An improved process for the synthesis of YG-056SP, a potent oxazolidinone candidate against multi-drug resistant bacteria, is developed. Compared with the original synthetic route, this new approach is two steps shorter, and all of the steps involve simple purifications without column chromatography. More importantly, it avoids the use of explosive azide compounds and expensive metal catalysts. The new reaction conditions are mild and safe, which is more suitable for the scalable synthesis of YG-056SP for preclinical studies.
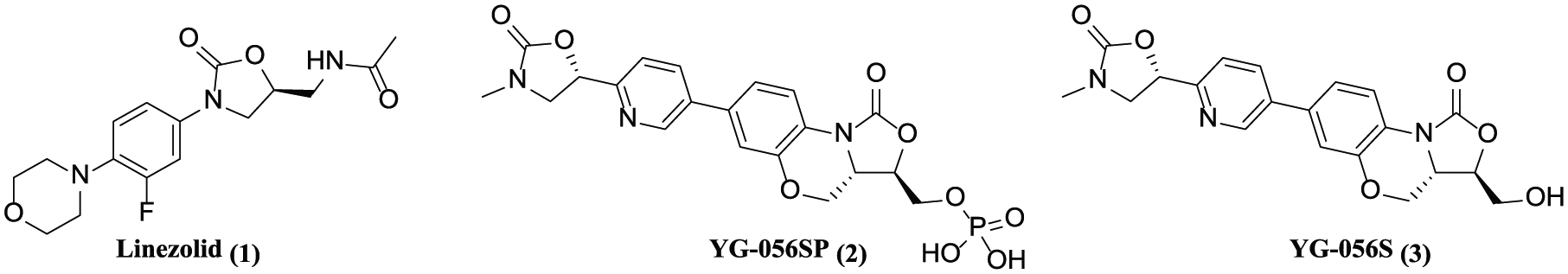
With the irrational use of antibiotics in different fields such as medical, agricultural, and food industries, the problem of bacterial resistance has become increasingly prominent and is a serious threat to human life.1,2 Recently, multi-drug resistant bacteria, especially Gram-positive bacteria including methicillin-resistant Staphylococcus aureus (MRSA) and Staphylococcus epidermidis (MRSE), vancomycin-resistant Enterococci (VRE), and penicillin-resistant Streptococcus pneumoniae (PRSP), have spread worldwide.3–5 Oxazolidinones, a new class of synthetic antibacterial agents, have shown good activity against a broad range of Gram-positive bacteria and little cross-resistance. Linezolid (1) (Figure 1) was the first member of this class to achieve Food and Drug Administration (FDA) approval and has achieved good results in the treatment of infections caused by Gram-positive pathogens.6 However, linezolid-resistant Staphylococcus aureus and Enterococcus faecium began to emerge shortly after the drug was launched.7–9 Meanwhile, the bone marrow toxicity of linezolid presents a number of limitations for its clinical use. Therefore, it is of great significance to develop new second-generation oxazolidinone agents with improved activity and safety.
Chemical structures of oxazolidinone antibiotics.
YG-056SP (2) is a new type of second-generation oxazolidinone antibacterial agent developed with linezolid as a lead compound.10 YG-056S (3), the parent compound of YG-056SP, shows excellent in vitro activity against a vast majority of susceptible and resistant Gram-positive organisms, including linezolid-resistant strains (Table 1). Furthermore, YG-056SP has a favorable pharmacokinetic profile and excellent in vivo antibacterial activity. In addition, YG-056SP exhibits no hERG inhibition or hepatocyte and bone marrow toxicity. For further preclinical studies, multi-gram qualities of YG-056SP is needed. With this background in mind, we have developed a novel and scalable synthetic process for the potent oxazolidinone antibacterial agent, YG-056SP.
