
Abstract
Background
Chemokine axis chemokine C-X-C motif ligand 12/C-X-C chemokine receptor type 4 (CXCL12/CXCR4) is an emerging pain modulator, but mechanisms for its involvement in neuropathic pain remain unclear. Here, we aimed to study whether CXCL12/CXCR4 axis modulated the development of neuropathic pain via glial mechanisms. In this study, two mouse models of neuropathic pain, namely partial sciatic nerve ligation (pSNL) model and chronic post-ischemia pain (CPIP) model, were used.
Results
In the dorsal horn of L3–L5 segment of spinal cord, CXCL12 and CXCR4 were expressed in both astrocyte and microglia in normal mice. In the pSNL or CPIP model, the expression level of CXCL12 in the ipsilateral L3–L5 segment of mice spinal cord was increased in an astrocyte-dependent manner on post-operative day (POD) 3. Intrathecal administration of CXCL12 with AMD3100 (CXCR4 antagonist) or minocycline (microglia activation inhibitor), but not fluorocitrate (astrocyte activation inhibitor), reversed CXCL12-indued mechanical allodynia in naïve mice. In these models, AMD3100 and AMD3465 (CXCR4 antagonist), administered daily from 1 h before surgery and up to POD 3, attenuated the development of mechanical allodynia. Moreover, AMD3100 administered daily from 1 h before surgery and up to POD 3 downregulated mRNA levels of tumor necrosis factor alpha, interleukin 1β, and interleukin 6 in the ipsilateral L3–L5 segment of spinal cord in the pSNL and CPIP models on POD 3.
Conclusion
This study demonstrates the crosstalk between astrocytic CXCL12 and microglial CXCR4 in the pathogenesis of neuropathic pain using pSNL and CPIP models. Our results offer insights for the future research on CXCL12/CXCR4 axis and neuropathic pain therapy.
Background
Pathological pain is a disease caused by abnormal processing in the nervous system under various pathological conditions, which lasts for more than three months clinically. 1 Neuropathic pain is induced by the lesion or the disease in the somatosensory nervous system and is the major subtype of pathological pain. 2 Central mechanisms contribute to the chronification of pain and have been taken as the target in the research of pathological pain, especially neuropathic pain.1,3 Mounting studies indicate that spinal glial cells (astrocyte and microglia) are activated in animal models of neuropathic pain 4 and in patients suffering from chronic pain.5,6 Then, these reactive glial cells release cytokines and chemokines, which contribute to the neuroinflammation and the central mechanisms of neuropathic pain. 7 Chemokines are small and secreted proteins which include four subfamilies (C-C, C-X-C, X-C, and C-X3-C). 8 Chemokines bind to chemokine receptors that belong to G-protein-coupled receptor (GPCR) family to exert the biology function. 9 Nowadays, increasing body of preclinical evidence suggests chemokine axis, such as chemokine C-C motif ligand 2/C-C chemokine receptor type 2 (CCL2/CCR2) axis and chemokine (C-X3-C motif) ligand 1 (CX3CL1)/C-X3-C chemokine receptor type 1 (CX3CR1) axis,10–13 as the novel neuromodulator in the pain perception.
Chemokine C-X-C motif ligand 12 (CXCL12) belongs to C-X-C subfamily of chemokine and is the ligand of C-X-C chemokine receptor type 4 (CXCR4). CXCL12/CXCR4 axis widely exists in the central nerve system (CNS) and serves multiple functions, from “well-known” migration regulation to “newly found” neuromodulation. 14 Knowledge from CXCL12 and CXCR4 knockout mice would be the best way for us to understand the role of CXCL12/CXCR4 axis in pathological pain. However, animals with the deficiency of CXCL12 or CXCR4 could not survive due to the abnormal tissue development. 15 The distribution of CXCL12/CXCR4 axis on the nociceptive structure and its pro-nociceptive property indicates the involvement of this chemokine axis in the pain perception. CXCL12/CXCR4 axis is distributed in different components in the nociceptor at the peripheral nervous system, such as dorsal root ganglion neuron, satellite glia, and Schwann cell,16–18 and at the CNS, such as neuron, astrocyte, and microglia.16,19,20 Furthermore, CXCL12 peptide has been shown to induce mechanical allodynia by peripheral 18 and by central administration.16,19 Moreover, CXCL12/CXCR4 axis was proved to function in the peripheral processing of pathological pain as shown in animal models of diabetes disease, sciatic nerve injury, and acquired immune deficiency syndrome.17,18,21–23 Recently, the role of CXCL12/CXCR4 axis in central mechanisms of pathological pain raised a growing concern. 11 For example, central CXCL12/CXCR4 axis contributed to the pathogenesis of opioid tolerance. 24 Moreover, the spinal blockade of CXCL12/CXCR4 axis showed analgesic effects in animal models of sciatic nerve injury 25 and bone cancer pain.19,26,27
Nowadays, CXCL12/CXCR4 axis has been considered an emerging pain modulator; 28 however, the common mechanisms accounting for roles of CXCL12/CXCR4 axis in various pathological pain states, especially neuropathic pain, remain unclear. As the glial mechanism is the common mechanism for neuropathic pain, 1 we hypothesized that the glial–glial crosstalk may account for the involvement of CXCL12/CXCR4 axis in a wide range of neuropathic pain conditions. To verify this, the roles of CXCL12/CXCR4 axis in the central mechanisms were explored in two different mouse models of neuropathic pain, with partial sciatic nerve ligation (pSNL) to induce peripheral neuropathic pain (PNP) 29 and chronic post-ischemia pain (CPIP) as a model for complex regional pain syndrome (CRPS). 30
Methods
Animals
Animal experiments in this study were approved by the Committee on the Use of Live Animals in Teaching and Research and performed according to the guidelines for the care and use of laboratory animals as established by the Laboratory Animal Unit at the University of Hong Kong. The experiments were conducted using adult male C57BL/6 wild-type mice (25–30 g). Animals were housed at 23 ± 3℃, with humidity ranges between 25% and 45% under a 12-h light/12-h dark cycle (lights on at 07:00). The mice were offered free access to water and food. They were fed with Lab Diet 5012 (1.0% calcium, 0.5% phosphorus, and 3.3 IU/g of vitamin D3).
pSNL model
The pSNL model is an animal model of PNP. 29 Mice were anesthetized with the inhalation anesthesia by isoflurane in O2. Under aseptic conditions, the right sciatic nerve was exposed by an incision from the right sciatic notch to the distal thigh. By using the femoral head as a landmark, the location of the sciatic nerve ligation was identified. Approximately half of the sciatic nerve was tightly ligated with a 7-0 silk suture. The incision was closed with 5-0 cotton suture and disinfected with ethanol. As the control treatments, the right sciatic nerve of sham-operated mice was only exposed, but not ligated. Furthermore, time after the operation was counted, when the nerve was ligated. The first day after the surgery was counted as post-operative day 1 (POD 1).
