail and is surrounded by hydrophobic amino acids. 10 There are several articles describing Sph binding sites in detail.7,9,10 Several sphingosine kinase inhibitors have been developed and many reviews have introduced them in detail.2,11,12 In this work, we constructed some pharmacophore models and then evaluated the pharmacophore models. We first used the pharmacophore and then used molecular docking to perform virtual screening of a compound database. A total of 12 molecules were selected as potential SphK1 inhibitors for in vitro kinase activity testing. Finally, we report a new SphK1 inhibitor, which has an IC50 value of 9.8 μM for SphK1 and demonstrates anticancer activity.

Structures of SphK inhibitors.
Results and discussion
Establishment and evaluation of the pharmacophore model
In this work, the computer-aided drug design software used was BIOVIA Discovery Studio2020 (DS). We built multiple pharmacophore models based on the co-crystal structures of SphK1 with PF-543 (PF-543 has the highest activity among the existing co-crystal structures) and selected the best pharmacophore model through evaluation (Figure 2). 9 We used the method based on the receptor–ligand complex to construct the pharmacophore and constructed 50 pharmacophores in total by utilizing the Receptor-Ligand Pharmacophore Generation module in the DS software. To further screen the optimum pharmacophore model, we selected 120 small molecules as a test set. Among these 120 compounds, there were 20 active SphK1 inhibitors and 100 inactive SphK1 inhibitors. We use the test set to validate the generated pharmacophore model. In 50 models, the area under the curve (AUC) is defined as the area enclosed by the receiver operating characteristic (ROC) curve and the coordinate axis. The closer the AUC is to 1.0, the higher the authenticity of the detection method) value of pharmacophore 38 reached 0.9, which can effectively distinguish SphK inhibitors (Figure 2). Therefore, we choose pharmacophore 38 as the optimum model for preliminary screening of compounds. The Supporting Information provides the detailed process of the pharmacophore model construction.
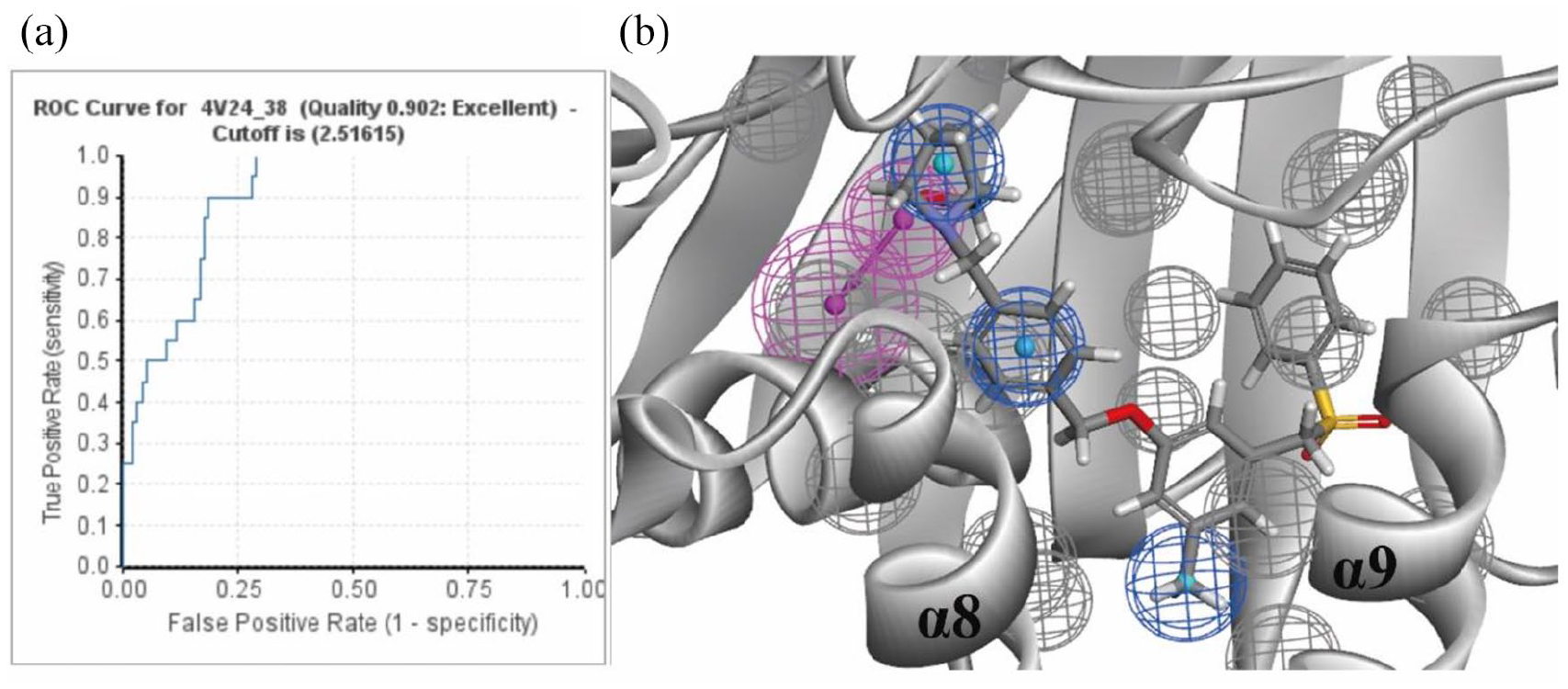
(a) ROC curve (The ROC curve graph is a curve reflecting the relationship between sensitivity and specificity) for pharmacophore 38. (b) Schematic diagram of pharmacophore 38 superimposed into the co-crystal structure of SphK1 and PF-543. 9 The gray balls represent the excluded volume, the magenta balls are the hydrogen bond acceptor, and the blue balls represent hydrophobic characteristics.