The original medicinal chemistry route to YG-056SP (2) used 2,5-dibromopyridine as a raw material, and involved acylation, Noyori asymmetric transfer hydrogenation, azide nucleophilic substitution, reduction, cyclization, methylation, Suzuki coupling, esterification, and hydrogenation—a total of nine steps, to obtain target product (Scheme 1).10 However, this route exhibits problems that have hampered its scale-up. First of all, sodium azide was used in the synthesis of intermediate 9 and the reaction needed to be carried out at high temperature conditions, which do not meet the standards of industrial production and had potential safety issues. Second, chiral intermediate 8 was synthesized by Noyori asymmetric transfer hydrogenation, which involves an expensive metal catalyst making production costly. In addition, post-treatment of many of the reactions in this route were commonly performed by column chromatography, which are not suitable for large-scale production. These problems prompted us to seek for an improved synthetic process toward YG-056SP (2).
Original medicinal chemistry route to YG-056SP. Reagents and conditions: (a) N,O-dimethylhydroxylamine hydrochloride, K2CO3, t-BuOMe, 0 °C, 85%; (b) n-BuLi, toluene, ‒78 °C, 73%; (c) HCOOH, Et3N, dichloro (p-cymene)ruthenium(II) dimer, (1S,2S)-(+)-N-(4-toluenesulfonyl)-1,2-ethane diamine, MTBE, r.t., 89%; (d) NaN3, DMF, 90 °C, 83%; (e) PPh3, THF/H2O, 45 °C to r.t., 81%; (f) CDI, DMAP, DMF, r.t., 87%; (g) MeI, NaH, THF, 0 °C to r.t., 91%; (h) Pd(PPh3)4, Cs2CO3, 1,4-dioxane/H2O, 80 °C, 71%; (i) (BnO)2PN(i-Pr)2, 4,5-dicyanoimidazole, CH2Cl2, r.t.; (j) 3-chloroperbenzoic acid, CH2Cl2, 0 °C, 72%; (k) 10% Pd/C, H2, r.t., 80%.
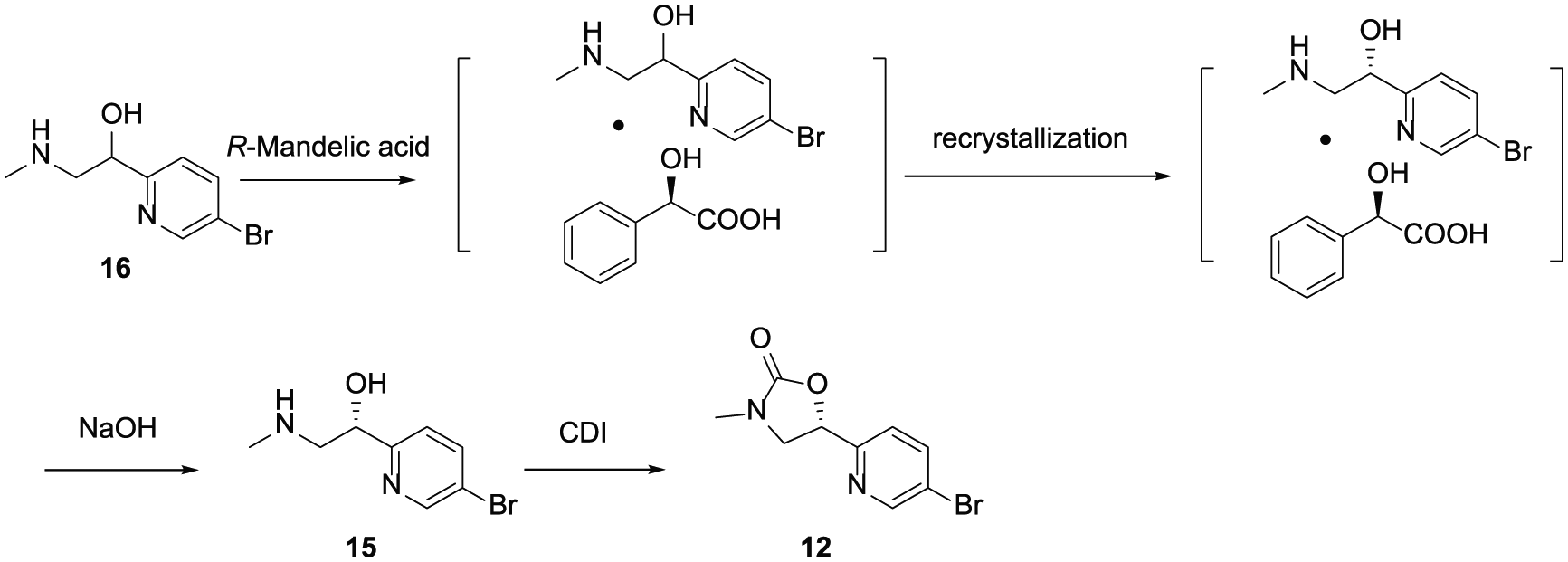
As shown in Scheme 2, retrosynthetic analysis suggests that compound 12 can be synthesized by cyclization of compound 15, which avoids methylation as used in the initial route. Compound 15 can be obtained by chiral resolution of compound 16, itself formed via reduction of compound 17, which avoids the expensive metal catalyst that was used in the Noyori asymmetric transfer hydrogenation. Compound 17 would be prepared by reaction between 2,5-dibromopyridine (6) with commercially available sarcosinate ester in the presence of n-BuLi.