CPIP model
In the CPIP model, ischemia-and-reperfusion injury (CPIP injury) caused the edema and mechanical allodynia in the mice hindpaw. 31 As the symptoms usually occur in the distal part of the affected limb in CRPS patients, the CPIP model was developed and used to study the pathology of CRPS-1. 30 Animals were anesthetized with the inhalation anesthesia by isoflurane in O2. After the induction of anesthesia, Durometer O-rings (O-rings West) were placed around the mice’s right hindlimbs just proximal to the ankle joint and caused the CPIP injury to ipsilateral hindpaws. As the control treatments, sham-operated mice were only anesthetized, but not placed by O-rings. These rings were removed 3 h later to induce the reperfusion. Furthermore, time after the operation was counted, when O-ring was removed. The first day after the surgery was counted as POD 1.
Study drugs
Rat CXCL12 peptide (Genscript) and two CXCR4 antagonists, AMD3100 (Sigma) and AMD3465 (Tocris), were prepared in saline on the day of the experiment. Recombinant tumor necrosis factor alpha (TNF-α) from Genscript was used. Astrocyte activation inhibitor fluorocitrate (Sigma) and microglia activation inhibitor minocycline (Sigma) were prepared in 1% dimethyl sulfoxide (DMSO) on the day of the experiment. Saline or 1% DMSO was taken as vehicle controls in this study.
Cell culture
DI TNC1 astrocyte cell line, derived from rat diencephalon type I astrocyte in a day 1 neonate, was used. 32 These cells were maintained with Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% (v/v) fetal bovine serum (Invitrogen) and 1% (v/v) penicillin/streptomycin (P/S) (Invitrogen) at 37℃ with 5% CO2. Astrocytes (1 × 105 cells/ml) were plated on six-well culture plate and treated with the medium (control) or TNF-α (10, 100 ng/ml) for 24 h.
Intrathecal injection
A single dose of the intrathecal injection was performed following previous protocol. 33 Before the injection, mice were anesthetized by gaseous anesthesia with isoflurane and O2 as mentioned above. Using a microliter syringe (Hamilton) with a 30-gauge needle (BD), a spinal cord puncture was made to deliver a total volume of 5 µl of drug(s) to the subarachnoid space between the L3 and L5 levels. Successful administration was indicated by a tail configuration of the “S” type or tail swinging immediately after the administration.
von Frey test
Paw withdrawal threshold (PWT) of mice was assessed by von Frey test, and the same protocol was used in our previous research. 34 Before the experiments, the mice were placed in a transparent plastic dome with a metal mesh floor for nearly 30 min. PWT was measured with a series of von Frey filaments (IITC). These filaments were applied perpendicularly to the plantar surface of mice hindpaws, with sufficient force to bend the filaments into an “S” shape. Licking of the paw or quick withdrawal was considered as a positive response.
Immunohistochemistry
Mice were deeply anesthetized with sodium pentobarbital and perfused with ice-cold 0.9% saline, followed by 4% paraformaldehyde in 0.1 M phosphate buffer via the cardiovascular system. The ipsilateral and contralateral L3–L5 spinal cord tissues were collected and post-fixed in 4% paraformaldehyde and then dehydrated overnight in 25% sucrose at 4℃. The tissue samples were frozen in tissue freezing medium (Jung) and sliced transversely at 15 µm, using a cryostat (Leica). The sections were blocked with 10% normal goat serum in phosphate-buffered saline (PBS) containing 0.1% Triton X-100 (PBST) at room temperature for 2 h and incubated with antibody(s), including CXCL12 (1:100, Abcam, #25117), CXCR4 (1:100, Abcam, #2074), glial fibrillary acidic protein (GFAP; 1:250, Abcam, #10062), and ionized calcium binding adaptor molecule 1 (IBA1; 1:100, Abcam, #15690), at 4℃ overnight. After washing with PBS, sections were incubated for 2 h with secondary antibody conjugated with Alexa Fluor 488 or/and 568 (Abcam) and followed by nuclear staining using DAPI (Vector). The immunoreactivity in these sections was detected with a confocal microscope LSM 710 (Zeiss), and the immunofluorescent images were analyzed by Image-Pro Plus (Media Cybernetics).
Rotarod test
To assess the potential side effects of treatments in this study, a rotarod test was conducted to measure the motor coordination and balance of the mice. 35 Animals were habituated to the rotarod apparatus (IITC) for two consecutive days at low-speed rotation (5 r/min) for 600 s each day before basal measurement. Mice that could not stay on the rod for 600 s were excluded from this experiment. During the experiment, the animals were tested in three accelerating trials of 300 s with the rotarod speed increasing from 5 to 40 r/min over first 120 s. There was an inter-trial interval of at least 20 min between each trial for the same mouse. The falling latency of mice was recorded for each trial with a cutoff time at 300 s.
Real-time polymerase chain reaction
After being euthanized with pentobarbital, both ipsilateral and contralateral L3–L5 segments of spinal cord were quickly removed from mice and stored at −80℃ until RNA extraction. These spinal cord segments were homogenized in 1 ml of ice-cold RNAiso Plus (Takara), and total mRNA was extracted following the manufacturer’s protocol. The quality and quantity of extracted RNA were determined with GeneQuant spectrophotometer (Pharmacia). cDNA library was prepared by the reverse transcription using 2 µg of the extracted total mRNA with the reverse transcriptase (Life Technologies). Then, these cDNA samples were diluted and stored at −20℃ until further tests. Real-time polymerase chain reaction (PCR) test was performed using Taqman chemistry method (Life Technologies), and the probes of targets, including TNF-α, interleukin 6 (IL-6), IL-1β, substance P (SP), calcitonin gene-related peptide (CGRP), prodynorphin (PDYN), and β-actin (as internal reference; Life Technologies), were used in this study.
Statistical analysis
The data were expressed as means ± SEM. Results from the immunohistochemical work and the real-time PCR test (on the samples from in vitro work) were tested using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. Two-way ANOVA followed by Tukey’s post hoc test were used to analyze data from the von Frey test, the rotarod test, and the real-time PCR test (on the samples from in vivo work). A p value < 0.05 was considered to be statistically significant in this study.
Results
Mechanical allodynia developed in the pSNL and CPIP models
In this study, the ipsilateral hindpaws of pSNL-injured mice showed decrease in the PWT from POD 1 to POD 21 in the pSNL model (p < 0.001, n = 6, Figure 1(a)). In CPIP-injured mice, the ipsilateral PWT was decreased from POD 2 to POD 21 comparing to that of POD 7 (p < 0.05, n = 6, Figure 1(b)). However, it was increased on POD 21 (p < 0.001). Moreover, CPIP-injured mice showed transient edema in the ipsilateral hindpaws from 1 h to POD1 (p < 0.001, n = 5, data not shown).
