
Abstract
The role of T cells in chemotherapy-induced peripheral neuropathy (CIPN) is complex and shaped by biological and experimental factors, including sex, hormonal status, genetic background, and cancer model. This complexity has contributed to inconsistent findings among studies, limiting therapeutic progress. In this study, we investigate how T cells contribute to painful paclitaxel (PTX)-induced peripheral neuropathy (PIPN). Adult male T cell-competent (RNU+/−) and T cell-deficient (RNU−/−) rats were subcutaneously inoculated with tumor cells and subsequently treated with intraperitoneal PTX (8 mg/kg total dose). Reflexive (mechanical, heat, cold) and non-reflexive (burrowing, gait) pain behaviors were assessed from baseline through week 6. Immunohistochemistry (CD68, CX3CR1, CD206) and flow cytometry (CD163, CD86, CD11b/c, CD3, CD161a, CD45RA) were used to assess macrophage and lymphocyte populations. T cell-competent, but not -deficient, rats developed and maintained cold hypersensitivity following PTX. T cells also reduced the onset intensity of PTX-induced mechanical hypersensitivity. In T cell-competent rats, PTX reduced T and B cell counts and increased the CD4+/CD8+ T cell ratio across DRG, sciatic nerve, and spleen. PTX shifted macrophage polarization toward the M1 phenotype and reduced the M2/M1 ratio, independent of T cells. However, M2 macrophages (M2γ and M2a) increased specifically in the sciatic nerves of T cell-deficient rats. Additionally, natural killer (NK) cells decreased in PTX-treated, T cell-deficient rats but remained unchanged in T cell-competent rats. These findings highlight the complex role of T cells in PIPN. In PIPN, T cells play a critical role in driving PTX-induced cold hypersensitivity. A decrease in their number worsens pain intensity, possibly by altering the CD4+/CD8+ T cell balance. In contrast, NK cell reductions in T cell-deficient rats may contribute to hypersensitivity in the absence of T cells.
Introduction
Taxanes are among the most effective antineoplastic agents for treating cancers of the breast, lung, pancreas, esophagus, ovary, and cervix. However, chemotherapy-induced peripheral neuropathy (CIPN) is a frequent and debilitating side effect of taxane therapy, with prevalence estimates varying widely depending on timing and assessment criteria. 1 Paclitaxel (PTX), a commonly used taxane, induces neuroinflammation and peripheral nerve injury, contributing to the onset and maintenance of peripheral neuropathy and gait impairment, which are debilitating neurotoxic side effects.2,3 Peripheral immune cells play a crucial role in the development, maintenance, and resolution of pain. Within the peripheral nervous system, immune cells, including T cells, B cells, natural killer (NK) cells, and macrophages, undergo activation, infiltration, and polarization during the transition from acute to chronic pain.4–8
T cells, a key component of adaptive immunity, influence chronic pain through the release of cytokines and endogenous peptides that modulate neuronal sensitivity. 9 Their role in biological systems, particularly in neuroimmune disorders such as CIPN, is complex and shaped by factors including sex, hormonal status, genetic background, and age. This complexity likely underlies the variable findings regarding T cell involvement in neuropathic pain and presents challenges to developing effective immune-based therapies for CIPN. While some studies suggest that T cell depletion facilitates the resolution of inflammatory and neuropathic pain,10,11 others report that T cell deficiency does not affect the onset of mechanical hypersensitivity but instead exacerbates the intensity and duration of mechanical and thermal hypersensitivity.12,13 Recent research has shifted toward understanding the roles of specific T cell subtypes. CD4+ T cells have been implicated in pain resolution,14,15 with regulatory CD4+ T cells inhibiting proinflammatory CD4+ T helper 1 cells as well as other proinflammatory immune cells. 16 CD8+ T cells have also been shown to contribute to pain resolution by promoting IL-10 production via macrophages and other immune cells,17,18 though some studies argue that CD8+ T cell depletion facilitates pain relief. 7
Injury to sensory neurons triggers monocyte and macrophage infiltration into the dorsal root ganglia (DRG) and sciatic nerve.1,8 A preferential shift in macrophage polarization toward the proinflammatory subtype (M1) rather than the anti-inflammatory subtype (M2) contributes to neuropathic pain development.8,19 T cell-mediated cytokine signaling may enhance M2 polarization, thereby promoting pain resolution.17,18 However, many studies rely on genetically modified models that lack both T and B cells, potentially contributing to conflicting findings on T cell function in neuropathic pain.17,20 The role of T cells in chemotherapy-induced peripheral neuropathy (CIPN) pain, as well as their interactions with B cells, NK cells, and macrophages in the DRG, sciatic nerve, and spleen, remains largely unexplored. Moreover, it is unclear how paclitaxel influences immune changes in the peripheral nervous system of T cell-competent versus T cell-deficient animals. Similarly, the influence of tumor presence on CIPN pathology has received limited investigation in preclinical models.
In this study, we hypothesized that T cells facilitate macrophage infiltration into the peripheral nervous system and modulate painful CIPN through interactions with B cells, NK cells, and macrophages. Using homozygous athymic Rowett nude (RNU) rats, which are markedly T cell deficient but retain other immune cell populations,21–24 and heterozygous euthymic rats, which are immunocompetent and serve as the wild-type counterpart, 22 we investigated the contribution of T cells development and maintenance of pain in a tumor-bearing PTX-induced peripheral neuropathy (PIPN) model. Furthermore, we evaluated T cell interactions with B cells, NK cells, and macrophages in the peripheral nervous system (DRG and sciatic nerve) and systemically (spleen) following PTX treatment, while also assessing the contribution of tumors to PIPN pathology.
Methods
Animals
Seven-week-old male homozygous (RNU−/− #316; T cell immunodeficient) and heterozygous (RNU+/− #118; T cell immunocompetent) Rowette nude rats (Crl: NIH-Foxn1rnu), were obtained from Charles River Laboratories (Wilmington, MA, USA). Homozygous rats have an almost complete absence of T cells while retaining other leukocyte populations comparable to normal rats. Heterozygous rats have normal T cell number and function and serve as the wild-type counterpart to the homozygous animals.21,22,24 The rats had an average initial weight of 211 ± 5 g and were housed in groups of three per cage under standard environmental conditions (25 °C ± 2 °C, 60% humidity, 12-h light/dark cycle) with ad libitum access to food and water. All animals were acclimated to their housing conditions and habituated to the experimental rooms for 7 days before the start of the study. Experimental protocols were approved by the Johns Hopkins University Animal Care and Use Committee and complied with the NIH Guide for the Care and Use of Laboratory Animals.