Virtual screening of databases
The compound library we used was Ambinter (http://www.ambinter.com/). We randomly selected 200,000 commercial ligands with drug-like properties from Ambinter for this screening. These ligands were optimized by the Prepare Ligand Procedure module in the DS software. We first used the pharmacophore and then used molecular docking to perform virtual screening of the compound database (Figure 3). The 200,000 commercial ligands were matched with optimal pharmacophore model 38. After this preliminary screening, the top 20,000 of molecules were selected according to the ranking of fitvules and were used for subsequent molecular docking screening. When using molecular docking for screening, we chose the CDOCKER module in the DS software. After the docking was complete, sorted by -CDOCKER_INTERACTION_ENERGY (The negative of the receptor–ligand interaction energy is reported as -CDOCKER_INTERACTION_ENERGY), and then the top 2000 of small molecules were manually screened. We selected compounds of suitable length that matched the shape of the substrate binding pocket and based on their interactions with the key amino acid residues Asp167 and Asp264, we selected 12 compounds as potential lead compounds for subsequent in vitro kinase activity testing (Figure 4). 7 The Supporting Information provides the -CDOCKER_INTERACTION_ENERGY data for the 12 selected compounds and the detailed process of the molecular docking.

Schematic diagram of the virtual screening process.
The structures of the 12 compounds selected for in vitro SphK1 kinase activity testing.
Kinase inhibitory activities
These 12 compounds (Figure 4) were tested for their inhibition activity against SphK1 at a concentration of 10 μM. The inhibitory activity of these compounds against SphK1 is shown in Table 1. At 10 μM, most of the compounds had no inhibitory effects against on SphK1. However, to our delight, compound SAMS10 (SAMS10 used in the experiments is a racemate), is excellent. Inhibition of SphK1 is at 73%.
a SphK1 inhibitory activity of the 12 selected compounds at a concentration of 10 μM.

NA: no activity.
The value is the percentage of inhibition against SphK1 (average of two independent experiments) with a standard deviation of ±5%.
