
Abstract
Two complexes of N1-(2-aminoethyl)propane-1,3-diamine (AEPD), [Ni(AEPD)2](NO3)2 (
Introduction
Ligand transfer reactions between two metallic complexes are interesting reactions which have been observed between Fe/Ru, 1 Th/Mn, 2 Mn/Ni, 3 Mn/Pd, 3 La/Mg, 4 Y/Mg, 5 Pt/Th, 6 Fe/Co, 7 Sn/Pt, 8 Cu/Fe, 9 Cu/Co, 9 Sn/Au, 10 Sn/Ni, 10 Ti/Pt, 11 and Ti/Ru couples. In some cases, the ligands transfer from a cluster species 9 or transferring of the ligand is accompanied with electron transfer between metals. 6 This reaction can occur as single or multiple ligand transfer1,8,12 and commonly occurs for organometallic species.1,4–6 The occurrence of the reaction seems to be driven thermodynamically if a mismatch of coordination in the starting complex is more unfavorable than in the product.
Studies on coordination modes of the organosulfur compounds and their transformations on bimetallic complexes and clusters are informative in gaining insights into the hydrodesulfurization (HDS) process (a catalytic process that is used to remove sulfur from organosulfur compounds in fossil fuel feedstocks). 13 These types of reactions have been employed for the synthesis of chiral complexes. 1 It is known that the catalytic activity of metals involves the mobility of species chemisorbed on metallic surfaces or the transfer of ligands between metal atoms, 14 thus ligand-transferring complexes may be a good choice for catalytically active compounds.
Herein, we report on the ligand transfer of N1-(2-aminoethyl)propane-1,3-diamine (AEPD, Figure 1) between two non-organometallic compounds, a complex of nickel(II), [Ni(AEPD)2](NO3)2, and copper(II) chloride, to produce [Cu2(μ-Cl)2(AEPD)2](NO3)2·2H2O. The complexes were characterized by elemental analysis, Fourier transform infrared spectroscopy (FTIR) and UV–Vis spectroscopy, and X-ray analysis (for
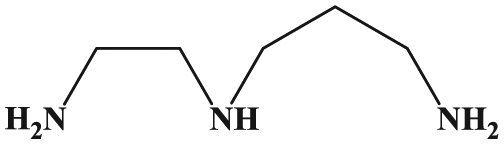
Structure of N1-(2-aminoethyl)propane-1,3-diamine (AEPD).
The biological properties of nickel15–17 and copper18–22 complexes make these complexes a good choice for biologically active compounds. For the study of the biological activities of the AEPD ligand and its complexes, docking calculations were run to investigate the possibility of an interaction between these compounds with 10 biomacromolecule targets:21,23,24 BRAF-kinase, cathepsin B (CatB), DNA-gyrase, histone deacetylase (HDAC7), recombinant human albumin (rHA), ribonucleotide reductases (RNR), thioredoxin reductase (TrxR), thymidylate synthase (TS), topoisomerase II (Top II), and B-DNA. These proteins were selected either due to their reported roles in cancer growth or as transport agents that affect drug pharmacokinetic properties (e.g. rHA). DNA-gyrase was included to study the possibility of anticancer properties and their activity as antimalarial agents. 25 The knowledge gained from docking on B-DNA should be useful for the development of potential probes for DNA structure and new therapeutic reagents for tumors and other diseases. 26
Experimental
Materials and instrumentation
All starting chemicals and solvents (Merck, Aldrich) were used as received without further purification. Infrared spectra (as KBr pellets) in the range 4000–400 cm–1 were recorded with a FTIR 8400-Shimadzu spectrometer. The carbon, hydrogen, and nitrogen contents were determined using a Thermo Finnigan Flash Elemental Analyzer 1112 EA. The melting points were measured with a Barnsted Electrothermal 9200 electrically heated apparatus. The electronic spectrum was recorded in water using a Shimadzu model 2550 UV–Vis spectrophotometer (190–900 nm).
Synthesis of bis[N1-(2-aminoethyl)propane-1,3-diamine]nickel(II) nitrate [Ni(AEPD)2](NO3)2 (1)
AEPD (0.40 g, 3.41 mmol) was added to a stirred solution of Ni(NO3)2·6H2O (0.50 g, 1.72 mmol) in EtOH (20 mL). The reaction mixture was heated under reflux for 5 h, cooled, and filtered. A violet precipitate formed which was collected by filtration. Yield: 0.34 g, 48%; m.p. 143 °C (decomposed). Anal. Calcd for C10H30N8NiO6 (%): C, 28.80; H, 7.25; N, 26.87. Found: C, 29.01; H, 7.29; N, 27.05. IR (KBr disk): 3299 m (νas NH2), 3249 s (νs NH2), 3161 m (ν N−H), 2923 m (νas CH2), 2874 m (νs CH2), 1598 m (δ NH2), 1447 m (δ
as
CH2), 1378 s (ν4
Synthesis of bis[(μ-chloro)N1-(2-aminoethyl)propane-1,3-diamine]nickel(II) nitrate dihydrate [Cu2(μ-Cl)2(AEPD)2](NO3)2·2H2O (2)
Complex
Crystal structure determination
Data were collected at 173 K using a Rigaku FRX/Pilatus P200 diffractometer (Mo radiation). Intensity data were collected using ω scans. All data were corrected for Lorentz and polarization effects. Absorption effects were corrected based on numerical absorption corrections. Structures were solved by direct methods (ShelxS) and refined by full-matrix least-squares against F2 using ShelxL. 27 Diagrams of the molecular structure and unit cell were created using Ortep-III28,29 and Diamond. 30 Crystallographic data and details of the data collection and structure refinement are listed in Table 1, selected bond lengths and angles in Table 2, and hydrogen bond geometries in Table 3.
Crystal data and structure refinement for