Mechanical allodynia developed in pSNL- or CPIP-injured mice. The PWT of mice was decreased in the pSNL model (a) and the CPIP model (b) as determined by the von Frey test. Results are means ± SEM (n = 5–6). ***p < 0.001 and *p < 0.05 versus the baseline and ###p < 0.001 versus data on POD 7 from the same group.
Expression and distribution of CXCL12/CXCR4 axis in the lumbar spinal cord of mice
Next, we used immunofluorescence co-staining to investigate whether CXCL12 and CXCR4 were co-expressed in glia cell of spinal cord of naïve mice. We found that CXCL12 and CXCR4 were co-expressed with GFAP and IBA1, indicating CXCL12 and CXCR4 were expressed in both astrocyte and microglial cells of spinal cord (Figure 2).
Expression and distribution of CXCL12/CXCR4 axis in L3–L5 segment of spinal cord in normal mice. In the spinal cord dorsal horn, CXCL12 (green) and CXCR4 (green) were co-expressed with IBA1 (red) and GFAP (red). High-resolution images were shown in boxes, the co-expression was marked with arrows, and nuclear was marked with blue (original magnification: 200×, scale bar = 100 µm).
Expression pattern of CXCL12 in the lumbar spinal cord in the pSNL and CPIP models
The expression pattern of CXCL12 axis in the spinal cord has not been reported in the pSNL or CPIP model. To explore this, animals were divided into three groups (sham group, pSNL POD 3 group, and CPIP POD 3 group, n = 3 in each group), which received sham, pSNL, and CPIP surgery, respectively. Then, the spinal cord tissues were harvested on POD 3 for the immunohistochemical study. In the ipsilateral lumbar spinal cord dorsal horn, the spinal expression of GFAP was increased on POD 3 in both pSNL and CPIP models (p < 0.05, Figure 3(a) and (b)). The expression of CXCL12 was also increased in the ipsilateral spinal cord dorsal horn of mice receiving the pSNL or CPIP surgery, comparing to that in sham-operated mice (p < 0.05, Figure 3(a) and (c)). Moreover, CXCL12 was mainly co-localized with GFAP in both models (Figure 3(a)). As the spinal expression of TNF-α was increased in the pSNL and CPIP models as mentioned below, TNF-α was applied to induce the pathological insult in vitro in the current study (n = 4 in each group). Our results showed that TNF-α increased mRNA levels of CXCL12 in astrocyte in a dose-dependent manner (p < 0.001, Figure 3(d)). However, there was no any change in the expression of CXCR4 at the ipsilateral lumbar spinal cord among mice receiving pSNL, CPIP, or sham operation (data not shown).
Expression pattern of CXCL12 in ipsilateral L3–L5 segment of spinal cord in the pSNL or CPIP model. (a) The immunoreactivity of GFAP (red) and CXCL12 (green) were increased in the ipsilateral lumbar spinal cord of pSNL-injured and CPIP-injured mice on POD 3, which was summarized in (b) and (c). Original magnification: 200× for all the confocal images (Bar = 100 µm). (d) TNF-α increased the mRNA levels of CXCL12 in astrocyte in vitro. Results are means ± SEM (n = 3). ***p < 0.001, **p < 0.01 and *p < 0.05 versus sham group. ###p < 0.001 versus TNF-α 0 ng group. CXCL12: chemokine C-X-C motif ligand 12; GFAP: glial fibrillary acidic protein; pSNL: partial sciatic nerve ligation; CPIP: chronic post-ischemia pain; POD: post-operative day; TNF-α: tumor necrosis factor alpha.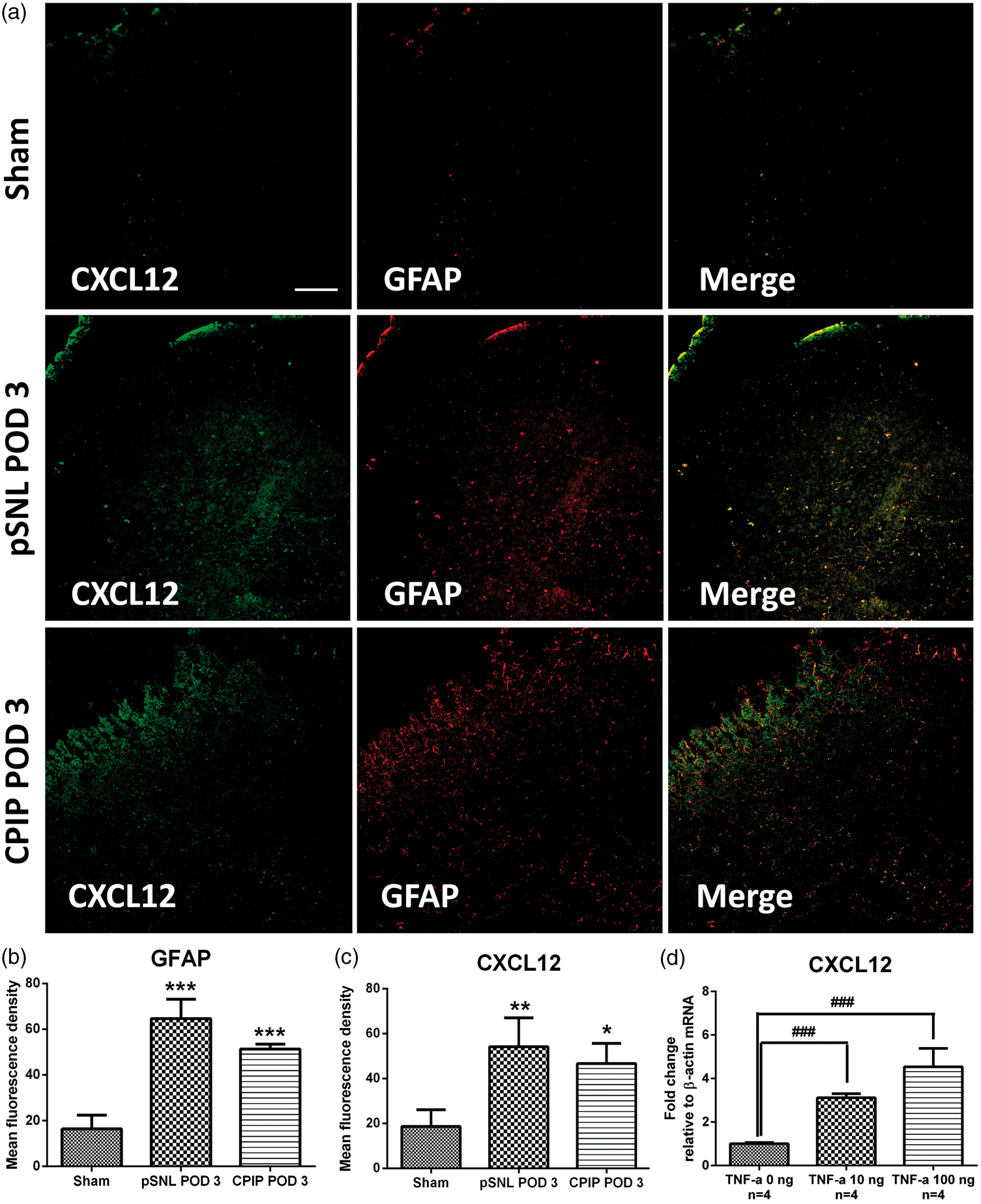
Cellular mechanisms accounting for CXCL12/CXCR4-mediated nociception in naïve mice
As CXCR4 was expressed in microglia and astrocyte as mentioned above, the cellular targets of astrocytic CXCL12 in the neuropathic pain were further assessed. Minocycline is a selective inhibitor of the microglial activation, whereas fluorocitrate is a selective inhibitor of the astrocytic activation at a relatively low concentration.