We further evaluated the IC50 value of SAMS10 (SAMS10 used in the experiments is a racemate) against SphK1 and SphK2. As shown in Table 2, SAMS10 has strong inhibitory activity against SphK1, with an IC50 value of 9.8 μM, and only exhibits a weak inhibitory effect against SphK2. This indicates that SAMS10 is a highly selective SphK1 inhibitor.
The inhibitory effects of Amb30572637 (SAMS10) against SphK1 and SphK2.
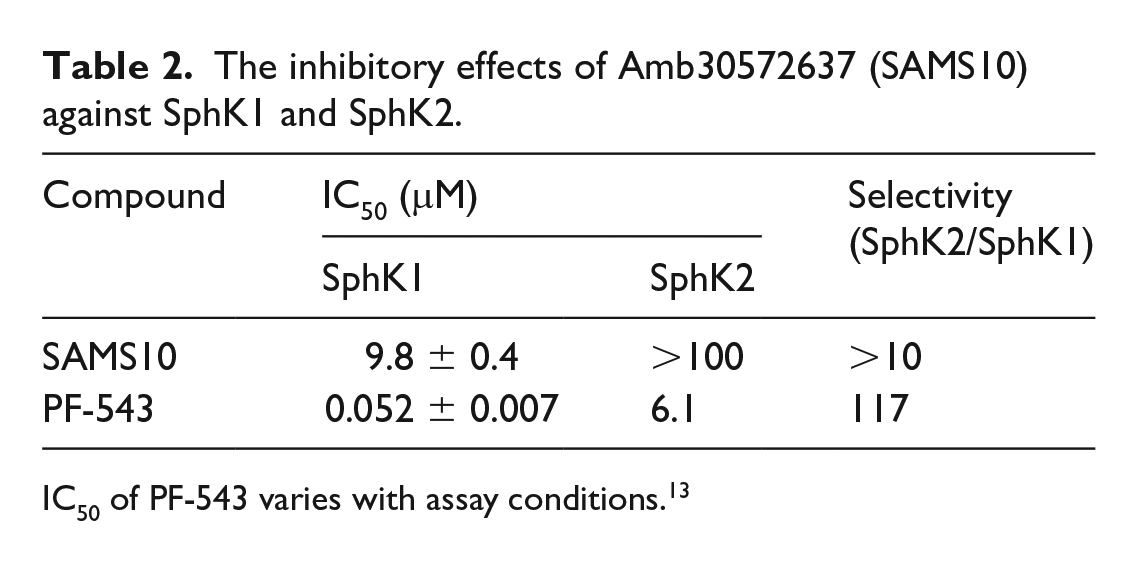
IC50 of PF-543 varies with assay conditions. 13
Cytotoxic activity
Cells with high SphK1 expression are distributed in liver cancer, prostate cancer, melanoma, lung cancer, and gliomas. Thus, it is necessary to test the cytotoxic activity of SAMS10 (SAMS10 used in the experiments is a racemate). For this purpose, we used four cell lines, A549, SKVO3, A375, and LOVO to test the cytotoxic activity of SAMS10 (Table 3). SAMS10 does not exhibit strong anticancer effects compared with cisplatin, but it had certain anticancer effects toward the SKVO3, A549, LOVO, and A375 cell lines. Among them, SAMS10 has a prominent inhibitory activity against the A549 and LOVO cell line, with an IC50 value of 14.64 and 14.48 μM. In healthy cells L929, SAMS10 is basically noncytotoxic and its IC50 value is 74.28 μM, which is beneficial to its future development.
Anti-proliferative effects of SAMS10 and cisplatin as a reference.

