
Abstract
Background
Paclitaxel is an important chemotherapeutic agent for the treatment of breast cancer. Paclitaxel-induced peripheral neuropathy (PIPN) is a major dose-limiting toxicity that can persist into survivorship. While not all survivors develop PIPN, for those who do, it has a substantial negative impact on their functional status and quality of life. No interventions are available to treat PIPN. In our previous studies, we identified that the HIF-1 signaling pathway (H1SP) was perturbed between breast cancer survivors with and without PIPN. Preclinical studies suggest that the H1SP is involved in the development of bortezomib-induced and diabetic peripheral neuropathy, and sciatic nerve injury. The purpose of this study was to identify H1SP genes that have both differential methylation and differential gene expression between breast cancer survivors with and without PIPN.
Methods
A multi-staged integrated analysis was performed. In peripheral blood, methylation was assayed using microarray and gene expression was assayed using RNA-seq. Candidate genes in the H1SP having both differentially methylation and differential expression were identified between survivors who received paclitaxel and did (n = 25) and did not (n = 25) develop PIPN. Then, candidate genes were evaluated for differential methylation and differential expression in public data sets of preclinical models of PIPN and sciatic nerve injury.
Results
Eight candidate genes were identified as both differential methylation and differential expression in survivors. Of the eight homologs identified, one was found to be differential expression in both PIPN and “normal” mice dorsal root ganglia; three were differential methylation in sciatic nerve injury versus sham rats in both pre-frontal cortex and T-cells; and two were differential methylation in sciatic nerve injury versus sham rats in the pre-frontal cortex.
Conclusions
This study is the first to evaluate for methylation in cancer survivors with chronic PIPN. The findings provide evidence that the expression of H1SP genes associated with chronic PIPN in cancer survivors may be regulated by epigenetic mechanisms and suggests genes for validation as potential therapeutic targets.
Keywords
Introduction
Paclitaxel is an important chemotherapeutic agent for the treatment of breast cancer. 1 Peripheral neuropathy is an adverse effect that occurs in 59% to 87% of patients who receive paclitaxel2,3 and can persist into survivorship. 4 Paclitaxel-induced peripheral neuropathy (PIPN) has a substantial negative effect on survivors’ functional status and quality of life.5–11
Not all patients develop this neurotoxicity, which supports the suggestion that molecular mechanisms may be involved in the development of PIPN. Because research with preclinical models of neuropathic pain have not yielded effective treatments, studies that use samples from patients and survivors with chemotherapy-induced peripheral neuropathy (CIPN)12,13 are needed to address this gap. In our previous studies that evaluated gene expression patterns in survivors with and without persistent PIPN,14–16 we identified perturbed pathways associated with mitochondrial dysfunction, 14 neuroinflamation, 15 and changes in cytoskeleton and axon morphology. 16 These findings are consistent with mechanisms identified in preclinical models of neuropathic pain including CIPN.17–23
One of the common pathways across these three mechanisms is the hypoxia-inducible factor 1 (HIF-1) signaling pathway. This pathway, a master regulator of cellular responses to hypoxia, 24 plays an important role in axon regeneration following peripheral nerve injury. 25 HIF-α is a transcription factor (TF) and global regulator of oxygen homeostasis that facilitates oxygen delivery and adaptation to oxygen deprivation.26,27 It mediates processes that protect against axonal degeneration 28 and stimulate axon regeneration. 29 While our recent studies were the first to report on an association between PIPN and the HIF-1 pathway,14–16 findings from preclinical studies suggest that this TF is involved in mechanisms that underlie the development of bortezomib-induced peripheral neuropathy, 30 diabetic peripheral neuropathy, 31 and sciatic nerve injury. 32
Given that our previous studies of PIPN in breast cancer survivors provide evidence for differential gene expression and pathway perturbations14–16 and that epigenetics modifications may be involved in the development of neuropathic pain33,34 as well as involved in the transition from acute to chronic pain,35,36 we sought to evaluate for epigenetic mechanisms 37 that may influence the expression of these products. 38 One potential epigenetic mechanism is DNA methylation, which regulates gene expression by adding or removing methyl groups at the 5′-position of DNA cytosine residues. 39 Methylation of DNA can impact transcription by recruiting or blocking TFs, which results in changes in gene expression. 40 Epigenetic variations can occur in response to environmental factors (e.g., CTX). 38 The identification of genes with both differential methylation (DM) and differential expression patterns associated with PIPN may provide useful information on loci that exhibit changes in function and regulation.