36
These two glial activation inhibitors were found to account for the analgesic effects in various neuropathic pain models,
37
and they were thus used in this study. Normal mice were randomly divided into five groups, and the pain behavioral responses (PWT) of these mice were assessed by the von Frey test. In these groups, mice received a single intrathecal injection of (1) CXCL12 rat peptide (250 ng, n = 6), (2) CXCL12 rat peptide (250 ng) and AMD3100 (5 µg) (n = 6), (3) CXCL12 rat peptide (250 ng) and minocycline (15 µg), (4) CXCL12 rat peptide (250 ng) and fluorocitrate (2 µg), and (5) 1% DMSO as the vehicle control (n = 6). As shown in Figure 2, the intrathecal delivery of 1% DMSO did not influence PWT in normal mice (p > 0.05). A single intrathecal injection of rat CXCL12 peptide (250 ng) decreased PWT from 1 h to one day after the treatment (p < 0.05), comparing to the baseline. The co-administration of CXCL12 peptide (250 ng) and AMD3100 (5 µg) also did not affect PWT in normal mice as compared to the baseline (p > 0.05). Similarly, the intrathecal administration of CXCL12 (250 ng) and minocycline (15 µg) did not affected the PWT. In contrast, the intrathecal administration of CXCL12 (250 ng) with fluorocitrate (2 µg) decreased PWT from 1 h to one day after the injection in normal mice (p < 0.05), comparing to the baseline (Figure 4).
Cellular mechanisms accounting for CXCL12/CXCR4 axis-mediated nociception in naïve mice. The co-treatment of CXCL12 with AMD3100 and minocycline, but not fluorocitrate, prevented CXCL12-induced mechanical allodynia. Results are means ± SEM (n = 6–8). ***p and ###p < 0.001, *p < 0.05 versus the baseline.
Effects of intrathecal CXCR4 antagonists on motor function in naïve mice
AMD3100 and AMD3465 are CXCR4-specific antagonists. AMD3465 exhibited nearly 10-fold affinity to CXCR4, comparing to AMD3100.
38
To explore the effects of these reagents on the motor function, normal mice were randomly divided into three groups. The study drugs were given intrathecally, and motor function was assessed by the rotarod test then. Animals respectively received intrathecal AMD3100 (10 µg, n = 6), AMD3465 (10 µg, n = 6), and saline (control, n = 6) for four days consecutively. Comparing to the control treatment, the intrathecal injection of AMD3100 or AMD3465 did not affect the falling latency (p > 0.05) (Figure 5).
Effects of intrathecal injection of two CXCR4 antagonists on motor function in naïve mice. The falling latency of mice were not affected by intrathecal AMD3100, AMD3465, or saline. Results are means ± SEM (n = 5–6).
Effects of intrathecal AMD3100 or AMD3465 on the development of mechanical allodynia in the pSNL model
The effects of intrathecal CXCR4 antagonists on mechanical allodynia in pSNL-injured mice were assessed by the von Frey test. Animals were randomly divided into three groups before receiving the study drugs and the von Frey test. In these groups, mice received a single intrathecal injection of AMD3100 (10 µg, n = 10), AMD3465 (10 µg, n = 7), or saline (vehicle, n = 6) daily from 1 h before the surgery and up to POD 3. Following the pSNL surgery, mice from the three groups exhibited decrease in the PWT from POD 1 to POD 14 (p < 0.001). Comparing to control group, intrathecal AMD3100 increased the ipsilateral PWT from POD 1 to POD 6 (p < 0.001) and intrathecal AMD3465 increased the ipsilateral PWT from POD 1 to POD 4 (p < 0.05, Figure 6(a)). The pSNL surgery or the intrathecal treatment of AMD3100 or AMD3465 did not affect the contralateral PWT (Figure 6(b)).
Effects of intrathecal AMD3100 or AMD3465 on the development of mechanical allodynia in the pSNL model. The PWT of pSNL-injured mice was increased by intrathecal AMD3100 and AMD3465 in the ipsilateral (a) but not the contralateral (b) hindpaws as compared to the saline group. Results are means ± SEM (n = 6–10). ***p, ###p, and ψψψp < 0.001 and #p < 0.05 versus the baseline. aaap and bbbp < 0.001, bbp < 0.01 and bp < 0.05 versus the saline group.
Effects of intrathecal AMD3100 on the spinal production of pain molecules in the pSNL model
The molecular mechanisms accounting for the anti-allodynia effects of CXCR4 antagonism on the development of PNP remain unclear. Therefore, we studied the effects of intrathecal AMD3100 on the spinal production of pain-related molecules in the pSNL models. After pSNL-injured mice had been received intrathecal AMD3100 (n = 8) or saline (n = 6) daily from 1 h before the surgery and up to POD 3, both ipsilateral and contralateral L3–L5 segments of spinal cord tissue were harvested on POD 3, and the mRNA levels of pain molecules were assessed by real-time PCR test. In saline group, the unilateral sciatic nerve injury increased the mRNA levels of TNF-α, IL-6, SP, CGRP, and PDYN (p < 0.05), but did not increase the mRNA levels of IL-1β, in the ipsilateral lumbar spinal cord as compared to their contralateral levels (p > 0.05). Comparing to the control group, intrathecal AMD3100 decreased the ipsilateral levels of TNF-α and IL-6 (p < 0.05) and increased the contralateral levels of PDYN in pSNL-injured mice (p < 0.05, Figure 7).