Selected bond lengths (Å) and angles (°) for

i: −x, −y+ 1, −z+ 2.
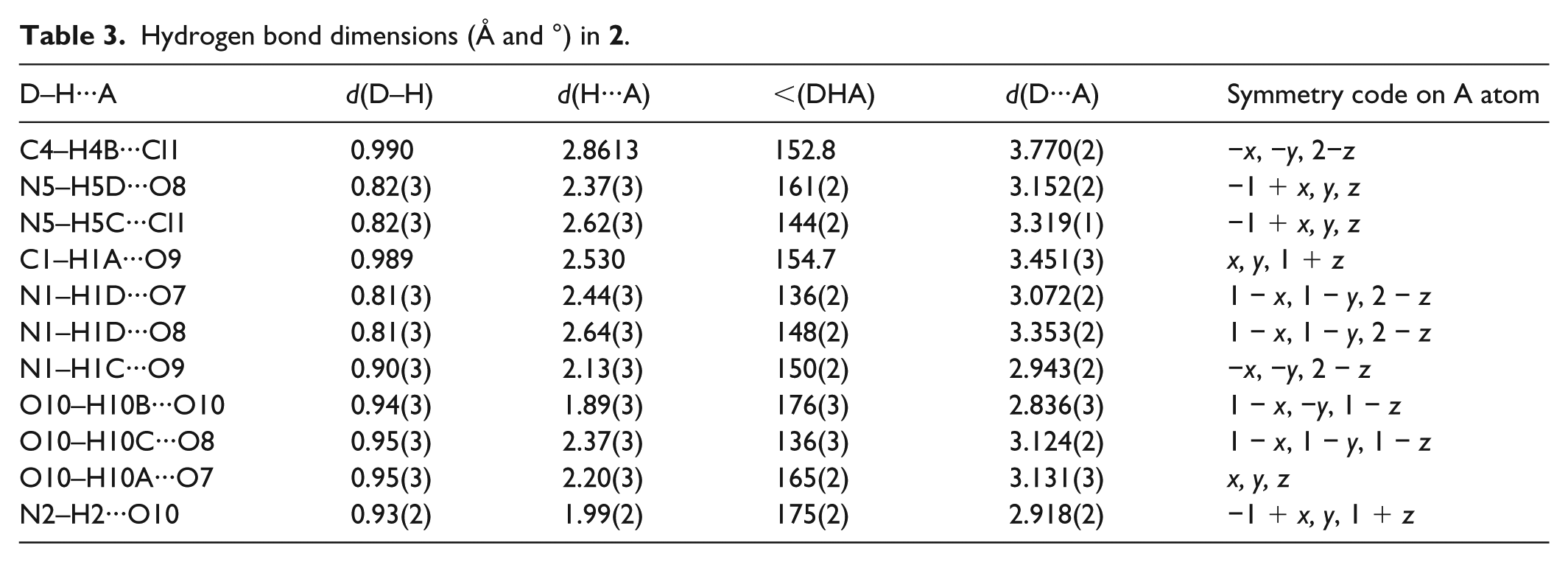
Hydrogen bond dimensions (Å and °) in
Computational details
All structures were optimized with the Gaussian 09 software 31 and calculated for an isolated molecule using DFT 32 at the B3LYP/6-31G(d, p) level of theory for ligand and B3LYP/LanL2DZ for complexes as well as by NBO analysis.
Docking details
The pdb files 4r5y, 3ai8, 5cdn, 3c0z, 2bx8, 1peo, 3qfa, 1njb, and 4gfh for the nine receptors, BRAF-kinase, CatB, DNA-gyrase, histone deacetylase (HDAC7), rHA, RNR, TrxR, TS, and Top II, respectively, used in this research were obtained from the Protein Data Bank (pdb). 33 The full version of Genetic Optimisation for Ligand Docking (GOLD) 5.5 34 was used for the docking. The Hermes visualizer in the GOLD Suite was used to further prepare the compounds and the receptors for docking. The optimized structure of compounds was used for docking studies. The region of interest used for GOLD docking was defined as all the protein residues within 6 Å of the reference ligand “A” that accompanied the downloaded protein. For B-DNA, the region of interest was defined on the DNA backbone within 10 Å of the O4, DT19 and O2, DT19 atoms for the major and minor grooves, respectively. All free water molecules in the structures of the proteins were deleted before docking. Default values of all other parameters were used and the compounds were submitted to 10 genetic algorithm runs using the GOLDScore fitness function.
Results and discussion
The reaction between AEPD and nickel(II) nitrate provides complex