Retrosynthetic analysis of compound 12.
Through the retrosynthetic analysis of intermediate 12 in Scheme 2, we designed a new synthetic route and verified its feasibility through experiments. We chose ethyl sarcosinate hydrochloride compound 19, which is inexpensive and of higher purity compared with methyl sarcosinate hydrochloride, as the starting material to prepare compound 17 by reaction with 2,5-dibromopyridine (6). However, compound 17 cannot be obtained by direct acylation of compound 6 with the sarcosinate ester due to the N-H effect. Therefore, it is necessary to protect the nitrogen atom of the sarcosinate ester in order to prepare compound 17. Since compound 23 is a solid and compound 24 is an oil which cannot be purified by recrystallization, it is feasible to protect the nitrogen atom of the sarcosinate ester with di-tert-butyl dicarbonate (Scheme 3).
Synthesis of compound 23.
The solvent and concentration strongly influence the selectivity of the acylation of 2,5-dibromopyridine (6) at either the 2-position or the 5-position (Scheme 4).11 By optimizing the solvent, reaction time, and concentration, the most favorable conditions were confirmed. In addition, all regioisomers could be removed by recrystallization, which was not covered in the initial route. Subsequently, compound 16 could be prepared in high yield through reduction and deprotection of resulting alcohol compound 23.
Synthesis of compound 16.
As shown in Scheme 5, the chiral compound 15 was prepared through chiral separation of compound 16. After screening, R-mandelic acid was identified as the preferred chiral acid to give a high yield and ee value. Finally, the key intermediate 12 was obtained by cyclization of alcohol 15 with CDI as the cyclizing agent.
Chiral separation of alcohol 16.