Experimental design
Rats from each strain (homozygous/heterozygous) were randomly assigned to one of four groups (N = 8/strain): no treatment (naïve), tumor only (Tumor), PTX only (PTX), and PTX with tumor (PTX + tumor; Figure 1(a)). Animal housing cages were randomly arranged in the facility. Baseline behavioral assessments – including the Hargreaves test, dry ice and acetone cold allodynia tests, von Frey mechanical hypersensitivity test, and gait analysis – were conducted at week 0 before tumor cell inoculation (Figure 1). To approximate key aspects of CIPN as experienced by patients in clinical settings, animals were implanted with tumors prior to the initiation of PTX treatment. A human non-small cell lung cancer (NSCLC) cell line was selected based on its relevance to clinical populations receiving taxane-based chemotherapy, where CIPN frequently occurs. This model builds upon our previous work demonstrating that NSCLC tumor-bearing rodents are a useful platform for investigating tumor-associated neuroimmune interactions and pain mechanisms in the context of chemotherapy exposure.19,25 NSCLC cells were implanted subcutaneously, and tumors were allowed to develop for 2 weeks before PTX or vehicle treatment.

Experimental protocol. (a) Rats were inoculated with tumor cells at week 0. After 2 weeks, PTX (2 mg/kg, green bars) was administered to rats intraperitoneally every other day for a total dose of 8 mg/kg. Pain behaviors were assessed at baseline (week 0) and weekly post-PTX (weeks 3–6). Sciatic nerves, DRG, and spleen were collected for immunohistochemistry and flow cytometry 3 weeks post-PTX. (b) Reflexive and non-reflexive behavioral tests.
PTX-treated groups received intraperitoneal (i.p.) injections of PTX (2 mg/kg) on days 0, 2, 4, and 6, for a total PTX dose of 8 mg/kg. Naïve and tumor-only groups received vehicle injections. Behavioral assessments were conducted on both right and left hind limbs, and their mean value was used for analysis. All animals were euthanized at week 6 via isoflurane overdose, and sciatic nerves, DRG, and spleen were harvested for further immune cell analysis. Tumors were excised at the end of sixth week and its size was estimated with the use of veiner caliper. Experiments and data analyses were conducted in a blinded manner. Biochemical and immunoassay experiments were repeated twice, incorporating data from all animals.
Sample size
Power analyses were performed using Sigma version 12.0 (Systat Software, Inc., San Jose, CA) to determine the minimum required sample size. Calculations were based on a significance level of α = 0.05, a power of 0.80, and an effect size derived from previous studies.19,25,26
Human NSCLC cell culture
NSCLC cell lines (A549-Luc2; ATCC, Manassas, VA) were cultured in growth medium (F-12K, cat: 21127030, ThermoFisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; cat: F2442, Millipore Sigma, St. Louis, MO, USA) and 1% antibiotic/antimycotic agent (cat: A5955, Millipore Sigma) as described previously. 25 Cells were incubated at 37 °C in a humidified 5% CO₂ environment. After two passages, NSCLC cells were harvested and prepared for inoculation. A total of 2 × 106 viable cells in 200 µL of medium were then injected subcutaneously into the right flank of each rat. 19
PTX administration
PIPN was induced using PTX (cat: 58055525; T7402, Millipore Sigma) following a previously established protocol. 25 A PTX stock solution was prepared by dissolving 25 mg of PTX in a 1:1 mixture of absolute ethanol and Cremophor EL (cat: 238470, Millipore Sigma), which was further diluted with normal saline (1:1:8) to a final concentration of 2 mg/mL. The vehicle control consisted of a mixture of absolute ethanol, Cremophor EL, and normal saline mixed at a 1:1:8 ratio. The vehicle control contained ethanol, Cremophor EL, and saline in the same ratio. To mimic low-dose clinical chemotherapy regimens, rats received i.p. injections of PTX (2 mg/kg) or vehicle every other day.
Mechanical sensitivity test
Mechanical hypersensitivity was assessed using von Frey filaments with the up-and-down method, as previously described.25,27 After acclimatization to the testing environment, filaments (2–15 g) were sequentially applied to the plantar hind paw for 5 s. A positive response (paw withdrawal, flicking, or licking) resulted in the application of the next weaker filament, while a negative response prompted the use of a stronger filament.
Hargreaves test
Rats were habituated to a Plexiglas chamber on a Hargreaves platform for 1 h. A radiant heat source (33% intensity; 55 °C ± 0.5 °C) was directed onto the plantar hind paw, and withdrawal latency was recorded. A cutoff time of 20 s was used to prevent tissue damage. Three trials were conducted and averaged per rat, with a 10-min interval between each trial.
Dry ice-cold hyperalgesia test
Cold hyperalgesia was assessed using granulated dry ice, as previously described. 26 Briefly, dry ice was granulated and placed in a top-cut 12 mL syringe. After 1 h of habituation on a glass platform, a dry ice-filled syringe was pressed against the glass beneath the animal. Withdrawal latency was recorded as the time taken by the animal to swiftly withdraw its paw. A cutoff time of 45 s was implemented to prevent tissue damage. Three trials were conducted and averaged per rat, with a 10-min interval between each trial.
Acetone cold allodynia test
Following 1 h of habituation on a mesh platform, a 50 µL droplet of acetone was applied to the plantar hind paw without directly touching the skin. The reaction frequency (licking or flinching within 120 s) was recorded. Three trials were conducted and averaged per rat, with a 40-min interval between each trial.
Gait assessment
Spatiotemporal gait analysis was performed using the CatWalk XT system (Noldus Information Technology, Netherlands), as previously described. 2 After 1 h of habituation, rats traversed a 1.3-meter illuminated glass walkway, with footprint data captured by a high-speed camera. Each rat completed three compliant runs (four to five paw placements per run), with data averaged across runs. Before the trial, rats were individually trained on the Catwalk walkway platform and performed at least three runs per day for 3 consecutive days.
The sciatic functional index was calculated with the following equation:
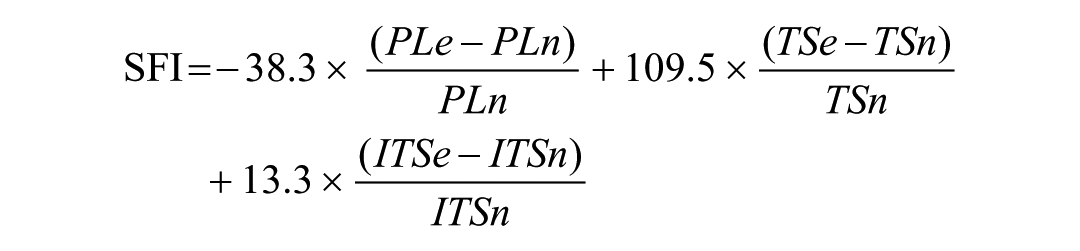
PL = print length; TS = toe spread; ITS = intermediate toe spread; n = normal (baseline value); e = experiment.