Effects of intrathecal AMD3100 on the spinal production of pain molecules in the pSNL model. After pSNL-injured mice receiving the intrathecal injection of AMD3100 (n = 8) or saline (n = 6), the lumbar spinal cord tissue was harvested on POD 3, and pain molecule levels were assessed by the real-time PCR test, including pro-inflammatory cytokines (a) and neuropeptides (b). Results are means ± SEM (n = 6–8). ###p < 0.001, aap < 0.01, ap < 0.05 versus the contralateral data in the saline group. bp < 0.05 versus the contralateral data in AMD3100 group. *p < 0.05 versus the ipsilateral data in the saline group.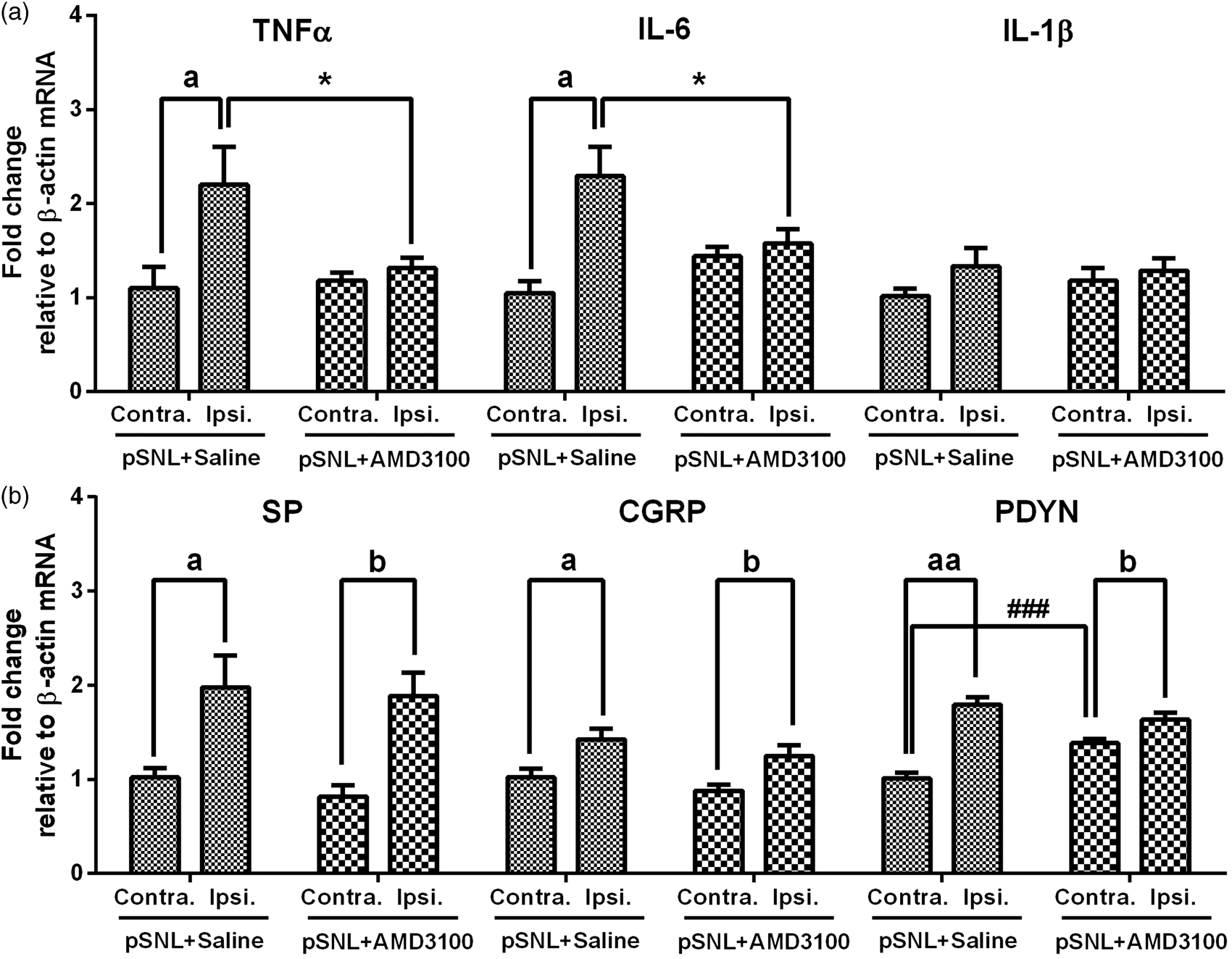
Effects of intrathecal AMD3100 or AMD3465 on the development of mechanical allodynia in the CPIP model
The behavioral effects after intrathecal CXCR4 antagonists were also studied in the CPIP model. Normal mice were randomly divided into three groups. In these three groups, animals received a single intrathecal injection of AMD3100 (10 µg, n = 9), AMD3465 (10 µg, n = 8), or saline as the vehicle (n = 6) daily from 1 h before the surgery and up to POD 3. Animals from control group showed decrease in the PWT from POD 2 to POD 14 (p < 0.001). Mice receiving intrathecal AMD3100 also showed decrease in the ipsilateral PWT from POD 4 to POD 14 (p < 0.001). Comparing to control group, intrathecal AMD3100 progressively and significantly increased ipsilateral PWT on POD 3 and 4 (p < 0.05). Mice receiving intrathecal AMD3465 exhibited decreased ipsilateral PWT from POD 5 to POD 14 (p < 0.001). Comparing to the control group, intrathecal AMD3465 increased the ipsilateral PWT on POD 4 (p < 0.001, Figure 8(a)). Moreover, CPIP injury or the intrathecal treatment of AMD3100 or AMD3465 did not affect the contralateral PWT (Figure 8(b)). These results indicated that central CXCL12/CXCR4 blockade also attenuated the development of mechanical allodynia in the CPIP model.