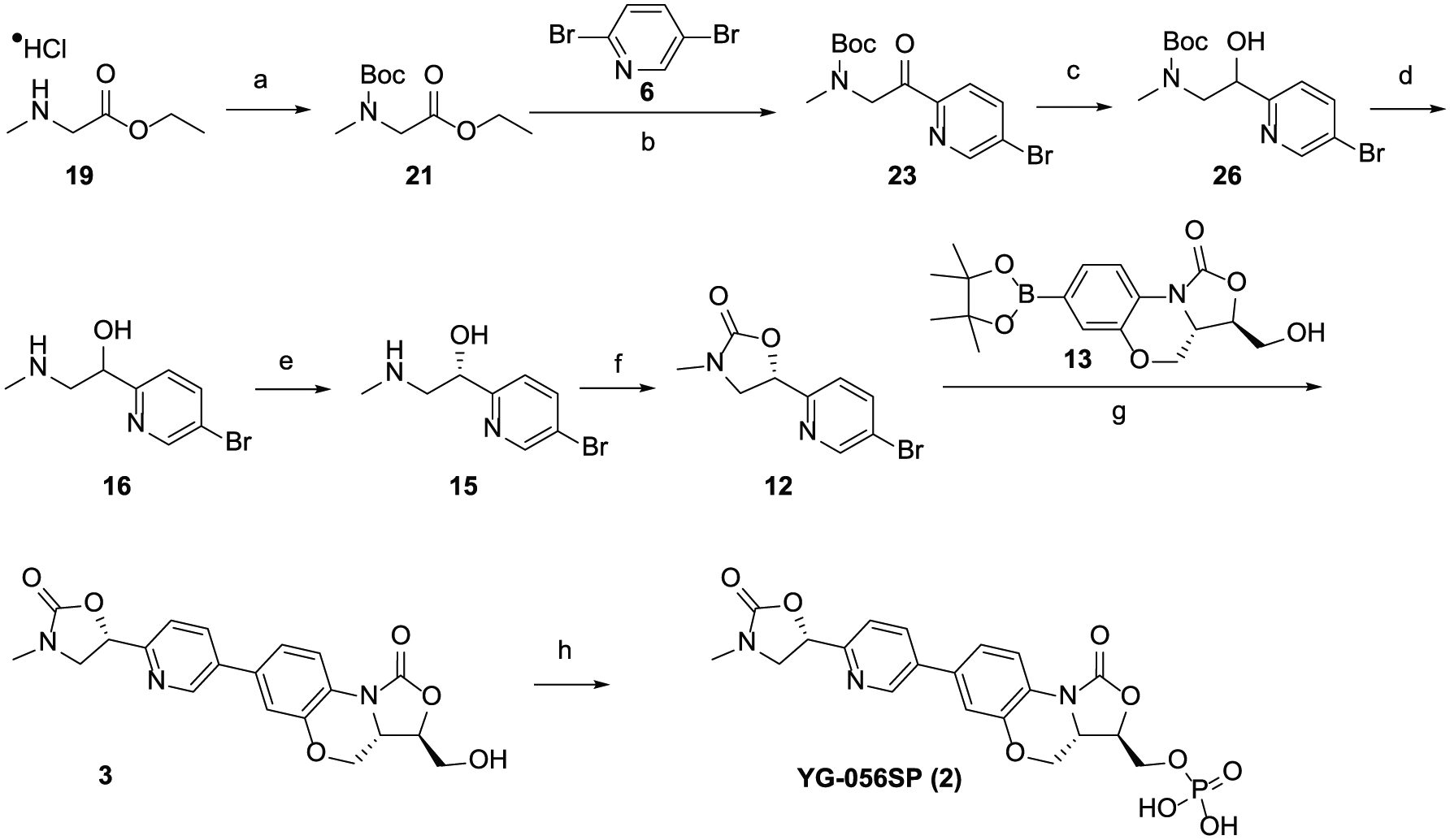
The final synthetic route to YG-056SP (2) is shown in Scheme 6. Sarcosine ethyl ester hydrochloride 19 was protected with Boc2O to give amide 21 in 92% yield. This was transformed into bromide 23 by reaction with 2,5-dibromopyridine (6) in the presence of n-BuLi in toluene. Recrystallization from ethanol/water (10/1, v/v) yielded the product with a purity of >98% and a regioisomer 24 of <1% in 50% yield. Alcohol 26 was prepared in 95% yield by reducing compound 23 with sodium borohydride. Deprotection of this alcohol with hydrochloric acid gave amine 16 in 91% yield. Chiral separation of 16 using the difference of solubility between diastereomeric organic salts gave 15 in 36% yield and 97% ee. Cyclization of compound 15 with CDI gave oxazolidinone 12 in 88% yield. Suzuki coupling of compound 12 with 13 gave product 3 in 85% yield. For the preparation of YG-056SP (2), the synthetic conditions were modified. The much cheaper POCl3 was selected as the phosphonating agent instead of dibenzyl N,N-diisopropylphosphoramidite, leading to YG-056SP (2) in 81% yield.