Docking of SAMS10 and SphK1
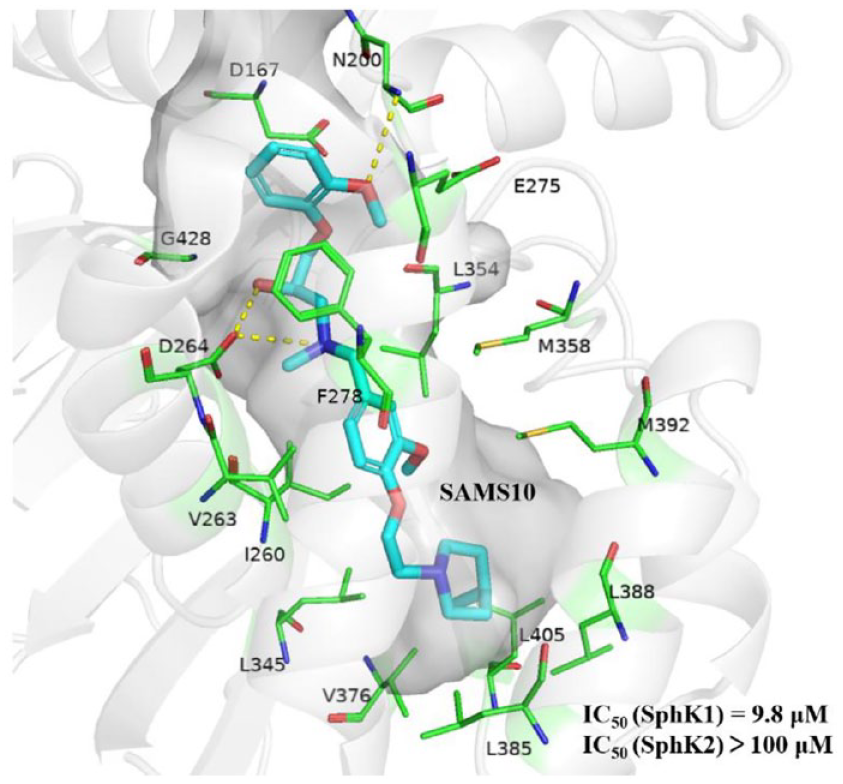
We performed molecular docking analysis on SAMS10 and the co-crystal structures of SphK1 with PF-543 (4V24). 9 According to the docking results (for the docking results of SAMS10, we chose the R configuration that matches the protein better), SAMS10 occupies in the “J-shaped” cavity of SphK1, and its hydroxyl group can form hydrogen bond with the key amino acid residues Asp264 with a bond length of 1.64 Å and a bond angle of 165.28°. The oxygen at the head of this compound forms a hydrogen bond with Asn200 with a bond length of 3.06 Å and a bond angle of 100.43°. In addition, the head of this compound forms unconventional hydrogen bonds with Asp167, Leu354, Glu275, and Gly428. The middles and tails of small molecules form a wide range of hydrophobic interactions with proteins, with the middle of SAMS10 forming interactions with Val263, Met358, Met392, and Ile260, while the tails form interactions with Leu345, Val376, Leu405, and Leu388 (Figure 5). Figure 5 also shows several inactive compounds. Although the hydroxyl group of SAMS8 forms a hydrogen bond with Gly428 and Ser254, the compound has a very weak interaction with the key amino acid Asp264. Similarly, SAMS9 and amino acid Asp264 only form weak carbon hydrogen bonds. Although the oxygen in SAMS8 forms a hydrogen bond with Asp264, its bond length is 2.04 Å, which is weaker than that of SAMS10. This may explain why only SAMS10 is active against SphK1.