Burrowing test
Burrowing activity, a behavioral measure indicative of ongoing spontaneous pain and reduced well-being in rodents, 28 was measured using burrowing tubes (32 × 10 cm) following the method described by Griffiths et al. 29 The tubes were sealed at one end and open at the other, with the entrance elevated 60 mm above the ground to prevent gravel loss. Each tube was placed in the rats’ home cage (600 × 340 × 200 mm), lined with absorbent paper, and filled with 1500 g of pea shingle gravel. Rats underwent a two-phase training protocol: social facilitation (SF) training followed by individual training (IT). Before training, all animals were habituated to the experimental room for 6 h. During SF training, rats were paired (typically with cage mates) and placed in a cage containing the burrowing tube for 2 h/day over two consecutive days. The amount of gravel displaced was recorded, and if a pair burrowed <1200 g, one rat from a more successful pair was swapped in on the second SF day. After training, rats were returned to their home cages.
Twenty-four hours later, IT training was conducted. Each rat was placed individually in the burrowing setup for 1 h/day over 3 consecutive days, and the average amount of gravel burrowed was recorded as the baseline. Rats that burrowed <500 g in total were excluded from further testing. 30 The final test was conducted in the same manner, with burrowing activity measured over 1 h.
Tumor size
Three weeks after PIPN induction (week 6), tumor weight and volume were assessed using a digital scale and caliper measurements, respectively. Animals were deeply anesthetized, and tumors were excised. Tumor length, width, and height were measured using a Vernier caliper, and volume was calculated using a widely accepted formula 31 :

Flow cytometry
Flow cytometry was used to assess leukocytes in the DRG (L4-L6), sciatic nerve (distal portion), and spleen. After being deeply anesthetized with isoflurane, rats underwent transcardiac perfusion with phosphate-buffered saline (PBS) to remove systemic blood.
DRG and sciatic nerve samples from two rats per group were pooled to obtain sufficient immune cell counts. Samples were diced into small pieces with scissors and digested in 1 mL of digestion media (HBSS, 10 µM HEPES, 5 mg/mL BSA, 1.6 mg/mL collagenase IV, 100 µg/mL DNase) at 37 °C with gentle agitation for 30 min. After 20 min of incubation, the mixture was triturated three times with a 1000 µL pipette in digestion media. The digested samples were then passed through a 70 µm cell strainer. Digestion was halted by adding 2 mL of fluorescence-activated cell sorting (FACS) buffer (PBS, 10% heat-inactivated FBS, 1 mM EDTA). The cell suspension was centrifuged at 300g for 10 min at 4 °C, then resuspended in 1 mL of FACS buffer and triturated.
After the spleen was diced into small pieces, the tissue was similarly digested. Gentle pressure was applied with a 1 mL syringe plunger to disaggregate the spleen, which was then passed through a 70 µm cell strainer. Digestion was stopped by adding 9 mL of FACS buffer to wash the spleen cells through the strainer. The resulting aliquot was centrifuged at 500g for 15 min at 4 °C. Red blood cells in the cell pellets were lysed by incubation in 2 mL of ammonium-chloride-potassium lysing buffer (cat: 118-156-101, Quality Biological, Gaithersburg, MD, USA) for 3 min at room temperature. The reaction was stopped with 3 mL of FACS buffer, and the suspension was centrifuged. The pellet was then resuspended in FACS buffer and passed through a 40 µm cell strainer.
For cell staining, single cell suspensions from each tissue were washed twice with PBS and incubated with live/dead fixable aqua dead cell stain (cat: 34957, ThermoFisher Scientific) for 30 min at room temperature. Cells were centrifuged, resuspended in FACS buffer, and treated with anti-rat CD32 to block Fc receptor binding prior to staining. We stained 100 µL of cells by incubating them for 30 min with combinations of the following mouse antibodies: CD45 (BD Bioscience, catalog: BD567734, clone: OX-1), CD11b/c (BD Bioscience, catalog: BD746165, clone: OX-42), CD163 (Abcam, catalog: MCA342B; clone: ED2, secondary: BV711 streptavidin; BD Bioscience, catalog: 563262), CD86 (BD Bioscience, catalog: BD743211, clone: 24F), CD4 (BD Bioscience, catalog: BD740723, clone: OX-35), CD8a (BD Bioscience, catalog: BD740041, clone: OX-8), and T/B/NK cell cocktail (BD Bioscience, catalog: BD558495, CD3: clone IF4, CD45RA: clone OX-33, CD161a: clone 10/78). Cells were stained in two sets. Set 1 included T cell subtypes: CD45, T/B/NK cell cocktail, CD4, and CD8a; set 2 included macrophages and lymphocytes: CD45, T/B/NK cell cocktail, CD11b/c, CD86, and CD163. Cells were washed, resuspended in FACS buffer, and analyzed with a BD LSR II flow cytometer at the Johns Hopkins University Bloomberg Flow Cytometry and Immunology Core Facility. Data from all cytometric evaluations were further analyzed with FlowJo 10.10.0. Before data analysis, flow data quality was assessed with flowAI. 32
Immunohistochemistry
After being deeply anesthetized with isoflurane, each rat underwent transcardiac perfusion with PBS to remove systemic blood followed by freshly prepared 4% paraformaldehyde. The L4-L6 DRG and distal sciatic nerves were quickly excised, fixed in 4% paraformaldehyde for 24 h, and subsequently transferred to 30% sucrose at 4 °C for 48–72 h. Tissues were embedded in paraffin wax, sectioned at 10 µm thickness, and mounted on glass slides. After the tissue sections were dewaxed, cleared, and rehydrated, antigen retrieval was performed by heating the sections at 90 °C in citric buffer solution (pH 6.0). Sections were then blocked and incubated with primary antibodies at 4 °C overnight, washed, incubated with secondary antibodies at room temperature for 2 h, and then washed again. Sections were imaged with a confocal microscope (Leica SP8), and micrographs were captured with Leica Application Suite X software version 3.5.7 under a 20× objective lens. Positive cells were counted with ImageJ software (version 1.53t; National Institutes of Health, Bethesda, MD, USA).
The primary antibodies used were mouse anti-CD68 (1:100; cat: MCA341R; Bio-Rad, Hercules, CA, USA), rabbit anti-CD206 (1:500; cat: AB64693; Abcam, Cambridge, UK), and rabbit anti-fractalkine receptor (CX3CR1; 1:250; cat: 13885-1-AP; Proteintech, Rosemont, IL, USA). Secondary antibodies included Alexa Fluor 488 (1:1000; cat: A11001; Invitrogen, Waltham, MA USA), Alexa Fluor 568 (1:1000; cat: A11036; Invitrogen, Waltham, MA, USA), and DAPI (1:1000; cat AS-83210; Invitrogen, Waltham, MA, USA).