Effects of intrathecal AMD3100 or AMD3465 on the development of mechanical allodynia in the CPIP model. The PWT of CPIP mice was increased by intrathecal AMD3100 and AMD3465 in the ipsilateral (a) but not the contralateral (b) hindpaws as compared to the saline group. Results are means ± SEM (n = 6–10). Results are means ± SEM (n = 6–9). ***p, ###p, and ψψψp < 0.001 and **p < 0.01 versus the baseline. bbbp < 0.001, aap < 0.01, and ap < 0.05 versus the saline group.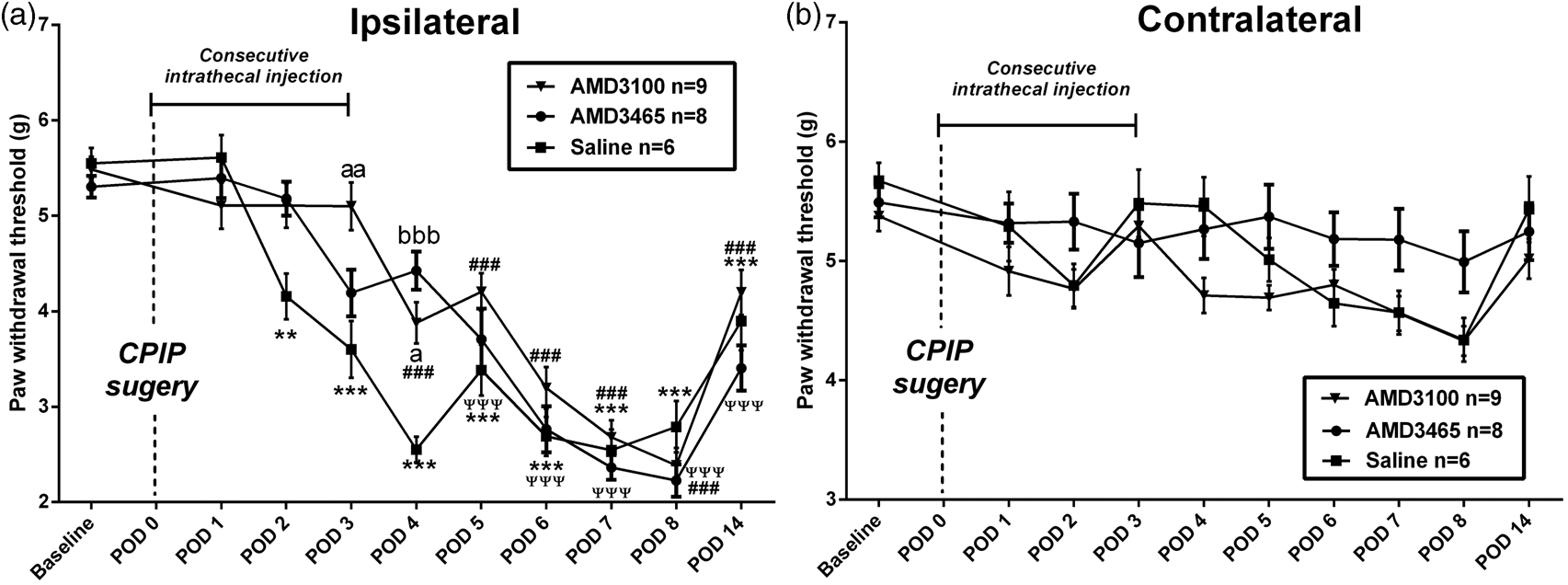
Effects of intrathecal AMD3100 on the spinal production of pain molecules in the CPIP model
The molecular mechanisms for intrathecal AMD3100 in pain were also studied in the CPIP model. After having received intrathecal injections of AMD3100 (10 µg, n = 8) or saline (n = 6) for four consecutive days, both ipsilateral and contralateral L3–L5 segments of spinal cord were harvested from the CPIP-injured mice on POD 3, and the levels of pain molecules were assessed by the real-time PCR test. In saline group, CPIP injury increased the ipsilateral levels of TNF-α, IL-6, IL-1β, SP, and PYDN (p < 0.05) and decreased the ipsilateral level of CGRP, comparing to their contralateral levels (p < 0.05, Figure 9). Comparing to saline group, intrathecal AMD3100 decreased the ipsilateral levels of TNF-α, IL-6, and IL-1β (p < 0.05) and the contralateral level of CGRP (p < 0.05, Figure 9).
Effects of intrathecal AMD3100 on the spinal production of pain molecules in the CPIP model. After IR-injured mice receiving the intrathecal injection of AMD3100 (n = 8) or saline (n = 6), the lumbar spinal cord tissue was harvested on POD 3, and the levels of pain molecules were assessed by real-time PCR test, including pro-inflammatory cytokines (a) and neuropeptides (b). Results are means ± SEM (n = 6–8). #p < 0.05 and ap < 0.05 versus the contralateral data in the saline group. bp < 0.05 versus the contralateral data in AMD3100 group. *p < 0.05 versus the ipsilateral data in the saline group.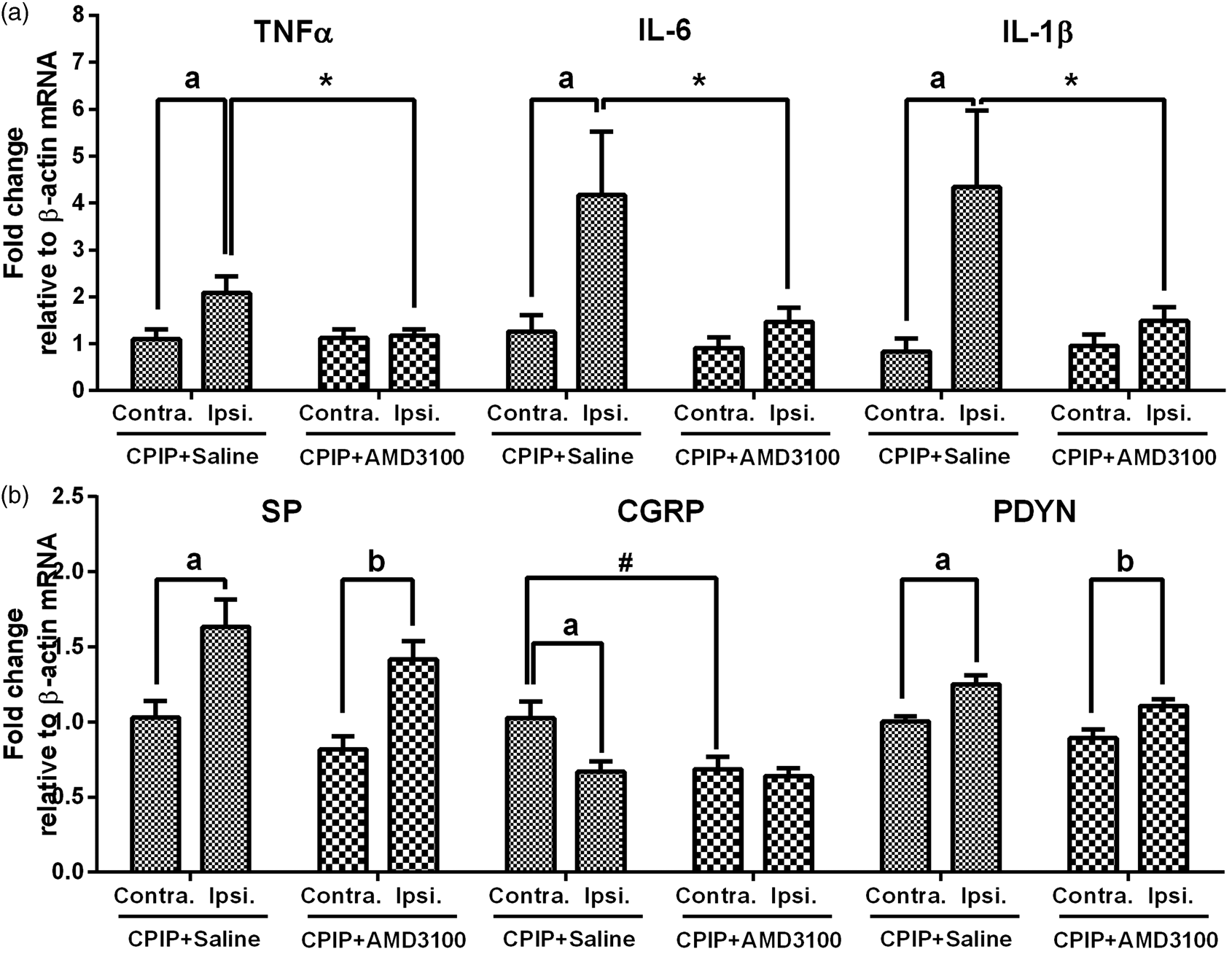
Discussion
In the present study, two different types of neuropathic pain models were used. The pSNL model is a classic PNP model. 29 In this model, pSNL-injured animals showed pain behavioral signs of patients with causalgia, and the mechanical allodynia induced in these animals lasted for weeks. The mechanisms accounting for CRPS are not fully understood, but neuropathy is considered as an essential factor in the pathogenesis of CRPS. 39 The CPIP model has been developed as an animal model of CRPS type 1. 30 In the CPIP model, the ischemia-and-reperfusion injury caused the transient edema and mechanical allodynia, two important features of CRPS type 1. In the current study, it was found that CXCL12 and CXCR4 were expressed in astrocyte and microglia. Therefore, it is highly possible that CXCL12/CXCR4 axis might be involved in the central mechanisms of pain through the glial–glial (astrocyte–astrocyte, microglia–microglia, or/and astrocyte–microglia) crosstalk. With the use of these two neuropathic pain models, we aimed to explore whether CXCL12/CXCR4 axis would be involved in the communication between glial cells and would contribute to the development of neuropathic pain.