Spectroscopic characterization
In the IR spectra of complexes, three frequencies at 3100−3300 cm−1, which can be assigned to the symmetric and asymmetric stretching vibrations of the N−H bonds, reveal the presence of an amine ligand in these structures. Comparing these peaks with those of the AEPD ligand 35 reveals that these peaks shift by 17−92 cm−1 to lower frequencies after complex formation, supporting N3-donor donation of AEPD toward metal atoms. No significant shift was observed for the δ (NH2).
Four bands in the IR spectra of the complexes in the range of 800−1400 cm–1 (ν4, ν1, ν2, and ν6) can be assigned to vibrations of the nitrate groups.36–38 The free nitrate ion has D3h symmetry and three infrared active vibrations, but this symmetry is lowered to C2v and Cs in metal complexes, to give up to six infrared active vibrations. In
The presence of a water molecule in
Metal–ligand interactions can be studied in the IR spectra below 600 cm−1.
44
In this region, a band at 505 and 450 cm–1 for
Description of the structures
The N1-(2-aminoethyl)propane-1,3-diamine unit is potentially tridentate and can bind to the metal centers in different coordination modes. A structural study of the Cambridge Structural Database (CSD) database reveals that there are three coordination modes for the title unit (Table 4). Among them the “mer-Two Chelates” coordination mode is common (89%) and there is one example of the “(Nb,Nc);Na” mode in which M is cadmium(II). 51
All coordination modes for the N1-(2-aminoethyl)propane-1,3-diamine-based ligands.