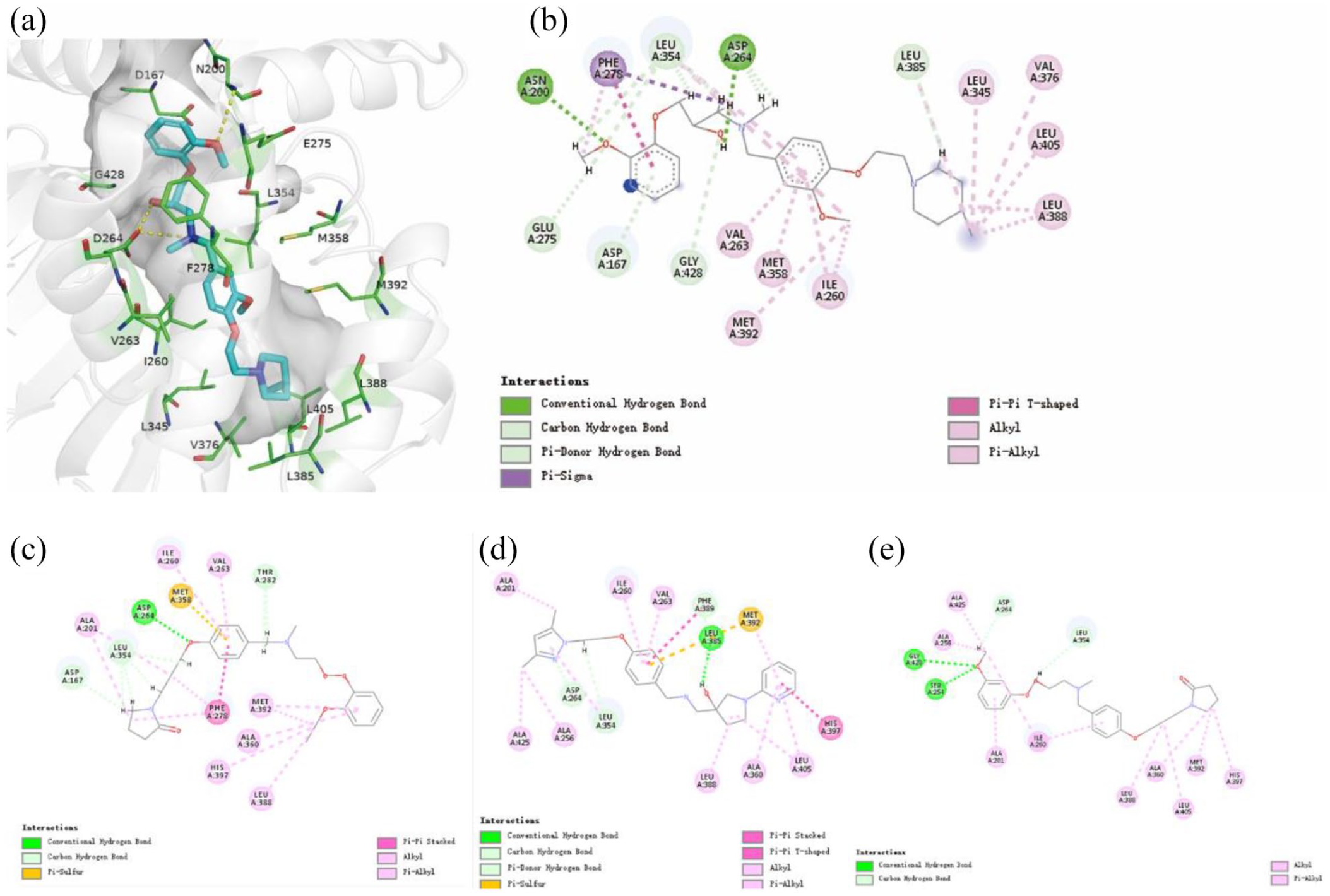
Schematic diagram of the interaction between SAMS7-10 and SphK. The green lines represent the hydrogen bonding interactions. The pink lines indicate hydrophobic interactions. (a) Three-dimensional schematic diagram of the interaction between SAMS10 and SphK. (b) Two-dimensional schematic diagram of the interaction between SAMS10 and SphK. (c) Two-dimensional schematic diagram of the interaction between SAMS7 and SphK. (d) Two-dimensional schematic diagram of the interaction between SAMS9 and SphK. (e) Two-dimensional schematic diagram of the interaction between SAMS8 and SphK.