In a recent review, 41 it was noted that opportunities may exist to develop or repurpose existing drugs that target the epigenome 42 to treat a variety of neuropathic pain conditions including PIPN. Therefore, an increased understanding of DM in biologic pathways associated with the occurrence of PIPN may provide new insights into how these pathways are regulated and suggest targets for drug development. Given the role that epigenetics may play in the development and maintenance of neuropathic pain,33,35 the purpose of this study was to evaluate for differentially methylated and differentially expressed genes in the HIF-1 signaling pathway in breast cancer survivors with (n = 25) and without (n = 25) chronic PIPN and evaluate these candidates in pre-existing data sets from animal models of neuropathic pain including PIPN.
Methods
Survivors and settings
The methods for this analysis, which is part of a larger study of CIPN, are described in detail elsewhere. 11 In brief, survivors were recruited from throughout the San Francisco Bay area and met pre-specified inclusion and exclusion criteria. The National Coalition for Cancer Survivorship’s definition of cancer survivor was used in this study (i.e., a person is a cancer survivor from the moment of diagnosis through the balance of life). 43 Of the 1450 survivors who were screened, 754 enrolled, and 623 completed the self-report questionnaires and study visit. Data from a randomly selected sample of breast cancer survivors with (n = 25) or without (n = 25) chronic PIPN were used in this analysis.
Study procedures
Research nurses screened and consented the survivors over the phone; sent and asked them to complete the self-report questionnaires prior to their study visit; and scheduled the in-person assessment. At this assessment, written informed consent was obtained, responses to questionnaires were reviewed for completeness, and objective measurements were obtained. Blood samples were drawn, processed, and stored for subsequent molecular analyses in PAXgene® Blood RNA tubes (Qiagen, Venlo, the Netherlands). This study was approved by the institutional review board of the University of California, San Francisco.
Study measures
Demographic and clinical characteristics
Breast cancer survivors provided information on demographic characteristics and completed the Alcohol Use Disorders Identification Test (AUDIT), 44 Karnofsky Performance Status (KPS) scale,45–47 and Self-Administered Comorbidity Questionnaire (SCQ).48,49
Pain measures
Survivors with PIPN rated their pain intensity using a 0 to 10 numeric rating scale and completed the pain interference scale from the Brief Pain Inventory 50 and the Pain Quality Assessment Scale. 51
Biospecimen processing, quantification of methylation status, and quality control
DNA samples from buffy coats were archived using the PUREGene DNA isolation kit (Invitrogen, Carlsbad, CA) as previously described.52,53 DNA samples were quantitated with a NanoDrop UV spectrophotometer (Thermo Fisher Scientific, Waltham, MA) and normalized to a concentration of 50 ng/μL. The DNA preparation and microarray work were performed at the UC Berkeley Vincent J. Coates Genomics Sequencing Laboratory. DNA was bisulfite converted using the Zymo EZ-96 DNA Methylation Kit (Catalog #D5004) Deep-Well Format (Zymo Research, Irvine, CA) and used as input for the Illumina Infinium HD Methylation Assay (Illumina, San Diego, CA). Processed DNA was dispensed onto the Infinium MethylationEPIC BeadChip and scanned on the Illumina iScan (Illumina). Preliminary analysis and quality control (QC) of the data were performed using GenomeStudio (Illumina). Target success rates were determined. Samples that had <90% of their targets methylated with a detection p-value of ≤0.01 were flagged for review. Sample replicates and Jurkat control replicates were checked to ensure an r2 value of >0.99.
Subsequent data analyses were done using well-established protocols in R (version 3.6.1).54,55 Corrections for Infinium I and II probes, balance correction, background correction, and quantile normalization were performed using the minfi package in R (version 1.30.0).56,57 Probes that contained a single nucleotide polymorphism at a CpG or flanking site and probes that aligned to multiple places on the genome were excluded. 58 Methylation scores were quantified as M-values. 59 Probes were annotated for genes based on mapping to the Genome Reference Consortium Human Build 38 (GRCh38) assembly. 60
Because DNA methylation differs among blood cell types,61–63 cell types were estimated using the estimateCellCounts2() function in the FlowSorted.Blood.EPIC R package (version 1.2.0). 64 Cell type deconvolution was performed using the IDOL L-DMR library for CD8 and CD4 T-cells, natural killer (NK) cells, B cells, monocytes, and neutrophils. Differences between PIPN groups in estimates of cell type composition were evaluated using t-tests and assessed for significance at a p-value of <0.05. Any cell type composition estimates that were significantly associated with PIPN group membership were included as covariates in the final model.