Proposed structure of [Ni(AEPD)2](NO3)2 (1)
Based on the spectral and physical properties of the complex
Crystal structure of [Cu2(μ-Cl)2(AEPD)2](NO3)2·2H2O (2)
X-ray analysis of

Ortep diagram of the molecular structure of complex
Searching the CSD database reveals that there are 10 analogues for complex

CSD average (coordinated bond lengths and angles) for analogues of complex
In this structure, the AEPD ligand is coordinated to the copper atom in its common mode (Table 4), N3-donor, by forming one five- and one six-membered non-planar chelate rings. The bond angle of the six-membered chelate ring (93.09(6)°) is larger than the five-membered one (84.01(6)°). A tridentate ligand can coordinate to the metal in facial or meridional forms. In the mer form, there are two angles of 90° and one of 180°, while in the fac form there are three angles of 90°.
42
In complex
In the crystal structure of
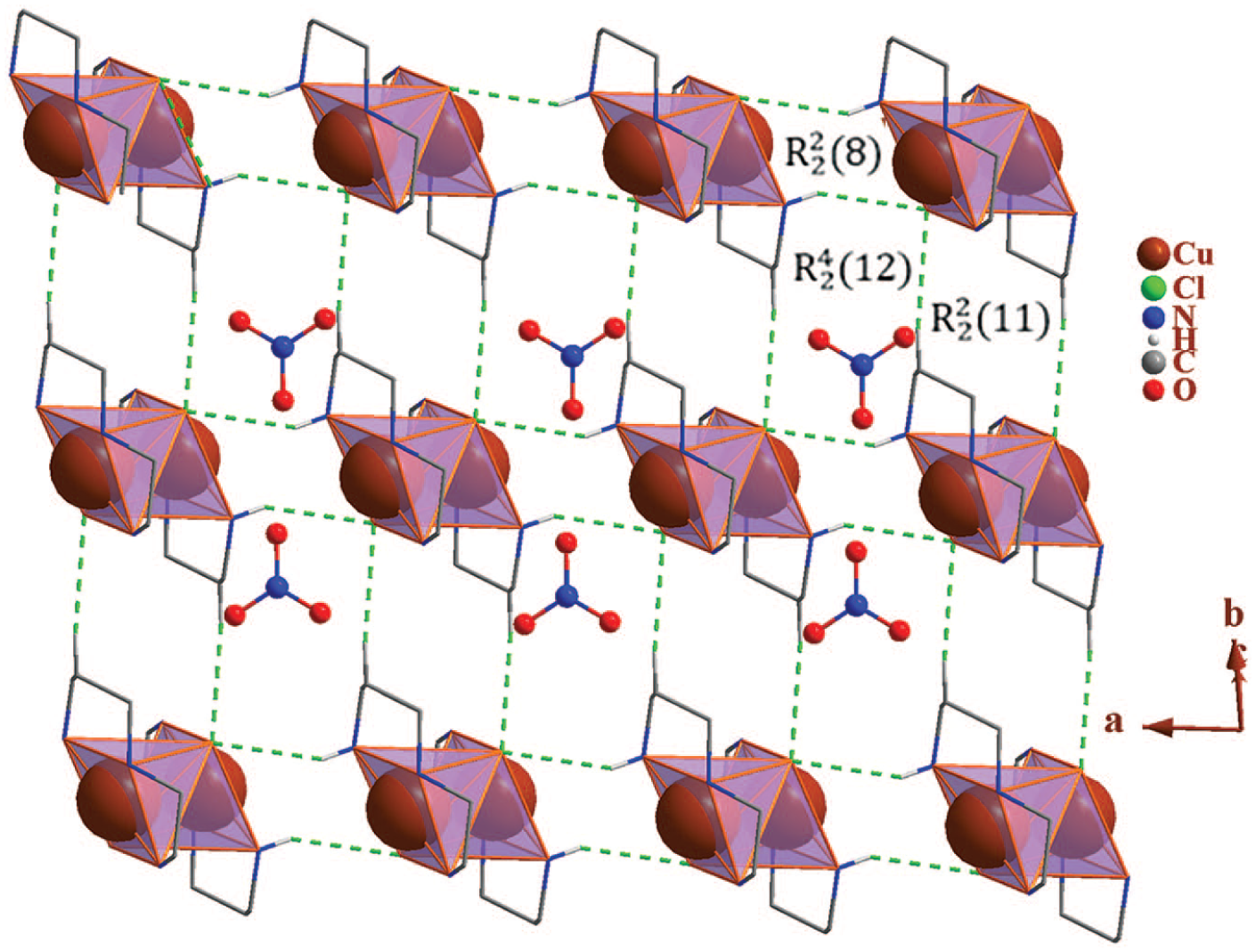
Packing of
Theoretical studies
In order to propose a structure for