Statistical analysis
Data were analyzed with GraphPad Prism 10.4.0 (GraphPad Software, La Jolla, CA). All data are presented as mean + standard error of the mean. Mechanical hypersensitivity, heat hypersensitivity, cold hyperalgesia, cold allodynia, burrowing activity, and gait data were analyzed using two-way ANOVA followed by Tukey’s post hoc test. Immunohistochemistry and flow cytometry data were analyzed using one-way ANOVA followed by the least significant difference post hoc test. A significance threshold of p < 0.05 was considered statistically significant.
Results
PTX reduces subcutaneous tumor size
To investigate PIPN in a tumor-bearing model, we implanted NSCLC cells subcutaneously into the right flank of both T cell-deficient and T cell-competent rats prior to PTX treatment. Consistent with their impaired immune status, tumor growth was observed only in T cell-deficient rats, with no tumors developing in T cell-competent animals throughout the study. PTX efficacy was confirmed in T cell-deficient rats, as treatment significantly reduced both tumor volume and weight at week 6 compared with untreated tumor-bearing controls (Supplementary Figure 1).
T cell-competent rats exhibit reduced early mechanical hypersensitivity and spontaneous pain following PTX treatment
PTX treatment, regardless of tumor presence, reduced mechanical withdrawal thresholds (von Frey) and burrowing activity in both T cell-competent and T cell-deficient rats (Figure 2(ai–ii) and (bi–ii)). However, at the onset of PIPN (week 3), T cell-competent rats showed significantly less mechanical hypersensitivity than T cell-deficient rats (Figure 2(aiii)). By weeks 5 and 6, thresholds were comparably reduced in both groups, indicating that T cells primarily influence early PIPN but not the maintenance phase. To investigate the effect of tumors, we compared tumor-treated and untreated groups within both T cell-deficient and -competent rats. In T cell-deficient rats, where tumors developed, tumor presence did not significantly affect mechanical hypersensitivity regardless of PTX administration (Figure 2(ai)). Similarly, although tumors did not grow in T cell-competent rats, their inoculation did not shift the level of mechanical hypersensitivity regardless of PTX administration (Figure 2(aii)).

T cells delay the onset of PTX-induced mechanical hypersensitivity and are required for cold hyperalgesia and allodynia development. (a) PTX induces mechanical hypersensitivity (von Frey test) in T cell-deficient and -competent rats irrespective of tumor presence. PTX-induced mechanical hypersensitivity was delayed in T cell-competent rats. (b) PTX and tumor decreased burrowing activity in both T cell-competent and -deficient rats. However, PTX-treated T cell-deficient rats exhibited a more pronounced decrease in burrowing activity compared to T cell-competent rats. (c) PTX induced cold hyperalgesia in T cell-competent, but not -deficient, rats. (d) PTX induced cold allodynia in T cell-competent, but not -deficient rats. PTX was administered during week 2 (green bars). Data represent mean + SEM. (i, ii) Two-way ANOVA and Tukey’s post hoc test; (iii) multiple unpaired t-tests with Welch’s correction.
Burrowing activity decreased in both groups following PTX, but the reduction was more pronounced in T cell-deficient rats at week 6 (Figure 2(biii)), suggesting that T cells also reduce the intensity of spontaneous pain in PIPN. Tumor inoculation reduced burrowing activity regardless of PTX treatment in both strains (Figure 2(bi–ii)). In T cell-deficient rats, PTX treatment, either alone or combined with tumor presence, impaired burrowing more than tumor alone (Figure 2(bi)). In T cell-competent rats, PTX treatment, either alone or with tumor inoculation, worsened burrowing but to a lesser degree than tumor inoculation alone (Figure 2(bii)).
Collectively, these findings indicate that T cells lessen the early development of mechanical hypersensitivity in PIPN but have limited influence on its later maintenance, while also lowering the intensity of ongoing spontaneous pain during the maintenance phase.
T cells are required for PTX-induced cold hypersensitivity but not heat hypersensitivity
Cold hypersensitivity was assessed using the dry ice test (cold hyperalgesia) and acetone spray test (cold allodynia), while the Hargreaves test evaluated heat hypersensitivity. PTX-treated, T cell-deficient rats displayed no changes in hind paw withdrawal thresholds to dry ice or flicking frequency to acetone compared to naïve rats (Figure 2(ci) and (di)). In contrast, PTX-treated T cell-competent rats exhibited cold hypersensitivity, with reduced paw withdrawal thresholds and increased flicking frequency (Figure 2(cii) and (dii)). Cold hypersensitivity in PTX + tumor-treated T cell-competent and -deficient rats was comparable to that observed in PTX-treated T cell-competent and -deficient rats (Figure 2(ciii) and (diii)). Heat hypersensitivity developed similarly in both PTX treated T cell-competent and -deficient rats (Supplementary Figure 2). These findings indicate that T cells are necessary for the development of cold hypersensitivity after PTX treatment, but not for heat hypersensitivity, regardless of tumor status.
T cells influence the development of spatial but not temporal gait impairment
Spontaneous pain-related gait abnormalities were assessed using the CatWalk system, a method for evaluating stimulus-independent pain via spatiotemporal gait properties.2,33 All PTX-treated rats, regardless of tumor status, exhibited gait impairments (Figure 3). T cell-competent rats showed increased single stance duration and swing rate, indices of temporal gait, beginning at week 3 (Figure 3(c) and (d)). In contrast, T cell-deficient rats showed similar changes only at weeks 5 and 6, and to a lesser extent (Figure 3(a) and (b)). However, single-stance duration and swing rate were similar when comparing PTX-treated T cell-competent and -deficient rats (Figure 3(e)). Tumor inoculation minimally affected temporal gait, as swing duration remained similar between T cell-competent and -deficient rats, while single-stance time increased only at week 6 in T cell-competent rats (Figure 3(f)). Taken together, these findings suggest that T cells have minimal impact on the development of temporal gait impairment in PIPN.

T cells delay the onset of spatial but not temporal gait impairment. (a) PTX increased single stance duration (ground contact duration for a single hind paw), swing duration, stride length, and sciatic functional index in T cell-deficient rats. (b) Gait representation of T cell-deficient rats at week 6, showing spatial gait properties (top panels, footprint view) and temporal gait properties (bottom panels, timing view). (c) Similarly, in T cell-competent rats, PTX induced temporal (single stance, swing) and spatial (stride length, sciatic functional index) gait impairment. (d) Gait representation of T cell-competent rats at week 6, depicting the footprint view (top panels) and timing view (bottom panels). (e) T cells delay the onset of spatial (stride length, sciatic functional index) but not temporal (single stance, swing duration) gait impairment in PTX-treated rats. PTX was administered during week 2 (green bars). Data represent mean + SEM. (a–d) Two-way ANOVA and Tukey’s post hoc test; (e) multiple unpaired t-tests with Welch’s correction.