Synthesis of SAMS10
SAMS10 has a prominent role in inhibiting SphK1 and a variety of cell lines. The synthetic process is show in Scheme 1. We first obtained compound 4 through a three-step reaction, and then obtained compound 6 through a one-step reaction, and finally compound 4 reacted with 6 to produce SAMS10 (Compound 6 and SAMS10 are both racemates). The nuclear magnetic resonance (NMR) data for SAMS10 and compounds 2, 3, 4, 6 was provided in the Supporting Information.

Synthetic route toward SAMS10.
Conclusion
In sum, through virtual screening methods and biological verification, we have discovered a new SphK1 inhibitor. In vitro enzyme activity experiments revealed that the compound Amb30572637 (SAMS10) has the potential to inhibit the activity of SphK1. SAMS10 proved to be a selective SphK1 inhibitor (IC50 9.8 μM). To our delight, SAMS10 showed anticancer effects toward the A549, SKVO3, A375, and LOVO cell lines. Thus, SAMS10 represents a promising lead compound for SphKs inhibition for future development.
Experimental
Materials
2-Ethoxyphenol, 4-hydroxy-3-methoxybenzaldehyde, 2-bromoethanol, 4-methylbenzenesulfonyl chloride, triethylamine, DMAP, 2-(bromomethyl)oxirane, potassium carbonate, tetrabutylammonium iodide, epibromohydrin, acetonitrile, pyridine, isopropanol, 4-methylpiperidine, tetrabutylammonium iodide, ethyl acetate, methylamine hydrochloride, sodium borohydride fetal bovine serum, SphK1 (Signalchem, Canada), SphK2 (Signalchem, Canada), and other materials were purchased from commercial sources (Aladdin, Macklin, Innochem) and were used without further purification.
Apparatus
High-resolution mass spectrometry (HRMS) was performed on an AB SCIEX X500R Accurate Mass Q-TOF by using electrospray ionization (ESI). The NMR spectra were recorded on a Bruker AVANCE-400 spectrometer with tetramethylsilane as the internal standard.
Synthesis
4-(2-Hydroxyethoxy)-3-methoxybenzaldehyde (2)
To a 500 mL reaction flask equipped with a stir bar was added compound 1 (5.0 g, 32.9 mmol, 1.0 equiv), 2-bromoethanol (5.8 g, 82.2 mmol, 2.5 equiv), CH3CN (260.0 mL), followed by the addition of K2CO3 (6.8 g, 49.3 mmol, 1.5 equiv). The reaction mixture was allowed to stir at 100℃ for 24 h. After cooling to room temperature, the mixture was poured into water then extracted into ethyl acetate. The combined organic layers were washed with brine and dried over Na2SO4. The organic layer was filtered and concentrated under reduced pressure to provide the crude product, which was purified by silica gel chromatography (CH2Cl2/MeOH 50:1) to give the desired product
4-(2-hydroxyethoxy)-3-methoxybenzaldehyde (
2-(4-Formyl-2-methoxyphenoxy)ethyl 4-methylbenzenesulfonate (3 )
To a 500 mL reaction flask equipped with a stir bar was added compound 2 (6.4 g, 32.6 mmol, 1.0 equiv), 4-methylbenzenesulfonyl chloride (18.7 g, 97.9 mmol, 3 equiv), CH2Cl2 (280.0 mL), triethylamine (22.6 mL, 163.1 mmol, 5 equiv) and DMAP (0.8 g, 6.524 mmol, 0.2 equiv). The reaction mixture was allowed to stir at room temperature for 24 h. After cooling to room temperature, the mixture was poured into water then extracted into ethyl acetate. The combined organic layers were washed with brine and dried over Na2SO4. The organic layer was filtered and concentrated under reduced pressure to provide the crude product, which was purified by silica gel chromatography (CH2Cl2/MeOH 20:1) to give the desired product
2-(4-formyl-2-methoxyphenoxy)ethyl 4-methylbenzenesulfonate (3): yield: 10.3g, (90%); white solid. 1H NMR (600 MHz, CDCl3): δ = 9.86 (s, 1H), 7.84 – 7.79 (m, 2H), 7.41 (d, J = 7.4 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H), 6.92 (d, J = 8.3 Hz, 1H), 4.45 – 4.40 (m, 2H), 4.32 (dd, J = 5.7, 4.2 Hz, 2H), 3.90 (s, 3H), 2.44 (s, 3H). 13C NMR (150 MHz, CDCl3): δ = 190.84, 152.90, 150.04, 145.05, 132.70, 130.91, 129.87 (2C), 128.01 (2C), 126.38, 112.52, 109.71, 67.58, 66.56, 56.01, 21.68. HRMS (ESI): m/z calcd for C17H19O6S [M + H]+: 351.0902. found: 351.0824.