In the pSNL or CPIP model, the spinal expression of GFAP was increased following the surgery. Spinal astrocyte is activated under various neuropathic pain conditions which contribute to the persistent pain by releasing cytokines and chemokines. 40 Therefore, reactive astrocyte might contribute to the central mechanisms for pain hypersensitivity in the pSNL or CPIP model. In these two models, the spinal expression of CXCL12 was increased and mainly expressed in reactive astrocyte on POD 3, an early stage of persistent pain. Furthermore, the expression pattern of CXCL12 in astrocyte was further substantiated by our in vitro study results. Previously, it was reported that CXCL12 was expressed in an astrocyte-dependent way in a bone cancer pain model, which was reversed by the intrathecal administration of the astrocyte inhibitor fluorocitrate. 19 Our results also supported that the spinal level of CXCL12 was increased in an astrocyte-dependent manner under various neuropathic pain conditions. In normal mice, intrathecal AMD3100 reversed CXCL12-induced mechanical allodynia, which confirmed the pro-nociceptive properties of central CXCL12/CXCR4 axis.16,19,26 It was further showed that intrathecal minocycline, but not fluorocitrate, attenuated CXCL12-induced allodynia, which implicated that microglial CXCR4 might play a major role in CXCL12-induced allodynia. Moreover, the intrathecal intervention with two CXCR4 antagonists, AMD3100 and AMD3465, delayed the development of chronic pain without impairing the motor function. Spinal blockade of CXCL12/CXCR4 axis signaling was reported to attenuate pain hypersensitivity in a PNP model 25 and a bone cancer pain. 19 Taken together, our immunohistochemical and pharmacological evidence uncovered the cellular and molecular pattern of CXCL12/CXCR4 axis-mediated glial crosstalk in the pSNL and CPIP models.
Intracellular elements downstream to GPCR modulate gene transcription through a signaling-dependent manner. 41 Since CXCR4 belongs to GPCR family, we thus evaluated whether the levels of pain molecules were regulated by central CXCL12/CXCR4 axis in neuropathic pain. TNF-α, IL-6, and IL-1β represented an important group of pro-inflammatory pain mediators, and they were mainly released by spinal microglia in the development of neuropathic pain.4,42,43 It has been found that pro-inflammatory cytokines contribute to the synaptic plasticity 44 and the glial–glial interaction 45 under different neuropathic pain conditions. In this study, spinal level of TNF-α and IL-6 is upregulated in the pSNL model, which is in line with their contributive role in the pain processing and consistent with previous findings in a model of peripheral neuropathy. 46 The expression pattern of spinal cytokines in the CPIP model has not been reported before, and we first found that the levels of TNF-α, IL-6, and IL-1β were increased in the ipsilateral lumbar spinal cord by the CPIP injury. In a model of sciatic nerve injury, pharmacological evidence has shown that microglia contributed to the development of pain hypersensitivity and neuroinflammation. 47 Previously, it was shown that CXCL12/CXCR4 axis modulates the production of proinflammatory cytokines (TNF-α and IL-6) in microglia in vitro.45,48 Here, our results first showed that the spinal blockade of CXCL12/CXCR4 axis downregulated levels of these pro-inflammatory cytokines in both the pSNL and CPIP models. Taken together, our results demonstrated the crosstalk between astrocytic CXCL12 and microglial CXCR4, which might contribute to pain hypersensitivity via modulating the neuroinflammation in the pSNL and CPIP models.
Among pain neuropeptides, SP was essential to the pain signal transduction at the lumbar spinal cord, while CGRP facilitated SP-mediated pain signaling. 49 Our results showed that the spinal level of SP was increased in two neuropathic pain models whereas that of CGRP levels was only increased in the pSNL model. Such difference in the expression pattern of CGRP and SP might represent the difference in the central mechanisms between PNP and CRPS potentially. Even CXCL12/CXCR4 axis was co-localized with SP and CGRP in rat spinal cord, 16 we could not find that the spinal blockade of CXCL12/CXCR4 axis affect the levels of SP and CGRP at the ipsilateral spinal cord in both models. These findings implicated that CXCL12/CXCR4 axis might not modulate SP or CGRP-mediated pain signaling in the pathogenesis of neuropathic pain. In both pSNL and CPIP models, we found that the ipsilateral level of PDYN was increased on POD 3. PDYN-containing neurons are widely distributed on the pain-related brain areas, and PDYN-derived peptides promote the abnormal pain perception. 50 Here, our results first showed the involvement of PDYN in the spinal cord in neuropathic pain development. The crosstalk between chemokine and opioid system has been considered as an emerging target for chronic pain therapy. 51 However, it was found that spinal level of PDYN was not affected by AMD3100 in the pSNL or CPIP model, suggesting that CXCL12/CXCR4 axis may not interact with PDYN at least in the current experimental settings. Additionally, our results also showed that other pain molecules might not contribute to CXCL12/CXCR4 axis-mediated pain processing in the pSNL or CPIP model, including proopiomelanocortin, proenkephalin, 50 endothelin-1, endothelin receptor A, endothelin receptor B,32,34,52 intercellular adhesive molecules-1, and vascular cell adhesive molecules-153 (data not shown).