Final synthetic route to YG-056SP (2). Reagents and conditions: (a) Boc2O, K2CO3, acetone/H2O, r.t., 92%; (b) n-BuLi, toluene, ‒78 °C, 50%; (c) NaBH4, methanol, r.t., 95%; (d) HCl, 1,4-dioxane, r.t., 91%; (e) R-mandelic acid, acetonitrile/H2O, 70 °C to 4 °C, 36%; (f) CDI, DMAP, THF, r.t., 88%; (g) Pd(PPh3)4, Cs2CO3, 1,4-dioxane /H2O, 95 °C, 85%; (h) POCl3, Et3N, CH2Cl2, 0 °C, 81%.
Conclusion
In summary, we have developed a novel and scalable process of the synthesis of the potent oxazolidinone antibiotic candidate YG-056SP. The key steps involved regioselective monolithiation of 2,5-dibromopyridine and chiral separation of racemic compound 16 with R-mandelic acid. This route avoided the use of explosive azide compounds and expensive metal catalysts by changing the starting materials. In addition, the new reaction route has the advantages of mild and safe reaction conditions, readily available reagents, low cost, avoids column chromatography, and should be applicable to scale-up.
Experimental
Unless otherwise mentioned, all reagents were purchased from commercial suppliers and used without further purification. All reactions were monitored by TLC (thin layer chromatography), using silica gel plates with fluorescence GF254 and UV light visualization. 1H NMR and 13C NMR spectra were obtained on a Bruker ARX-400, ARX-500, or ARX-600 spectrometer. Melting points (uncorrected) were determined on an X-4 melting point apparatus. The purity of the compounds was determined by high performance liquid chromatography on an Agilent 1100 series LC system. The ee values of the chiral compounds were determined by high performance liquid chromatography on a Shimadzu LC-10A VP. Analytical RP-HPLC was conducted for purity using UV detection and the following conditions: (a) Kromasil 100-5-C18 (250 × 4.6 mm, 5 μm) using a gradient of 30% v/v MeOH in buffer solution (0.1% CF3COOH and 0.1% NH4OH in water, pH 3.5) to 70% v/v MeOH in buffer solution (0.1% CF3COOH and 0.1% NH4OH in water, pH 3.5) with detection at 270 nm. (b) Platisil ODS (250 × 4.6 mm, 5 μm) using a gradient of 40% v/v MeOH in buffer solution (0.1% CF3COOH and 0.1% NH4OH in water, pH 3.5) to 60% v/v MeOH in buffer solution (0.1% CF3COOH and 0.1% NH4OH in water, pH 3.5) with detection at 265 nm. (c) Diamonsil plus 5 μm C18-A (250 × 4.6 mm, 5 μm) using a gradient of 10% v/v MeOH in buffer solution (0.1% CF3COOH and 0.1% NH4OH in water, pH 3.5) to 90% v/v MeOH in buffer solution (0.1% CF3COOH and 0.1% NH4OH in water, pH 3.5) with detection at 254 nm. (d) Kromasil 100-5-C18 (250 × 4.6 mm, 5 μm) using a gradient of 85% v/v CH3CN in water to 20% v/v CH3CN in water with detection at 254 nm. (e) Diamonsil plus 5 μm C18-A (250 × 4.6 mm, 5 μm) using a gradient of 0.025 mol/L NH4HCO3 and CH3CN with detection at 234 nm. Analytical NP-HPLC was conducted for ee% determination using UV detection and the following conditions: (f) Chiral ID (250 × 4.6 mm, 5 μm) using a gradient of 10% v/v EtOH with 0.1% diethylamine (DEA) in hexane with 0.1% DEA to 90% v/v EtOH with 0.1% diethylamine in hexane, 0.1% DEA with detection at 265 nm. (g) Chiral ID (250 × 4.6 mm, 5 μm) using a gradient of 20% v/v EtOH with 0.1% DEA in hexane, 0.1% DEA to 90% v/v EtOH, 0.1% DEA in hexane, and 0.1% DEA with detection at 265 nm.