1-{3-Methoxy-4-[2-(4-methylpiperidin-1-yl)ethoxy]phenyl}-N-methylmethanamine (4 )
To a 250 mL reaction flask equipped with a stir bar was added compound 3 (1.56 g, 4.45 mmol, 1.0 equiv), 4-methylpiperidine (662.5 mg, 6.68 mmol, 1.5 equiv), tetrabutylammonium iodide (328.74 mg, 0.89 mmol, 0.2 equiv), and CH3CN (135 mL). The reaction mixture was allowed to stir at 50℃ for 24 h under nitrogen protection. After cooling to room temperature, the mixture was filtered and concentrated under reduced pressure. Next, to the mixture was added MeOH (200 mL) followed by the addition of methylamine hydrochloride (1.5 eq) under the protection of nitrogen. After the reaction was stirred at room temperature for 24 h, sodium borohydride (253.5 mg, 6.7 mmol, 1.5 equiv) was slowly added in an ice bath. The reaction mixture was quenched with saturated sodium bicarbonate after 30 min. The mixture was poured into water then extracted into ethyl acetate. The combined organic layers were washed with brine and dried over Na2SO4. The organic layer was filtered and concentrated under reduced pressure to provide the crude product, which was purified by silica gel chromatography (CH2Cl2/MeOH 15:1) to give the desired product
1-{3-methoxy-4-[2-(4-methylpiperidin-1-yl)ethoxy]phenyl}-N-methylmethanamine (
2-[(2-Ethoxyphenoxy)methyl]oxirane (6 )
To a 25 mL glass tube equipped with a stir bar was added 2-ethoxyphenol (414.3 mg, 3.0 mmol, 1.0 equiv), K2CO3 (1.2 g, 9 mmol, 3.0 equiv), tetrabutylammonium iodide (221.6 mg, 0.2 mmol, 0.6 equiv) and CH3CN (16 mL), followed by the addition of epibromohydrin (1.0 mL, 12.0 mmol, 4.0 equiv) dropwise under the protection of nitrogen. The reaction mixture was allowed to stir at 50 ℃ for 36 h. After cooling to room temperature, the mixture was poured into water then extracted into ethyl acetate. The combined organic layers were washed with brine and dried over Na2SO4. The organic layer was filtered and concentrated under reduced pressure to provide the crude product, which was purified by silica gel chromatography (PE/EtOAc 20:1) to give the desired product
2-[(2-ethoxyphenoxy)methyl]oxirane (
1-(2-Ethoxyphenoxy)-3-({3-methoxy-4-[2-(4-methylpiperidin-1-yl)ethoxy]benzyl}(methyl)amino)propan-2-ol (SAMS10)
To a 25 mL glass tube equipped with a stir bar was added compound 6 (194.2 mg, 1.0 mmol, 1.0 equiv),
1-(2-ethoxyphenoxy)-3-({3-methoxy-4-[2-(4-methylpiperidin-1-yl)ethoxy]benzyl}(methyl)amino)propan-2-ol (SAMS10): Yield: 413.6 mg, (85%); colorless oil. 1H NMR (400 MHz, CDCl3): δ = 6.98–6.75 (m, 7H), 4.16 (dt, J = 18.6, 5.3 Hz, 3H), 4.10–3.95 (m, 4H), 3.84 (s, 3H), 3.59 (d, J = 13.0 Hz, 1H), 3.47 (d, J = 13.0 Hz, 1H), 3.05 (d, J = 11.4 Hz, 2H), 2.90 (t, J = 6.3 Hz, 2H), 2.70–2.51 (m, 2H), 2.28 (s, 3H), 2.26–2.14 (m, 2H), 1.66 (d, J = 10.2 Hz, 2H), 1.48–1.29 (m, 6H), 0.94 (d, J = 5.6 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ = 149.44, 149.39, 148.72, 147.33, 131.62, 122.12, 121.27, 121.09, 115.80, 113.84, 113.16, 112.49, 72.91, 66.61 (2C), 64.51, 62.45, 59.60, 57.25, 55.94, 54.37 (2C), 42.43, 33.84 (2C), 30.39, 21.76, 14.93. IR (KBr, cm-1): 2924, 2871, 2851, 2794, 2360, 2340, 1592, 1512, 1494, 1460, 1419, 1368, 1321, 1272, 1217, 1138, 1035, 980, 863, 803, 772, 670. HRMS (ESI): m/z [M + H]+ calcd for C28H43N2O5: 487.3172. found: 487.3199.