Surrogate variable analysis (SVA, R package version 3.32.1)65,66 was used to identify technical variations that contributed to heterogeneity in the sample (e.g., batch effects) that were not due to the variable of interest (i.e., PIPN), significant demographic (i.e., age, employment status) and clinical (i.e., AUDIT score, body mass index (BMI), KPS score) characteristics. 66 SVA can control for cell type heterogeneity in the methylation analysis.62,63 The “be” method was used to identify surrogate variables.65,66 Two surrogate variables associated with RIN, extraction date, and cell type composition estimates were included in the analysis. 67
Overlapping DM and gene expression
To understand the association of PIPN and biological variation in HIF-1 pathway genes, we performed a multi-staged integrated analysis using complementary layers of molecular data.68,69 In the first stage, DM analysis of probes between cases and non-cases was performed using the limma R package (version 3.40.6). 70 A linear model was fit using the “ls” method that included significant demographic (i.e., age, employment status) and clinical (i.e., AUDIT score, BMI, KPS score) characteristics, significant cell type composition associated with PIPN (i.e., CD4T cells), and surrogate variables as covariates in the models. We estimates that a minimum of 25 replicates in each group would provide at least 80% power to detect a mean difference in M-values of ≥0.09 at a nominal single locus threshold (type I error rate of 0.01). 71
In the second stage, differential gene expression (DGE) was evaluated for each candidate gene using a generalized linear model with edgeR 72 as previously described. 14 Briefly, these DGE analyses were adjusted for demographic (i.e., age, employment status) and clinical (i.e., AUDIT score, BMI, KPS score) characteristics that differed between the PIPN groups, as well as for technical variability (e.g., potential batch effects) using SVA. We estimated 14 that, at a type I error rate of 0.01, 73 we were powered to detect 1.5-fold changes for 83% of genes across the whole transcriptome.
Overlapping gene expression and methylation probe pairs were identified at the gene level using the annotated Human Genome Organisation (HUGO) approved symbols using the HUGO Gene Nomenclature Committee (HGNC) database. 74 By using a systems genomics design integrating data from multiple biological sources, we would have increased power to identify and better interpret the omics-phenotype relationships relative to an analysis that used only a single source of omics data.69,75 For this exploratory study, candidate genes were defined by a significant overlap assessed at a p-value <0.05 for both DM and expression tests. No minimal fold-change was utilized.
Functional analyses of candidate genes
To characterize potential functional roles and potential interactions among candidate genes with differentially methylated probes and DGE beyond the HIF-1 signaling pathway, we evaluated for protein–protein interaction (PPI) network connectivity using the Search Tool for the Retrieval of Interacting Genes (STRING). 76 The functional enrichment of the PPI was evaluated using the Kyoto Encyclopedia of Genes and Genomes (KEGG) 77 and Reactome 78 pathway databases. We assessed for significance of functional enrichment tests using a false discovery rate of 5 under the Benjamini–Hochberg (BH) procedure.79,80 In addition, we evaluated for similar expression patterns of our candidate genes in various cell types in previously identified peripheral neuroimmune interactions 81 from RNA-seq data sets using the “NIPPY—Neuro-Immune Interactions in the Periphery” database (https://rna-seq-browser.herokuapp.com/).
Differential expression of our candidate genes in DRG of mice with and without PIPN
Differential expression of our candidate genes was evaluated using a publicly available data set generated from mouse dorsal root ganglia (DRG) (NCBI GEO GSE113941). Data were collected from nine pools of mice 10 days after treatment with paclitaxel (n = 5) in a dose that was sufficient to induce PIPN (i.e. “CIPN model”) compared to not treated mice (n = 4) (i.e., “normal”). For our study, FASTQ files of the RNA-seq data from these experiments were downloaded from the NCBI Short Read Archive (GSM3124261, GSM3124262, GSM3124263, GSM3124264, GSM3124269, GSM3124270, GSM3124271, GSM3124272, and GSM3124273).
Following our previously described protocols, 14 these sequences were trimmed with Trimmomatic and aligned to the mouse genome assembly (GRCm38.p6) annotated by GENCODE vM23 with the STAR aligner. 82 Gene level counts were generated using the featureCounts tool in the subread R package (version 2.0.0). 83 QC and DGE analyses were performed as previously described. 14 The “be” method was used to identify surrogate variables.65,66 No outlier samples were identified and all samples were included in subsequent analyses. Genes with ≤1 read per million in at least four samples were excluded. Two surrogate variables were identified. Neither surrogate variable was associated with neuropathy and both were used as covariates in the final model. DGE was evaluated between the mouse DRG sample pools who did (n = 5) and did not (n = 4) received paclitaxel. These findings were compared to the differentially methylated and expressed candidate genes identified in our cancer survivors. Homologous mouse gene Ensemble ID was determined by the HUGO Symbol using the HGNC Database. 74 Significant DGE was assessed at a type I error rate of 0.05. No minimal fold-change was evaluated.