Optimized structures of the two complexes
To study the charge distribution pattern of the AEPD ligand before and after complexation, an NBO analysis was performed (Table 5). In the optimized complexes, the calculated charge on the metal atoms (Ni: +0.94, Cu: +0.67) is lower than the formal charge (+2) owing to electron donation of the ligand upon complexation. These calculations reveal that the charges on the carbon atoms in
The NBO analysis results for optimized complexes

AEPD: N1-(2-aminoethyl)propane-1,3-diamine.
The values are the average charge on similar atoms.
In the optimized structure of the ligand, the lowest unoccupied molecular orbital (LUMO) is delocalized on the side amine group, while the highest occupied molecular orbital (HOMO) is on the centered amine unit (Table 6). In
HOMO and LUMO orbitals for complexes

AEPD: N1-(2-aminoethyl)propane-1,3-diamine; HOMO: highest occupied molecular orbital; LUMO: lowest unoccupied molecular orbital.
Docking studies
To predict the biological activities of the AEPD ligand and its complexes, interactions of these compounds with 10 macromolecular receptors were studied using GOLD 34 docking software. The GOLD docking results are reported in terms of the values of fitness, which means that the higher the fitness, the better the docking interaction of the compounds. 25 The results of the docking presented in this work are the best binding results out of 10 favorites predicted by GOLD.
The general features from the GOLD docking prediction (Table 7) show that all the studied structures can be considered as biologically active compounds. Based on the calculated values, the best predicted targets for AEPD,
The calculated fitness values for AEPD and complexes

AEPD: N1-(2-aminoethyl)propane-1,3-diamine; CatB: cathepsin B; HDAC7: histone deacetylase; rHA: recombinant human albumin; RNR: ribonucleotide reductases; TrxR: thioredoxin reductase; TS: thymidylate synthase; Top II: topoisomerase II.

Docking study results showing the interaction between the AEPD and B-DNA (minor groove).

Docking study results showing the interaction between complex
Conclusion
In this work, two new complexes of AEPD, [Ni(AEPD)2](NO3)2 (
Supplemental Material
checkcif_3 – Supplemental material for A novel ligand transfer reaction: Transferring an N3-donor amine ligand from Ni(II) to Cu(II)—structural, spectral, theoretical, and docking studies
Supplemental material, checkcif_3 for A novel ligand transfer reaction: Transferring an N3-donor amine ligand from Ni(II) to Cu(II)—structural, spectral, theoretical, and docking studies by Zahra Mardani, Sima Dorjani, Keyvan Moeini, Majid Darroudi, Cameron Carpenter-Warren, Alexandra MZ Slawin and J Derek Woollins in Journal of Chemical Research
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.