The in vitro kinase test
The SphKs assays were carried out as described previously. 13 We used the Kinase-Glo Plus Luminescent Kinase Assay Kit purchased from Promega to measure the kinase activity by quantifying the amount of ATP remaining in the solution after the kinase reaction. The reaction volume is 50μM. PF-543 purchased from Selleck was used as a positive control. SAMS10 was dissolved in pure DMSO to prepare 10 mM stock solutions and diluted with kinase buffer (pH = 7.4, composition: 40 mM/L Tris, 10 mM/L MgCl2, 0.1 g/L BSA, 1 mM/L DTT, 10 µM/L ATP). SphK1 or SphK2 was added to 96-well plates and treated with the appropriate concentration of SAMS10 (0.01, 0.1, 1, 10 or 100 µM) at 30°C for 40 min. ATP test solution was added and the mixture was incubated at room temperature for 5 min. The luminescence was immediately measured using a microplate spectrophotometer (AD 340, Beckman, USA). Graphpad Prism 9 software was used for data analysis.
MTT cell viability assay
Here we used cisplatin as a positive control and used the MTT method to test the effect of compound SAMS10 on the survival of five cell lines A549, SKVO3, A375, LOVO and L929 (purchased from the Cell Bank of the Chinese Academy of Sciences, Shanghai, China). First, five cell lines were seeded at a density of 6 × 103 per well in 96-well plates and cultured for 24 h. Compounds were dissolved in pure DMSO and diluted into 10 mM stock solution with the culture medium. Then different concentrations of SAMS10 (0.01, 0.1, 1, 10, 100 µM) and cisplatin was added to a 96-well plate and incubated at 37°C for 48 h. After 48 h, 10 µL of a solution of MTT (5 mg/mL in PBS) was added to each well in the 96-well plates. After 4 h, the OD value of each well was measured at 570 nm in a microplate reader (Bio-Rad Laboratories). Finally, the IC50 values were calculated.
Supplemental Material
sj-docx-1-chl-10.1177_17475198221089222 – Supplemental material for Discovery of an SphK1 inhibitor: A hybrid approach involving a receptor–ligand-complex-based pharmacophore and docking-based virtual screening
Supplemental material, sj-docx-1-chl-10.1177_17475198221089222 for Discovery of an SphK1 inhibitor: A hybrid approach involving a receptor–ligand-complex-based pharmacophore and docking-based virtual screening by Tiandi Ding, HaiJiao Chen, Yan Li, Ying Li, Ying Zhi, Zhiqiang Qu, Qiang Sun, Qingqiang Yao and Bo Liu in Journal of Chemical Research
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China (Grant No. 81903473) and the Academic Promotion Program of Shandong First Medical University (No. 2019LJ003).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.