DM of our candidate genes in pre-frontal cortex and T-cells of rats with and without SNI
DM of our candidate genes was evaluated in two publicly available data sets generated from rat pre-frontal cortex (PFC) in sham (n = 8) vs. sciatic nerve injury (SNI) 84 (n = 7) animals (GEO GSE70006) and from T-cells in sham (n = 8) vs. SNI (n = 8) animals (GEO GSE70007) that were measured on a customized array (Aglient Technologies, Santa Clara, CA) nine months after the procedure. The PFC and T-cells were harvested from the same animals. Methylation levels were downloaded from GEO as the quantile normalized log2 ratio of the bound (Cy5) and input (Cy3) microarray channel intensities. DM in both experiments was evaluated using limma. Homologous rat genes were identified from their HUGO symbols using the HGNC Database 74 and Rat Genome Database. 85 Significant DM was assessed at a type I error rate of 0.05. No minimal fold-change was utilized.
Results
Differences in demographic, clinical, and pain characteristics
Cancer survivor characteristics were reported previously. 14 In brief, breast cancer survivors with chronic PIPN were significantly older (p = 0.006) and were more likely to be unemployed (p = 0.022) (Supplemental Table 1). In terms of clinical characteristics, survivors with PIPN had a lower AUDIT score (p = 0.012), a higher BMI (p = 0.011), and a lower KPS score (p < 0.001) (Supplemental Table 2). Of note, no between-group differences were found in the number of years since cancer diagnosis, the total dose of paclitaxel received or in the percentage of patients who had a dose reduction or delay due to PIPN. Worst pain severity was 6.3 (±2.1) and duration of PIPN was 3.8 (±3.9) years (Supplemental Table 3).
DM and DGE associated with PIPN in survivors
For the 100 genes that were identified in the KEGG HIF-1 signaling pathway, we mapped 732 probes that had a regulatory feature group classified as “promoter associated” and evaluated for DM. Only CD4+ T-cell composition estimates were associated with PIPN group membership (Supplemental Table 4). Eleven surrogate variables were identified. One was associated with PIPN group membership and four were identified as significantly associated with cell type composition. The final regression model included five significant demographic and clinical characteristics (i.e., age, employment status, AUDIT score, BMI, KPS score) and four surrogate variables. We had methylation and expression data for 81 genes in the HIF-1 signaling pathway that were candidates for evaluation for both DGE and DM. Twelve probes across eight genes were identified as both differentially methylated and expressed between survivors with and without PIPN (Table 1).
Genes in the HIF-1 pathway that were differentially methylated at promoter associated sites and differentially expressed between breast cancer survivors with and without paclitaxel-induced peripheral neuropathy.

Gx: gene expression; HIF: hypoxia-inducible factor; HGNC: HUGO Gene Nomenclature Committee; HUGO: Human Genome Organisation; logFC: log2 fold change; MAPK: mitogen-activated protein kinase; Mt: methylation.
Functional analysis identified seven KEGG and three Reactome pathways that were enriched for the eight genes listed in Table 1 (excluding the HIF-1 signaling pathway itself; Table 2). These eight genes represent a significantly enriched PPI network (p = 0.002) and the network identified evidence of protein–protein interactions between six genes (Figure 1).
Functionally enriched pathways associated from eight genes in the HIF-1 signaling pathway with overlapping differential methylation and differential expression between breast cancer survivors with and without paclitaxel-induced peripheral neuropathy.
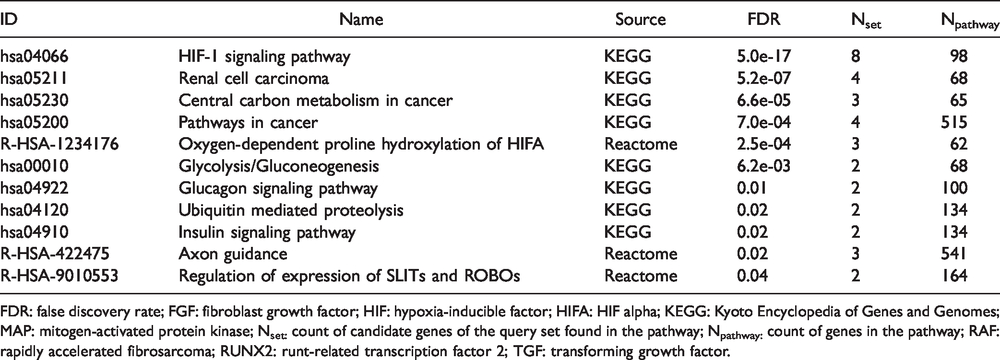
FDR: false discovery rate; FGF: fibroblast growth factor; HIF: hypoxia-inducible factor; HIFA: HIF alpha; KEGG: Kyoto Encyclopedia of Genes and Genomes; MAP: mitogen-activated protein kinase; Nset: count of candidate genes of the query set found in the pathway; Npathway: count of genes in the pathway; RAF: rapidly accelerated fibrosarcoma; RUNX2: runt-related transcription factor 2; TGF: transforming growth factor.