Spatial gait parameters, stride length and sciatic functional index, were similarly impaired in PTX treated, T cell-deficient and -competent rats at weeks 5 and 6 (Figure 3(a)–(d)). However, T cells appear to delay early deficits, as sciatic functional index was worse at the onset of PIPN (weeks 3 and 4) in PTX-treated T cell-deficient rats compared with T cell-competent rats (Figure 3(e)). In addition, stride length was increased in PTX-treated, T cell-deficient rats relative to T cell-competent rats at week 4 (Figure 3(e)). Tumor inoculation minimally affected spatial gait, as stride length and SFI remained similar between PTX + tumor-treated and PTX-only T cell-competent and -deficient rats (Figure 3). These findings suggest that T cells modulate spatial gait deficits by delaying their onset in PIPN.
PIPN decreases CD8+ T cell frequency more than CD4+ T cell frequency in the peripheral nervous system and systemically
In T cell-competent rats, PTX treatment reduced the frequency of total T cells (live CD45+CD3+) in the DRG and sciatic nerve of regardless of tumor presence, but not in the spleen (Figure 4(a)). Furthermore, PTX treatment markedly increased the CD4+/CD8+ T cell ratio, driven by greater depletion of CD8+ T cells compared with CD4+ T cells in the DRG, sciatic nerve, and spleen of T cell-competent rats, irrespective of tumor presence (Figure 4(b)).

PTX decreases total T cell frequency and selectively alters CD4+ and CD8+ T cell populations in the DRG, sciatic nerve, and spleen. (a) Representative flow cytometry plots of total T cells (live CD45+CD3+) in the DRG and spleen. PTX reduced total T cell frequency in the DRG and sciatic nerve but had no effect in the spleen. Results represent T cell frequencies relative to the total live CD45+ cell population within each tissue. (b) Representative flow cytometry plots of CD4+ and CD8+ T cells in the DRG and spleen. PTX increased the frequency of CD4+ T cells (live CD45+CD3+CD4+) and decreased the frequency of CD8+ T cells (live CD45+CD3+CD8+) in the DRG, sciatic nerve, and spleen. As a result, PTX increased the CD4+/CD8+ T cell ratio in the DRG, sciatic nerve, and spleen. Results represent CD4+/CD8+ T cell ratios derived from the total live CD45+CD3+ cell population within each tissue. (c) Representative flow cytometry plots comparing total T cells (live CD45+CD3+) between heterozygous (T cell-competent) and homozygous (T cell-deficient) naïve rats in the DRG, sciatic nerve, and spleen. Data represent mean + SEM. One-way ANOVA and least significant difference post hoc test.
We verified T cell deficiency in DRG, sciatic nerve, and spleen by comparing T cell-competent and T cell-deficient rats. T cell-deficient rats had a pronounced reduction in T cell frequencies across all tissues. In contrast, T cell-competent rats exhibited higher T cell frequencies in all tissues (Figure 4(c)). This distinction is critical for interpreting the observed reduction in total T cells and the concurrent increase in the CD4+/CD8+ T cell ratios across the DRG, sciatic nerve, and spleen.
T cells support NK cell maintenance but do not prevent PTX-induced B cell reduction
PTX treatment reduced B cell (live CD45+CD45RA+) frequency in the DRG of both T cell-competent and T cell-deficient rats, regardless of tumor presence (Figure 5(a) and (b)). However, in the sciatic nerve and spleen, PTX reduced B cell frequency only in T cell-competent rats, while T cell-deficient rats maintained their B cell levels, independent of tumor presence.
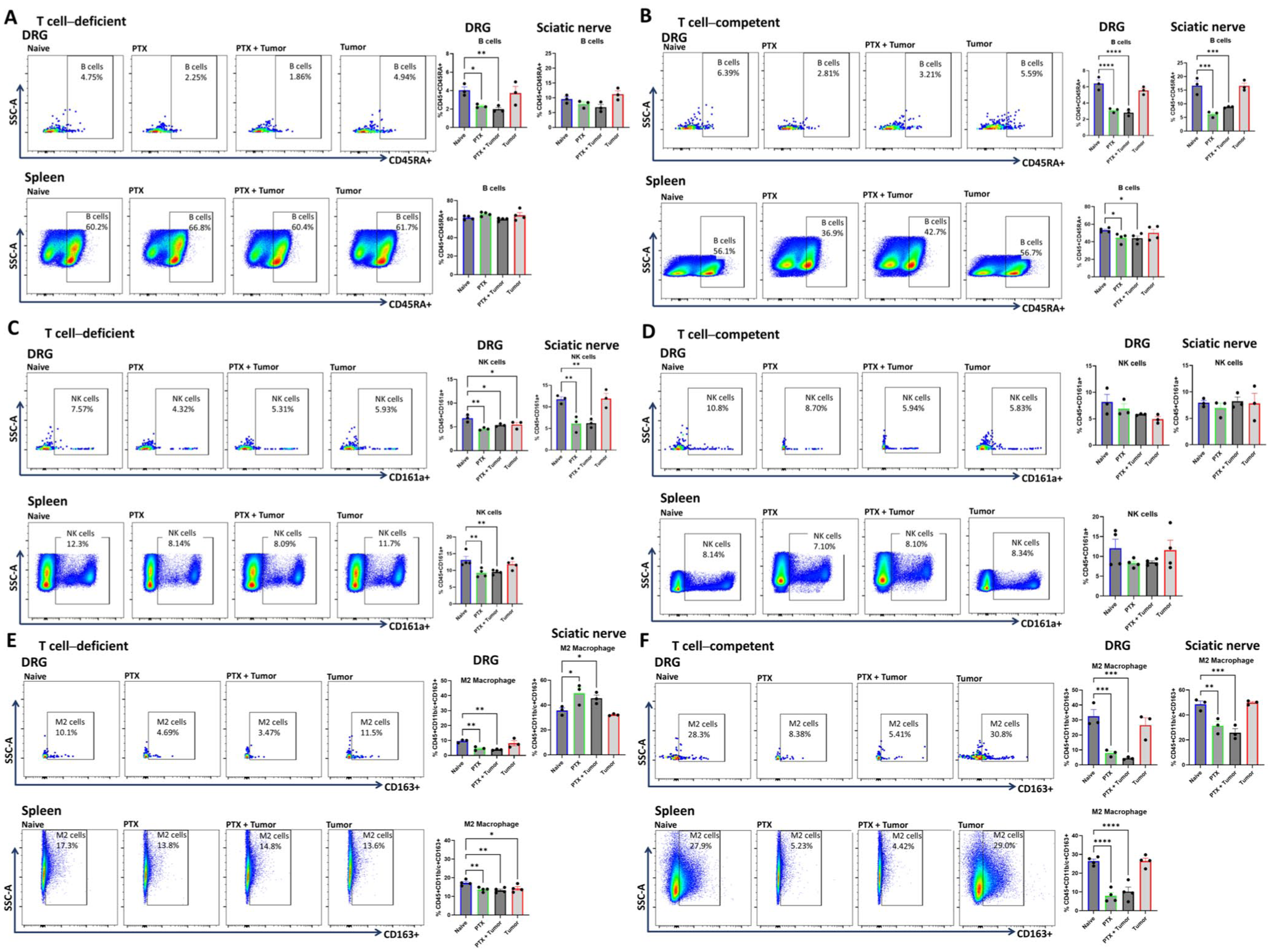
T cells are crucial for maintaining NK cells but do not prevent PTX-induced reductions in B cell and M2 macrophage frequency. (a) Representative flow cytometry plots of B cells (live CD45+CD45RA+) in the DRG and spleen of T cell-deficient rats. PTX decreased B cell frequency in the DRG but not in the sciatic nerve or spleen. (b) Representative flow cytometry plots of B cells in the DRG and spleen of T cell-competent rats. PTX decreased B cell frequency in all three tissues. (c) Representative flow cytometry plots of NK cells (live CD45+CD161a+) in the DRG and spleen of T cell-deficient rats. PTX decreased NK cell frequency in in all three tissues. (d) Representative flow cytometry plots of NK cells in the DRG and spleen of T cell-competent rats. PTX did not alter NK cell frequency in any of the tissues. (e) Representative flow cytometry plots of M2 macrophages (live CD45+CD11b/c+/CD163+) in the DRG and spleen of T cell-deficient rats. PTX decreased M2 macrophage frequency in the DRG and spleen but increased it in the sciatic nerve. (f) Representative flow cytometry plots of M2 macrophages in the DRG and spleen of T cell-competent rats. PTX decreased M2 macrophage frequency in all three tissues. Results represent B and NK cells frequencies relative to the total live CD45+ cell population, and M2 macrophage frequencies relative to the total live CD45+CD11b/c+ cell population within each tissue. Data represent mean + SEM. One-way ANOVA and least significant difference post hoc test.