To a solution of sarcosine ethyl ester hydrochloride (19) (100 g, 0.65 mol) in 500 mL of a mixed solution of acetone and water (volume ratio: ketone/water = 1/1) was added potassium carbonate (224.5 g, 1.62 mol) under ice-bath conditions, and the mixture was stirred for 1 h at room temperature. Di-tert-butyl dicarbonate (128 g, 0.58 mol) was added, and the reaction was complete after 12 h at room temperature. The reaction solution was evaporated under reduced pressure to remove most of the solvent. The organic phase was separated, and the aqueous phase was extracted with dichloromethane 3 times. The combined organic layer was washed with water and brine, dried over anhydrous Na2SO4, filtered, and then evaporated under reduced pressure to give 21 as a colorless oil, yield: 117 g (92%).1H NMR (500 MHz, CDCl3): δ as a 1:1 mixture of rotamers: 4.19–4.10 (m, 2H), 3.92 (s, 2H, rotamer), 3.84 (s, 2H, rotamer), 2.89 (s, 3H, rotamer), 2.87 (s, 3H, rotamer), 1.43 (s, 9H, rotamer), 1.38 (s, 9H, rotamer), 1.27–1.19 (m, 3H); 13C NMR (126 MHz, CDCl3): δ (ppm) as a 1:1 mixture of rotamers: 169.8, 156.0, 155.4, 80.0, 60.9, 51.0, 50.2, 35.5, 28.2, 14.2; MS (EI): m/z 217 [M]+. HRMS (EI): m/z [M]+ calcd for C10H19O4N: 217.1309, found: 217.1304.
A solution of 2,5-dibromopyridine (6) (20 g, 84.5 mmol) in dry toluene (800 mL) was cooled to ‒78 °C and then n-BuLi (2.5 M in n-hexane, 37.2 mL, 92.9 mmol) was added dropwise. The mixture was stirred for 5 h at ‒78 °C. Next, amide 21 (20.2 g, 93.0 mmol) was added dropwise. The reaction was completed after 2 h and was quenched by adding 43 mL of 2 M aqueous HCl solution. The organic phase was separated, and the aqueous phase was extracted with dichloromethane 3 times. The combined organic layer was washed with water and brine, dried over anhydrous Na2SO4, filtered, and then evaporated under reduced pressure. The residue was purified by recrystallization from a 10:1 (v/v) mixture of ethanol and water, and filtered off. The product was dried at 40 °C under reduced pressure to provide compound 23 as a white solid; yield: 13.9 g (50%); m.p. 87–89 °C. HPLC purity (conditions A): 99%.1H NMR (400 MHz, CDCl3): δ as a 1:1 mixture of rotamers: 8.71 (d, J = 2.2 Hz, 1H), 8.70 (d, J = 2.3 Hz, 1H, rotamer), 8.00 (dd, J = 8.3, 2.3 Hz, 1H), 7.97 (dd, J = 8.3, 2.2 Hz, 1H, rotamer), 7.93 (d, J = 8.3 Hz, 1H), 7.91 (d, J = 8.3 Hz, 1H, rotamer), 4.87 (s, 2H), 4.80 (s, 2H, rotamer), 2.96 (s, 3H), 1.49 (s, 9H), 1.36 (s, 9H, rotamer). 13C NMR (126 MHz, CDCl3): δ as a 1:1 mixture of rotamers: 195.7, 195.4, 156.3, 155.8, 150.7, 150.2, 139.8, 139.7, 125.8, 125.7, 123.1, 123.0, 79.9, 79.8, 55.6, 55.2, 35.9, 35.7, 28.4, 28.2. MS (EI): m/z 330 [M]+. HRMS (EI): m/z [M]+ calcd for C13H17O3N2Br: 328.0417, found: 328.0410.