STRING connectivity network analysis identified protein–protein interactions between cullin 2 (CUL2), egl-9 family hypoxia-inducible factor 1 (EGLN1), ring-box 1 (RBX1), lactate dehydrogenase A (LDHA), transferrin receptor (TFRC), and phosphofructokinase, liver type (PFKL). Nodes represent all proteins produced by a single protein coding gene locus. Edges represent specific or meaningful associations. Known or predicted 3D structures are presented within the nodes. Color of the edges connecting the nodes represents the types of evidence supporting the connections: predicted gene neighborhood (green), predicted gene fusions (red), known interactions from experimental evidence (pink), co-expression (black), and text-mining (green).
Differential expression of candidate genes in mouse DRG associated with PIPN
Of the eight candidate genes identified as both differentially methylated and expressed in breast cancer survivors, eight homologs were identified in the mouse DRG data set (Table 3). Of these eight candidates, one gene (i.e., Mknk1) was found to be differentially expressed between PIPN and “normal” pools of mice DRG.
Test for differential expression of candidate genes in the HIF-1 pathway in DRG between pools of mice treated with paclitaxel and normal controls.
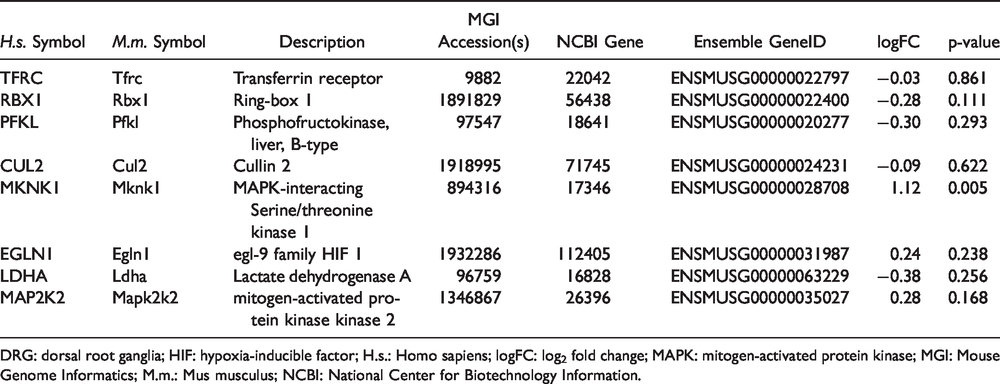
DRG: dorsal root ganglia; HIF: hypoxia-inducible factor; H.s.: Homo sapiens; logFC: log2 fold change; MAPK: mitogen-activated protein kinase; MGI: Mouse Genome Informatics; M.m.: Mus musculus; NCBI: National Center for Biotechnology Information.
DM of candidate genes in rat PFC and T-cells associated with SNI
Of the eight candidate genes with overlapping DM and DGE in breast cancer survivors, seven homologs were identified in the rat PFC and T-cell data set (i.e., Mknk1, Map2k2, Egln1, Rbx1, Pfkl, Cul2, Ldha). For these seven genes, we identified 72 probes (Egln1 n = 14, Map2k2 n = 10, Mknk1 n = 8, Pfkl n = 12, Rbx1 n = 5, Cul2 n = 12, Ldha n = 11) in the promoter regions and evaluated them for DM. Of these seven candidates, three genes (i.e., Mknk1, Ldha, Egln1) were differentially methylated between SNI and sham rats in both PFC and T-cells (Table 4). While no genes were differentially methylated only in T-cells, two genes (i.e., Cul2, Map2k2) were differentially methylated only in the PFC.
Differential methylation of probes in promoter regions of candidate genes in pre-frontal cortex and T-cells of rats with spared nerve injury versus sham.
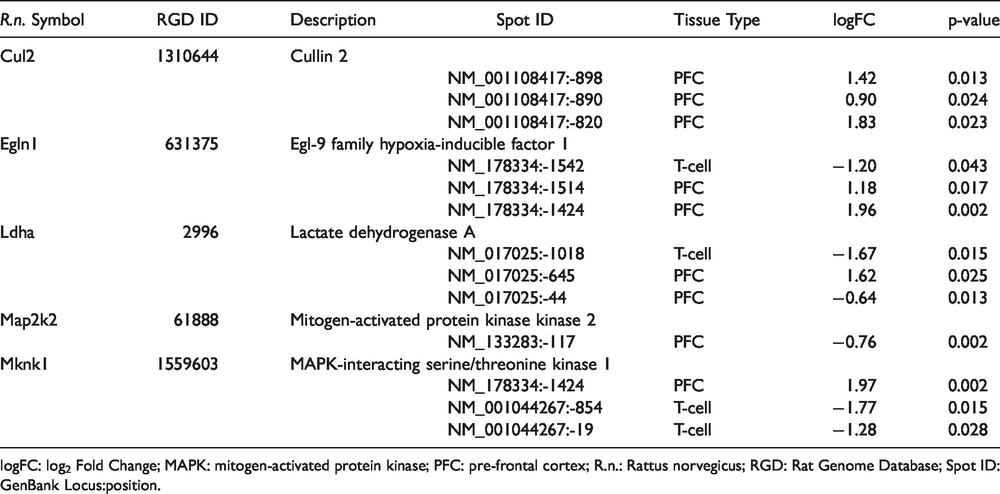
logFC: log2 Fold Change; MAPK: mitogen-activated protein kinase; PFC: pre-frontal cortex; R.n.: Rattus norvegicus; RGD: Rat Genome Database; Spot ID: GenBank Locus:position.