NK cell (CD45+CD161a+) frequency was reduced in the DRG, sciatic nerve, and spleen of PTX-treated, T cell-deficient rats with and without tumor (Figure 5(c)). In the DRG, PTX treatment and tumor presence independently contributed to decreased NK cell frequency in T cell-deficient rats. In contrast, PTX did not alter NK cell frequency in T cell-competent rats, regardless of tumor inoculation (Figure 5(d)).
These findings suggest that T cells are critical for the maintenance of NK cells after PTX treatment, whereas they do not prevent PTX-induced B cell reductions. The loss of NK cells in T cell-deficient rats may contribute to their altered pain behaviors compared to T cell-competent rats.
PTX treatment reduces the M2/M1 macrophage ratio independently of T cells
In T cell-deficient rats, PTX reduced M2-like macrophages (CD45+CD11b/c+CD163+) in the DRG and spleen but increased them in the sciatic nerve, irrespective of tumor presence (Figure 5(e)). Simultaneously, PTX increased M1-like macrophage (CD45+CD11b/c+CD86+) frequency in the DRG, sciatic nerve, and spleen, independent of tumor status (Supplementary Figure 4(A)). As a result, the overall M2/M1 macrophage ratio decreased across these tissues in PTX-treated, T cell-deficient rats.
In T cell-competent rats, PTX similarly reduced M2-like macrophages in the DRG, sciatic nerve, and spleen (Figure 5(f)), but decreased M1-like macrophages only in the DRG and sciatic nerve (Supplementary Figure 4(B)). Notably, tumor-bearing rats not treated with PTX also exhibited a modest decline in M2 macrophage frequency. As observed in T cell-deficient rats, PTX-treated, T cell-competent rats showed a reduced M2/M1 ratio in the DRG and spleen.
These findings suggest that PTX induces a proinflammatory shift by decreasing the M2/M1 macrophage ratio in the DRG and spleen, regardless of T cell presence, while its effects on M2 macrophages in the sciatic nerve is T cell-dependent, indicating a potential role for T cells in modulating local macrophage polarization in PIPN.
PTX induces distinct changes in M2 macrophage subtypes in a tissue-specific, T cell-dependent manner
We further characterized macrophage subtypes using immunohistochemistry by distinguishing proinflammatory M1-like macrophages (CD68+CD206−) from anti-inflammatory M2-like macrophages, which highly express CD206.17,34,35 M2-like macrophages were subclassified into M2a (CD68−CD206+) and M2γ (CD68+CD206+) phenotypes. PTX treatment reduced both M2a and M2γ macrophages in the DRG of T cell-competent and T cell-deficient rats, independent of tumor presence (Figure 6). In the sciatic nerve, however, PTX increased M2a and M2γ macrophages in T cell-deficient rats but decreased M2γ macrophages in T cell-competent rats (Supplementary Figure 5).

T cells do not influence PTX-induced reductions in M2/M1 ratios but are essential for maintaining CX3CR1 expression in the DRG following PIPN induction. (a) Representative confocal images of CD68+CD206− (M1), CD206+ (M2a), and CD68+CD206+ (M2γ) macrophages in the DRG of T cell-deficient rats. (b) PTX and tumor increased M1 but reduced M2a macrophages in the DRG of T cell-deficient rats. PTX decreased M2γ macrophages and CX3CR1 expression, whereas Tumor increased M2γ macrophages. (c) Representative confocal images of M1, M2a, and M2γ macrophages in the DRG of T cell-competent rats. (d) PTX increased M1 and decreased M2a macrophages in the DRG of T cell-competent rats. PTX, but not PTX + tumor, decreased M2γ macrophages and did not influence CX3CR1 expression. One-way ANOVA and Fisher’s least significant difference post hoc test. Data represent mean + SEM.
To assess macrophage trafficking, we examined CX3CR1 expression. 36 In T cell-deficient rats, PTX reduced the frequency of CX3CR1-expressing cells in the DRG but increased them in the sciatic nerve, regardless of tumor presence (Figure 6(a) and (b) and Supplementary Figure 5(A) and (B)). In contrast, T cell-competent rats showed no change in CX3CR1 expression in the DRG but exhibited an increase in CX3CR1+ cells in the sciatic nerve following PTX treatment (Figure 6(c) and (d) and Supplementary Figure 5(C) and (D)). Across all conditions, T cell-competent rats generally exhibited a higher frequency of CX3CR1+ infiltrating macrophages in the sciatic nerve compared to T cell-deficient rats, as well as a higher frequency of CX3CR1+ infiltrating M1 macrophages in both the DRG and sciatic nerve (Figure 7).