Based on the results presented in the current study, the mechanisms for central CXCL12/CXCR4 axis in the development of pain hypersensitivity in the pSNL or CPIP model are proposed at Figure 10. pSNL or CPIP injury leads to the increase of CXCL12 in the spinal dorsal horn through an astrocyte-dependent manner. By binding to microglial CXCR4, CXCL12 increases the production of pro-inflammatory cytokines in microglia, which contributes to the development of pain hypersensitivity. In this study, we found that CXCL12/CXCR4 axis contributed to the development of the neuroinflammation and pain hypersensitivity in two neuropathic pain models.
Proposed mechanisms for CXCL12/CXCR4 axis in the pathogenesis of pain hypersensitivity in the pSNL or CPIP model. pSNL injury or CPIP injury might facilitate the increase of CXCL12 in the spinal cord dorsal horn through an astrocyte-dependent manner. By binding to microglial CXCR4, CXCL12 might increase the production of pro-inflammatory cytokines in microglia and contribute to the development of neuropathic pain.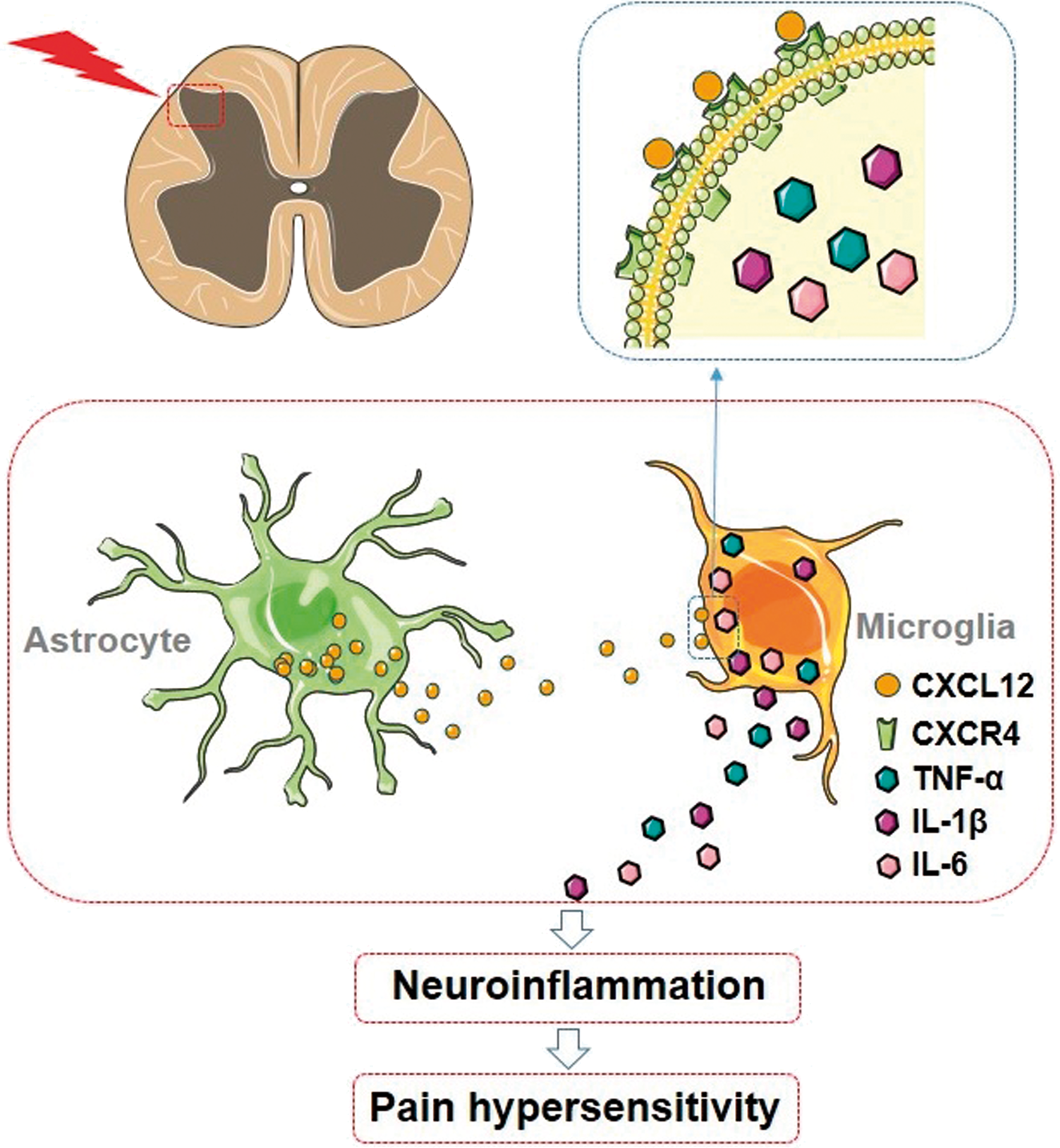
In conclusion, our study demonstrates that CXCL12 is mainly expressed in astrocyte, while CXCR4 in both astrocyte and microglia in the mouse spinal cord. In the pSNL or CPIP model, the spinal expression of CXCL12 is upregulated in an astrocyte-dependent manner at the early stage of the development of neuropathic pain. It is further found that intrathecal CXCL12 induces mechanical allodynia via microglial CXCR4. In both pSNL and CPIP models, the spinal blockade of CXCL12/CXCR4 axis delays the development of mechanical allodynia and neuroinflammation. This study suggests that the crosstalk between astrocytic CXCL12 and microglial CXCR4 is involved in the pathogenesis of neuropathic pain hypersensitivity using the pSNL and CPIP models. Our results provide new insights for the future research on CXCL12/CXCR4 axis and neuropathic pain.
Footnotes
Author Contributions
XL, ZX, SKC, and CWC have made substantial contributions to the experiment design; XL and WLT have made substantial contributions to the acquisition of data; XL, LS, ZP, ZX, SKC, and CWC have made substantial contributions to the analysis and interpretation of data. All authors have been involved in drafting the manuscript or revising it critically for important intellectual content. All authors have given final approval for the manuscript to be submitted.
Acknowledgment
The authors would like to thank Dr. Chaoliang Tang, Department of Anesthesiology, Anhui Provincial Hospital, Anhui Medical University, for help in the animal experiment.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Small Project Fund (201409176065) from the University of Hong Kong and the department fund, Department of Anaesthesiology, the University of Hong Kong.