Discussion
This study is the first to use a multi-staged integrated analysis to identify genes in the HIF-1 signaling pathway that were both differentially methylated and differentially expressed in breast cancer survivors with PIPN. In addition, this study is the first to identify a subset of these genes as being differentially methylated or differentially expressed in preclinical models of PIPN 86 or SNI. 84 Pre-clinical animal models are needed to increase our understanding of the fundamental biological mechanisms that underlie neuropathic.87–89 Given that these types of experiments cannot be done in humans, a comparison of our findings in cancer survivors with those from pre-clinical models allows us to gain insights into their translatability. Of note, one of these HIF-1 signaling pathway candidate genes (i.e., MKNK1/Mknk1) was associated with both DGE in mouse DRG and DM in rat PFC and T-cells. The remainder of this discussion focuses on these genes and evaluates their potential role in the mechanisms that underlie PIPN and the implications of these findings with respect to epigenetic regulation of gene expression.
HIF-1 signaling pathway genes associated with DM and DGE
The mitogen-activated protein kinase (MAPK) 1 interacting serine/threonine kinase 1 (MKNK1) gene is involved in the control of the cellular proteome 90 and in inflammatory responses. 41 MAPK interreacting protein kinases (Mnks) are broadly expressed across different tissues types. In terms of neuroinflammation, Mnks play critical roles in cytokine receptor signaling. 41 MKNK1 mediates cytokine production in macrophages through post-transcriptional regulation and can mediate TNF-α translation. 41
In terms of effecting translation, MKNK1 phosphorylates the eukaryotic initiation factor 4E (eIF4E) and is activated downstream of the p38 or MAPK pathways. 41 The regulation of the eIF4E complex is a key signaling pathway that controls the cellular proteome. 91 We found that the MKNK1 gene was differentially methylated and expressed in peripheral blood from breast cancer survivors with PIPN, differentially methylated in PFC and T-cells of rats with SNI, and differentially expressed in DRG of mice with PIPN. Recent work suggests that changes in Mknk1 expression and phosphorylation of elF4E in mice contribute to nociceptor plasticity92–94 and that inhibition or elimination of Mkn1 (Mknk1) attenuates PIPN in a murine model. 93 In addition, inhibition of Mkn1 and Mnk2 reduces spontaneous pain in a SNI murine model. 95 Interestingly, the phosphorylation of elF4E by Mknk1 was identified in a neurological model animal, Aplysia californica, which suggests that this regulatory mechanism for neuroplasticity is highly conserved 96 across Bilateria. 97
In terms of neuroinflammation, Mknk1 is expressed in both DRG and peripheral blood and across peripheral and neuroimmune cell lines (Supplemental Figures 1 and 2) which is consistent with our findings of expression across tissue types. Compared to survivors without PIPN, MKNK1 had lower expression and higher methylation in survivors with PIPN. In contrast to our findings in survivors, Mknk1 had higher expression in mice with PIPN compared to normal mice. One possible explanation for this difference is the timing of the measures. For the mice with PIPN, gene expression was measured 10 days after CTX compared to approximately four years in our cancer survivors (Supplemental Table 2). Similarly, in a recent preclinical study 81 of traumatic nerve injury, findings from macrophage and T-cells dissected from DRG suggests that Mknk1 expression levels decline 10 weeks after sciatic nerve injury (Supplemental Figure 4). Based on the fact that inflammation persisted unresolved for 3.5 months (the duration of the study) in these animals, the authors argued that the categorization of pain as either inflammatory or neuropathic may not be mechanistically appropriate. Given that DNA methylation reprogramming was observed in preclinical models of chronic neuropathic pain following nerve injury, 36 future research should evaluate for changes in the methylation and expression levels of Mknk1 over time.