T cells regulate macrophage infiltration in the peripheral nervous system. (a) Immunohistochemistry analysis of macrophage subtypes in the DRG of T cell-deficient and T cell-competent rats. Macrophage subtypes were defined by CX3CR1, CD68, and CD206 expression as follows: infiltrating M1 macrophages (CX3CR1+CD68+CD206−), infiltrating M2a macrophages (CX3CR1+CD206+CD68−), and infiltrating M2γ macrophages (CX3CR1+CD68+CD206+). PTX reduced M2a macrophage infiltration in T cell-deficient and -competent rats. M2γ macrophage infiltration decreased following PTX treatment alone in T cell-competent rats, and after PTX + tumor treatment in T cell-deficient rats. Infiltrating M1 macrophages were nearly absent in T cell-deficient rats under all conditions but increased after PTX treatment in T cell-deficient and -competent rats. (b) Immunohistochemistry analysis of macrophage subtypes in the sciatic nerve. PTX increased M2a macrophage infiltration in T cell-deficient and -competent rats but decreased M2γ macrophages only in T cell-competent rats. Infiltrating M1 macrophages were nearly absent in T cell-deficient rats but increased following PTX treatment in T cell-deficient and -competent rats. Data are presented as mean + SEM. Statistical analysis was performed using one-way ANOVA followed by Fisher’s LSD post hoc test.
Infiltrating M2a macrophages (CX3CR1+CD206+CD68−) were reduced in the DRG of both T cell-competent and T cell-deficient rats following PTX treatment but increased in the sciatic nerve of both groups (Figure 7). In the DRG, infiltrating M2γ macrophages (CX3CR1+CD206+CD68+) decreased following PTX treatment alone in T cell-competent rats, whereas in T cell-deficient rats, a reduction was observed only with combined PTX and tumor treatment. In the sciatic nerve, M2γ macrophages decreased following PTX treatment in T cell-competent rats but were unchanged in T cell-deficient rats. Infiltrating M1 macrophages (CX3CR1+CD206−CD68+) increased in the DRG of both groups following PTX treatment but increased in the sciatic nerve only in T cell-competent rats (Figure 7).
Together, these findings suggest that T cells modulate macrophage trafficking and polarization in a tissue-specific manner during PIPN. T cell deficiency reduces CX3CR1-mediated macrophage infiltration into the DRG but enhances anti-inflammatory M2-like macrophage presence in the sciatic nerve.
Discussion
This study investigated the influence of T cells on the development and maintenance of PIPN in tumor-bearing rats. Specifically, we examined the impact of T cell deficiency on PTX-induced hyperalgesia and allodynia. Our findings suggest that T cells shape neuroimmune responses to PTX, contributing to observed differences in pain behaviors. Notably, T cell-competent rats exhibited reduced onset intensity of mechanical hypersensitivity and spontaneous pain, while T cells played a crucial role in the development and onset of cold hyperalgesia and allodynia. Additionally, PTX-induced depletion of T cells, particularly CD8+ T cells, in the DRG and sciatic nerve led to an increased CD4+/CD8+ ratio, which may serve as a potential biomarker for PIPN progression.
Although non-neuronal immune cells are well-established mediators of chronic pain,4,6,8,9 the contribution of T cells to neuropathic pain remains unsettled. Some studies report that T cells worsen neuropathic pain,4,10,11,37 while others show that loss of T cells prolongs pain, intensifies hypersensitivity, or delays recovery.12,13,18,38 In our study, T cell deficient rats showed an earlier and more pronounced onset of mechanical hypersensitivity, but this effect narrowed as PIPN progressed, suggesting that T cells shape early but not late mechanical responses. Tumor presence or inoculation did not alter mechanical hypersensitivity in either T cell-deficient or T cell-competent rats. This suggests that mechanical hypersensitivity in PIPN is driven mainly by PTX rather than tumor burden or T cell status in later phases. Paclitaxel treatment also produced gait impairments in both T cell-competent and T cell-deficient rats, consistent with previous reports.2,19,33 T cells shaped spontaneous pain behaviors as reflected in spatiotemporal gait and burrowing measures. Although T cells had little effect on temporal gait parameters, they lessened spatial gait deficits during the early stages of PIPN. This pattern paralleled the reduced mechanical hypersensitivity seen at PIPN onset in T cell-competent rats. Tumor presence or inoculation did not affect gait impairment or mechanical hypersensitivity, but it was associated with ongoing spontaneous pain regardless of PTX treatment, indicating that tumor burden preferentially influences spontaneous rather than evoked pain.
Cold hypersensitivity, including both cold hyperalgesia and cold allodynia, is a well-established hallmark of PIPN in both human and preclinical studies.3,39,40 Our previous work demonstrated PTX-induced cold hypersensitivity in T cell-competent Sprague–Dawley rats, 26 which we now extend to Rowett nude rats. Interestingly, PTX-induced cold hyperalgesia and allodynia developed exclusively in T cell-competent rats, while T cell-deficient rats failed to exhibit cold hypersensitivity. Prior studies of cold hypersensitivity have largely relied on T cell-competent models.41,42 Notably, tumor presence or inoculation did not influence cold hypersensitivity regardless of T cell status.
Our findings contrast with earlier reports suggesting that T cells promote hypersensitivity, 37 likely due to differences in species, immune backgrounds, chronic pain models, and the phases of neuropathy being examined. For instance, chronic constriction injury-induced neuropathy involves widespread inflammation and axonal degeneration.15,43 Additionally, some CIPN studies used adoptive T cell transfer into mice lacking both T and B cells, while our model focuses on the effects of T cell deficiency in the presence of other immune cell types in rats.17,18,20 Aside from T cell deficiency, homozygous RNU rats have been reported to retain an intact immune system with white blood cell populations that are similar to those of normal rats.23,24 In contrast, our study focuses on the development and maintenance phases of PIPN, where we observed a reduction in T cell frequencies in the peripheral nervous system. These phase-specific differences, along with differences in experimental design, likely account for the contrasting observations regarding T cell dynamics in the DRG.
T cells were selectively reduced within the peripheral nervous system but remained largely unchanged systemically. These reductions were associated with increased pain severity, in line with other models in which T cell loss worsens neuropathic pain.12,13,18,38,44 PTX increased the CD4+ to CD8+ T cell ratio in both peripheral nerves and systemic tissues, driven mainly by preferential loss of CD8+ T cells. This pattern suggests that the CD4+ to CD8+ ratio may be a useful biomarker of PIPN progression. Overall, our findings indicate that T cell presence or absence does not prevent PIPN but affects its timing, severity, and the pain-related behaviors that emerge.
Several mechanisms may account for T cell dependent cold hypersensitivity. One possibility is cross-talk between anti-inflammatory CD4+ T cell subsets and MHCII-expressing DRG neurons. A recent study showed that CD4+ T cells interact with DRG neurons, and mice lacking neuronal MHCII display reduced anti-inflammatory CD4+ T cells in the DRG and increased cold sensitivity. This suggests that regulatory CD4+ T cells may suppress cold-sensing receptors such as TRPA1 and TRPM8. 3 Another possibility is that an imbalance between CD4+ and CD8+ T cells enhances cold sensitivity. In our model, paclitaxel reduces T cells in the DRG and sciatic nerve, with a more pronounced loss of CD8+ than CD4+ T cells, leading to an increased CD4+ to CD8+ ratio that may contribute to heightened cold sensitivity. In contrast, T cell deficient rats lack sufficient T cell populations to initiate cold hypersensitivity. Notably, T cells did not influence heat hypersensitivity, indicating a selective role in cold sensitivity.