To a solution of 26 (10.0 g, 30.2 mmol) in 1,4-dioxane (50 mL) was added 2 M aqueous HCl (91 mL) dropwise at 0 °C. The mixture was stirred for 6 h at room temperature. Under ice bath conditions, the pH of the solution was adjusted to 11~12 with 1 M NaOH solution. The organic phase was separated, and the aqueous phase was extracted with ethyl acetate 6 times. The combined organic layer was washed with water and brine, dried over anhydrous Na2SO4, filtered, and then evaporated under reduced pressure to give 16 as a white solid, which can be used without further purification; yield: 6.35 g (91%); m.p. 85–87 °C. HPLC purity (conditions C): 99%. 1H NMR (400 MHz, CDCl3): δ 8.60 (d, J = 2.3 Hz, 1H), 7.82 (dd, J = 8.4, 2.4 Hz, 1H), 7.37 (d, J = 8.4 Hz, 1H), 4.77 (dd, J = 7.9, 3.8 Hz, 1H), 2.99 (dd, J = 12.2, 3.8 Hz, 1H), 2.77 (dd, J = 12.1, 7.9 Hz, 1H), 2.46 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 160.2, 149.7, 139.4, 121.8, 119.2, 71.1, 57.8, 36.2. MS (EI): m/z 231 [M]+. HRMS (EI): m/z [M]+ calcd for C8H12O1N2Br: 231.0128, found: 231.0121.
To a solution of 16 (16.0 g, 69.2 mmol) in acetonitrile (200 mL) was added R-mandelic acid (5.28 g, 34.6 mmol) at 70 °C. The reaction was stirred for 8 h at 70 °C and then evaporated under reduced pressure to give a white solid. A solution of the obtained white solid in acetonitrile (100 mL) and water (4 mL) was heated at 70 °C, kept at this temperature for 3 h, and then cooled slowly to 4 °C to precipitate a white solid (11.4 g). A solution of 11.4 g of the white solid obtained from the above reaction was dissolved in acetonitrile (100 mL) and water (4 mL), and then the above experiment was repeated to give 7.62 g of a white solid. The white solid obtained was dissolved in water, and the pH of the solvent was adjusted to 12 with 1 M NaOH solution. The solution was extracted with dichloromethane 3 times. The organic solution was dried over anhydrous Na2SO4 and concentrated in vacuo. The product 15 was dried at 40 °C under reduced pressure to provide a white solid; yield: 5.82 g (36%); m.p. 85–87 °C. HPLC purity (conditions C): 99%, ee (conditions F): 97%. 1H NMR (400 MHz, CDCl3): δ 8.59 (d, J = 2.3 Hz, 1H), 7.82 (dd, J = 8.4, 2.3 Hz, 1H), 7.39 (d, J = 8.4 Hz, 1H), 4.78 (dd, J = 7.9, 3.8 Hz, 1H), 2.98 (dd, J = 12.1, 3.8 Hz, 1H), 2.77 (dd, J = 12.1, 7.9 Hz, 1H), 2.49 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 160.2, 149.8, 139.5, 121.9, 119.3, 71.3, 57.9, 36.3. MS (EI): m/z 231 [M]+. HRMS (EI): m/z [M]+ calcd for C8H12O1N2Br: 231.0128, found: 231.0109.