MAP2K2 is a kinase which phosphorylates and activates MAPK1 and MAPK3. 98 The MAPK1 gene is involved in eukaryotic signal transduction 99 and is associated with the development of neuropathic pain.100,101 We found that MAP2K2/Map2k2 was differentially methylated and differentially expressed in peripheral blood from breast cancer survivors with PIPN and differentially methylated in PFC of rats with SNI. ERK2 (MAPK1) activation by the three MAPK family pathways (i.e., phosphoinositide 3-kinases (PI3Ks), adenosine monophosphate-activated protein kinase (AMPK), mammalian target of rapamycin (mTOR)) promotes inflammatory and neuropathic pain conditions (reviewed in Wood et al. 102 ). Evaluations of the effects of paclitaxel on cancer cell lines found that paclitaxel activated these MAPK family pathways.103–105 Thus, the regulation of MAPK1 was an early therapeutic target for the treatment of neuropathic pain induced by peripheral nerve injury. 106 More recently, inhibition of a number of MAPK family pathways (e.g., p38, MAPK, and JNK) was shown to attenuate neuropathic and inflammatory pain in preclinical models (reviewed in Ji et al. 101 ). While no studies have examined associations between PIPN and changes in gene expression or methylation of MAP2K2, expression of Map2k2 increased in rat L4 and L5 DRGs cells with sciatic nerve lesions as compared with sham controls three days after injury. 107
The transferrin receptor (TFRC) gene encodes for a protein important for cellular iron uptake and homeostatis. 108 TFRC (TFR1) is a key iron metabolism gene that fine tunes cellular iron access, storage, and utilization. 109 Iron overload can lead to increased oxidative stress 110 and may be regulated by HIF-1 TFs. 111 We found that TFRC was differentially methylated and expressed in peripheral blood of breast cancer survivors with PIPN. However, data were not available to test for DM in T-cells or PFC of rats with SNI or DGE in DRG of mice treated with paclitaxel. While no studies have examined associations between PIPN and changes in gene expression or methylation of TFRC, polymorphisms in TFRC decreased the risk for distal neuropathic pain in HIV-infected patients on combination antiretroviral therapy. 112 In addition, iron overload is a risk factor for diabetic neuropathy. 113 In a preclinical central pain model, mRNA expression of TFR1 (an alias for TFRC) was upregulated in activated microglia after spinal cord injury. 114 Finally, our previous finding of perturbations in iron homeostasis pathways (i.e., Ferroptosis) in this sample 14 suggests that future studies should investigate the role of TFRC in the regulation of iron homeostasis in survivors with PIPN.
The phosphofructokinase, liver type (PFKL) gene codes for the liver subtype of an enzyme that catalyzes the phosphorylation of D-fructose 6-phosphate to fructose 1,6-bisphosphate. Therefore, PFKL is a key metabolic gene in glycolysis. 115 We found that PFKL was differentially methylated and expressed in peripheral blood from breast cancer survivors with PIPN. However, it was not DM in T-cells or PFC of rats with SNI or differentially expressed in DRG of mice treated with paclitaxel. No studies were found that evaluated for associations between gene expression or methylation of PFKL and CIPN. However, in a preclinical model of diabetic neuropathy, 116 PFKL was induced in IMS32 Schwann cells following chronic treatment with high levels of glucose (>8 weeks). Given that oncology patients with diabetes are at increased risk for developing CIPN, 117 future studies should evaluate this mechanism in patients with both comorbidities.
The lactate dehydrogenase A (LDHA) gene catalyzes the forward and backward conversion of lactate to pyruvate in glycolysis. LDHA is activated by HIF118,119 and the reduction of LDHA can induce oxidative stress. 120 We found that LDHA was differentially methylated and expressed in peripheral blood of breast cancer survivors with PIPN and differentially methylated in both T-cells and PFC of rats with SNI. Although no studies were found on associations between gene expression or methylation of LDHA and PIPN, LDHA-driven aerobic glycolysis was associated with pain in bortezomib-induced CIPN. 121 Given that we found an enrichment in the glycolysis/gluconeogenesis KEGG pathway for these eight candidate genes, future research should investigate the role of LDHA in oxidative stress and cellular respiration in survivors with PIPN.
The Egl nine homolog 1 (EGLN1) gene produces a protein which acts as a cellular oxygen sensor that catalyzes the formation of 4-hydroxyproline in HIF alpha proteins and hydroxylates HIF1A and HIF2A. Then, hydroxylated HIFs are degraded through a ubiquination complex. The ring box 1 (RBX1) and cullin 2 (CUL2) genes encode for proteins that act as an E3 ubuquitin-protein ligase, which are involved in mediating the ubiquitination and degradation of proteins. We found that EGLN1/Egln1 was differentially methylated and expressed in peripheral blood of breast cancer survivors with PIPN and differentially methylated in both T-cells and PFC of rats with SNI. In addition, RBX1 and CUL2 were differentially methylated and expressed in peripheral blood of breast cancer survivors with PIPN. However, although Cul2 was differentially methylated in PFC of rats with SNI, we did not find DM of Rbx1 in T-cells or PFC of rats with SNI. In addition, we did not find Rbx1 or Cul2 to be DGE in DRG of mice treated with paclitaxel. In a pharmacological network-based analysis of drug-induced peripheral neuropathy (including Paclitaxel), 122 Cul2 was identified as a highly connected significant intermediator between drugs and their pharmacological targets. Although no studies were found on associations between gene expression or methylation of RBX1 and neuropathy, in oxaliplatin-treated embryonic kidney cells, the overexpression of PHD2 (EGLN1) inhibits cold-induced hTRPA1 activation. 123 Future studies should evaluate this mechanism in patients having received taxanes, platinums, or both.