B cells remain relatively understudied in chronic pain, with most existing work centered on autoimmune models in which B cells generate pro-nociceptive autoantibodies.43,45,46 Our findings suggest that B cell involvement in PIPN is site-specific and partially dependent on T cells. B cell reductions in the DRG may be partly T cell independent, whereas changes in the sciatic nerve and spleen appear more closely tied to T cell status. The unchanged frequency of B cells in the sciatic nerve of T cell deficient rats coincided with an increased frequency of M2 macrophages, suggesting a possible B cell and macrophage interaction. Regulatory B cells are known to support regulatory T cell formation and IL-10 production, both of which contribute to pain resolution.43,47,48 Therefore, the observed reduction in B cells, particularly in T cell competent rats, may indicate a loss of IL-10 producing B cells that could amplify pro-inflammatory signaling and increase pain sensitivity. Overall, B cell activity during PIPN likely reflects complex interactions among T cells, macrophages, and local tissue microenvironments.
Additionally, T cell deficiency led to NK cell depletion in the peripheral nervous system and systemically, whereas T cell competent rats retained NK cell populations after PTX treatment. A prior study in T cell competent mice suggested that PIPN does not alter NK cells, 7 but our findings indicate a T cell dependent role in NK cell maintenance. Increased NK cytotoxic activity has been associated with reduced chronic neuropathic hypersensitivity,49,50 and clinical studies have linked low NK cell frequency to greater mechanical pain sensitivity and central sensitization. 51 T cells are necessary for preserving NK cell populations, and loss of NK cells under T cell deficient conditions contributes to worsened pain. Taken together, while T cell depletion promotes PIPN hypersensitivity in T cell competent rats, the additional loss of NK cells in immunocompromised conditions further amplifies PIPN severity.
Macrophages are well-established contributors to CIPN.1,5,8,17,19,25,43 Our previous work showed that PTX drives macrophage infiltration and polarization toward pro-inflammatory M1 phenotypes in the sciatic nerve. 19 Other studies have demonstrated that M2 macrophage repopulation is critical for pain resolution,17,52 consistent with our earlier finding that M2γ macrophage polarization in peripheral nerves is essential for resolving PIPN-associated pain. 19 The current study adds further complexity: T cell deficiency produced divergent macrophage patterns across tissues. In the DRG, reductions in anti-inflammatory M2-like macrophages (M2a and M2γ) occurred independently of T cells, contributing to a pro-inflammatory state that heightened hypersensitivity during PIPN maintenance. In contrast, in the sciatic nerve, T cell deficiency increased M2a and M2γ macrophages, whereas T cell competence reduced M2γ macrophages. These observations suggest that T cells modulate macrophage polarization in a tissue-specific manner. Prior work indicates that T cells can regulate macrophage infiltration, polarization, and cytokine production in neuropathic pain.17,18,52 Our results extend these findings by showing that T cells may restrict M2-like polarization in peripheral nerves but not in the DRG. Conversely, the loss of anti-inflammatory macrophages in the DRG occurred regardless of T cell status, supporting a model in which PTX directly alters the DRG microenvironment toward a pro-inflammatory state.
A limitation of this work is the exclusive use of male rats, as T cells may play a more prominent role in females.3,17 Future studies should evaluate sex-specific T cell contributions and perform T cell reconstitution in T cell-deficient rats to verify causality. Although tumor presence and tumor inoculation did not change PIPN hypersensitivity regardless of T cell status, the stage and anatomical location of tumor growth may limit interpretation in this study.
In summary, T cells play a critical role in PIPN-related cold hyperalgesia and allodynia, while their influence on mechanical hypersensitivity and gait impairment is most pronounced during the early stages of PIPN. Additionally, T cells are essential for maintaining NK cells, and their depletion may exacerbate PIPN symptoms. The impact of PTX on macrophage polarization is both tissue-specific and T cell-dependent. PTX reduces B and T cell populations while increasing the CD4+/CD8+ ratio, a potential biomarker for assessing PIPN risk and treatment efficacy.
Supplemental Material
sj-docx-1-mpx-10.1177_17448069261418431 – Supplemental material for T cells modulate the development and maintenance of painful paclitaxel-induced peripheral neuropathy in RNU rats
Supplemental material, sj-docx-1-mpx-10.1177_17448069261418431 for T cells modulate the development and maintenance of painful paclitaxel-induced peripheral neuropathy in RNU rats by Ahmed Olalekan Bakare, Gerard Limerick, Vasudha Goel, Ratan K Banik, Lei Zheng, Andrew J Shepherd, Kristine Glunde, Qin Zheng and Eellan Sivanesan in Molecular Pain
Footnotes
Acknowledgements
The authors thank Tricia Niles, Worod Allak, and Josh Monts for technical assistance with flow cytometry (Bloomberg Flow Cytometry and Immunology Core, Johns Hopkins University School of Medicine, Baltimore, MD, USA), George McNamara for technical assistance with immunohistochemistry imaging (Ross Fluorescence Imaging Core, Johns Hopkins University School of Medicine), and Claire Levine and Brittni Delmaine for editorial assistance (Johns Hopkins University).
Author contributions
Conceptualized and designed the study: AOB, ES. Conducted or assisted with experiments: AOB, ES. Analyzed and visualized the results: AOB, ES. Data interpretation: AOB, GL, VG, RB, LZ, AJS, KG, QZ, ES. Wrote the original draft: AOB, ES. Critical intellectual revision of the manuscript: AOB, GL, VG, RB, LZ, AJS, KG, QZ, ES. All authors read and approved the final manuscript.
Data availability statement
The data that support the findings of this study are available, upon reasonable request, from the corresponding author.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: GL is a consultant for Averitas Pharma. LZ receives grant support from Bristol-Meyer Squibb, Merck, AstraZeneca, iTeos, Amgen, NovaRock, Inxmed, Halozyme and Abmeta. LZ is a consultant/Advisory Board Member at Biosion, Alphamab, NovaRock, Ambrx, Akrevia/Xilio, QED, Novagenesis, Snow Lake Capitals, Amberstone, Pfzer, Tavotek, and Mingruizhiyao. LZ holds shares at Alphamab, Amberstone, Mingruizhiyao, and Cellaration. AOB, VG, RB, AJS, KG, QZ, and ES declare no competing interests.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was conducted at the Johns Hopkins University and supported by grant CA255428 from The National Cancer Institute–National Institutes of Health (Bethesda, MD, USA) and by funding from The Department of Anesthesiology and Critical Care Medicine, Johns Hopkins University School of Medicine (ES). Funders had no role in study design, data collection, or data interpretation, or in the decision to submit the work for publication.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.