To a solution of 15 (10.0 g, 43.3 mmol) in tetrahydrofuran (80 mL) was added CDI (8.42 g, 51.9 mmol) and DMAP (527 mg, 4.33 mmol). The mixture was stirred for 5 h at room temperature. Ethyl acetate (100 mL) was added, and the combined organic layer was washed with 10% KHSO4 solution, water, and brine; dried over anhydrous Na2SO4; filtered and then evaporated under reduced pressure. Purification by recrystallization from hexane and ethyl acetate afforded a solid, which was dried at 40 °C under reduced pressure to provide compound 12 as a white solid; yield: 9.78 g (88%); m.p. 68–70 °C. HPLC purity (conditions B): 99%. ee (conditions G): 100%. 1H NMR (400 MHz, CDCl3): δ 8.64 (d, J = 2.2 Hz, 1H), 7.89 (dd, J = 8.4, 2.3 Hz, 1H), 7.45 (d, J = 8.3 Hz, 1H), 5.51 (dd, J = 9.1, 6.0 Hz, 1H), 4.00 (t, J = 9.0 Hz, 1H), 3.71 (dd, J = 8.9, 6.0 Hz, 1H), 2.91 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 157.8, 156.9, 139.8, 121.7, 120.5, 52.3, 31.2. MS (EI): m/z 257 [M]+. HRMS (EI): m/z [M]+ calcd for C9H9O2N2Br: 255.9842, found: 255.9835.
To a suspension of 3 (5.0 g, 12.6 mmol) and triethylamine (5.2 mL, 37.8 mmol) in dry dichloromethane (100 mL) was added POCl3 (3.5 mL, 37.8 mmol) dropwise at 0 °C. The mixture was stirred for 3 h at 0 °C. Water (100 mL) was added, and stirring was continued for 6 h. The mixture was filtered, and the filter cake was slurried with methanol. The product was dried at 40 °C under reduced pressure to provide compound YG-056SP (2) as a white solid; yield: 4.87 g (81%); m.p. 211–214 °C. HPLC purity (conditions E): 98%. 1H NMR (600 MHz, DMSO-d6): δ 8.91 (s, 1H), 8.13 (dd, J = 8.1, 2.4 Hz, 1H), 7.96 (d, J = 9.0 Hz, 1H), 7.53 (d, J = 8.1 Hz, 1H), 7.42–7.39 (m, 2H), 5.64–5.60 (m, 1H), 4.70–4.65 (m, 1H), 4.65–4.61 (m, 1H), 4.22–4.08 (m, 4H), 3.95 (t, J = 8.9 Hz, 1H), 3.68 (dd, J = 8.8, 6.2 Hz, 1H), 2.79 (s, 3H).13C NMR (151 MHz, DMSO-d6): δ 157.22, 156.40, 153.34, 147.38, 144.70, 134.87, 134.50, 132.47, 123.53, 121.24, 119.81, 119.14, 115.03, 74.33, 73.31, 65.73, 64.62, 51.43, 51.19, 30.63. HRMS (ESI): m/z [M+H]+ calcd for C20H21O9N3P: 478.1010, found: 478.1024.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Youth Innovation Promotion Association of the Chinese Academy of Sciences (2016262).
ORCID iD
Bin Guo
References
1.
SpellbergBGuidosRGilbertD, et al. Clin Infect Dis2008; 46: 155.
2.
AlanisAJ.Arch Med Res2005; 36: 697.
3.
KhanSNKhanAU.Front Microbiol2016; 7: 174.
4.
TacconelliECarraraESavoldiA, et al. Lancet Infect Dis2018; 18: 318.
5.
WoodfordNLivermoreDM.J Infect2009; 59: S4.
6.
NorrbyR.Expert Opin Pharmacother2001; 2: 293.
7.
AucklandCTeareLCookeF, et al. J Antimicrob Chemother2002; 50: 743.
8.
TsiodrasSGoldHSSakoulasG, et al. Lancet2001; 358: 207.