Epigenetic regulation of transcription
Translational control is an essential component in the regulation of gene expression 124 and may be a core mechanism for the development and maintenance of persistent pain. 125 Translational control of molecular processes associated with neuropathy allows for localized translation outside of the cell body in the axons126,127 (i.e., activity-dependent regulation of mRNA translation 128 ) including the regulation of the eIF4E complex by MNKN1. However, the Mnks are effectors of other biological processes (i.e., receptor tyrosine kinase activity, TNF-α mRNA translation, arachidonate release). 90
Alternative splicing is an important step in post-transcriptional regulation of gene expression 129 and the diversity of various transcript isoforms of a gene are associated with environmental perturbations. 130 Alternative splicing is known to occur in the nucleus either during (co-transcriptional splicing) or after transcription (post transcriptional splicing) 131 and DNA methylation can play a role in the regulation of alternative splicing.132,133 Recent work describing variations in splicing across immune cells found that the distribution of isoforms was on/off, which suggests switch-like regulatory control. 134 Although not currently identified in CIPN, in a model of pre-diabetic polyneuropathy, errors in splicing factors were associated with neuronal dysfunction. 135 In addition, alternative splicing appears to play an important role in the development and function of the nervous system, 136 including axon guidance. 137 In terms of PIPN, two isoforms of the MKNK1 gene can occur through alternative splicing (i.e., MNK1a and MNK1b). However, the MKN1b isoform is not activated by either p38 MAPK or MAPK1.138,139 This finding suggests that the regulation of MKN1b occurs through other mechanisms. 41 Given that gene expression can be modulated at multiple levels, from chromatin folding to mRNA translation, future research should continue to evaluate the relative contributions of epigenetic regulation of translation to the development and maintenance of PIPN.
Conclusions
Several limitations warrant consideration. While our sample size was relatively small, we have an extremely well-characterized sample of breast cancer survivors with and without PIPN. While the integration of data from multiple sources we have increased the power to identify omics-phenotype relationships and enabled a more sophisticated interpretation of the findings,69,75 future research with larger sample sizes may improve the resolution of the methylation and gene expression signals. Our findings warrant validation in an independent sample. Of note, no differences were found in the total cumulative dose of paclitaxel that the two groups of survivors received. Given that methylation and gene expression are not independent processes, our findings must be verified in other samples and with other neurotoxic drugs. The utility of peripheral blood as a biomarker or surrogate for neuronal tissue13,140 or as a direct signal (e.g., peripheral neuro-immune interactions 81 ) is still an active area of research. 141 Future research should evaluate for peripheral neuro-immune interactions as well as for the utility of peripheral blood as a surrogate for neuronal tissue.
Our findings suggest that the expression of HIF-1 signaling pathway genes associated with chronic PIPN in cancer survivors may be regulated by epigenetic mechanisms. Future studies need to evaluate for epigenetic changes associated with gene expression and alternative splicing in other pathways associated with PIPN, other CTX drugs, and other forms of neuropathy.
Supplemental Material
sj-zip-1-mpx-10.1177_1744806920936502 - Supplemental material for Differential methylation and expression of genes in the hypoxia-inducible factor 1 signaling pathway are associated with paclitaxel-induced peripheral neuropathy in breast cancer survivors and with preclinical models of chemotherapy-induced neuropathic pain
Supplemental material, sj-zip-1-mpx-10.1177_1744806920936502 for Differential methylation and expression of genes in the hypoxia-inducible factor 1 signaling pathway are associated with paclitaxel-induced peripheral neuropathy in breast cancer survivors and with preclinical models of chemotherapy-induced neuropathic pain by Kord M Kober, Man-Cheung Lee, Adam Olshen Yvette P Conley, Marina Sirota, Michael Keiser, Marilyn J Hammer, Gary Abrams, Mark Schumacher, Jon D Levine and Christine Miaskowski in Molecular Pain
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the National Cancer Institute (NCI, CA151692) and the American Cancer Society (ACS, IRG-97–150-13). Dr. Miaskowski is supported by grants from the ACS and NCI (CA168960). Dr. Olshen is partially supported by the NCI Cancer Center Support Grant to UCSF (CA082103). This project was also supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through UCSF-CTSI Grant Number UL1 TR000004. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. Recruitment was facilitated by Dr. Susan Love Research Foundation’s Army of Women® Program.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.