
Abstract
Background:
Lung cancer is the leading cause of cancer-related deaths worldwide. Copy number variation (CNV) in several genetic regions correlate with cancer susceptibility. Hence, this study evaluated the association between CNV and non-small cell lung cancer (NSCLC) in the peripheral blood.
Methods:
Blood samples of 150 patients with NSCLC and 150 normal controls were obtained from a bioresource center (Seoul, Korea). Through an epigenome-wide analysis using the MethylationEPIC BeadChip method, we extracted CNVs by using an SVS8 software-supplied multivariate method. We compared CNV frequencies between the NSCLC and controls, and then performed stratification analyses according to smoking status.
Results:
We acquired 979 CNVs, with 582 and 967 copy-number gains and losses, respectively. We identified five nominally significant associations (ACOT1, NAA60, GSDMD, HLA-DPA1, and SLC35B3 genes). Among the current smokers, the NSCLC group had more CNV losses and gains at the GSDMD gene in chromosome 8 (P=0.02) and at the ACOT1 gene in chromosome 14 (P=0.03) than the control group. It also had more CNV losses at the NAA60 gene in chromosome 16 (P=0.03) among non-smokers. In the NSCLC group, current smokers had more CNV gains and losses at the ACOT1 gene in chromosome 14 (P=0.003) and at HLA-DPA1 gene in chromosome 6 (P=0.02), respectively, than non-smokers.
Conclusion:
Five nominally significant associations were found between the NSCLC and CNVs. CNVs are associated with the mechanism of lung cancer development. However, the role of CNVs in lung cancer development needs further investigation.
Introduction
Lung cancer is the main cause of cancer-related deaths worldwide. 1 In Korea, lung cancer accounts for 34 deaths per 100,000 people, making it the country’s leading cause of cancer deaths. 2 Although many prognostic factors have been identified, 3 the biomarker associated with lung cancer development remains unclear. Hence, this biomarker needs to be determined.
Single-nucleotide polymorphism (SNP) is a type of mutation in nucleic acids whereby the nucleotide polymer modifies only one base. Copy-number variation (CNV) is a structural variation in regions where copy-number differences are observed between two or more genomes. 4 As with SNP, the CNVs of genomic segments contribute considerably to the submicroscopic genomic diversity in humans. 5 Understanding the relationship between genetic changes, such as SNP or CNV, and biological functions on the genome scale provides fundamental new insights into human pathophysiology and association with diseases.6, 7 Moreover, further evaluating the functional mechanism of SNP or CNV in patients with lung cancer may identify biomarkers.
CNVs are unexpectedly frequent, dynamic, and complex forms of genetic diversity that could possibly gain or lose genomic DNA. 8 CNVs contribute to disease development.9, 10 They are increasingly known as an alternative source of genetic variation that may affect cancer-associated genes or pathways and increase the cancer risk. 9 The association of lung cancer incidence with CNV, 11 a genetic diversity that varies from person to person, has been extensively investigated. However, to date, the meaningful association between CNV and lung cancer has remained poorly known; knowing their association is necessary to understand lung cancer susceptibility and CNVs.
To evaluate the relationship between CNVs and lung cancer, we conducted a genome-wide association analysis in the peripheral blood of patients with non-small cell lung cancer (NSCLC) and normal controls, and analyzed the association between CNVs and NSCLC.
Materials and methods
The methods for subjects, preparation of blood samples, preparation of genomic DNA and DNA methylation profiling, and epigenome-wide association study were conducted on the basis of a previous study. 12 Study subjects are briefly described below, as well as the methods for CNV identification and real-time quantitative polymerase chain reaction (qPCR) for CNV validation.
Study subjects
This study was conducted using the blood samples of 150 patients with NSCLC and 150 normal controls who underwent medical checkups between 2012 and 2014. These blood samples were obtained from a bioresource center (Seoul, Korea). Frequency matching by age, sex, and smoking status was performed between the NSCLC and control groups. Smoking status was determined using a questionnaire. A past smoker was defined in this study as one who stopped for more than 1 year, whereas a non-smoker was defined as one who smoked with less than one pack throughout their lifetime. All subjects gave appropriate informed consents, and the Institutional Review Board of the Asan Medical Center (Seoul, South Korea) approved this study (IRB no. AMC IRB 2011-0883).
DNA methylation profiling and CNV identification
The epigenome-wide methylation profiles were obtained using the MethylationEPIC BeadChip kit, which covers 850,000 cytosine-phosphate-guanine (CpG) sites. 12 We preprocessed the methylation data through background correction, adjustment of probe type differences, batch effect removal, and probe exclusion. To normalize the probe intensity of the entire 850K CpG site, we utilized the R software EnrichmentBrowser. Thereafter, we extracted the CNVs by using the multivariate method supplied by the SVS8 software. We found a region with three or more markers showing the same CNV pattern in succession. Permutation (2000 times) of the P-value of less than 0.01 indicated CNV.
After merging all the segments, a logistic regression model was used with the response variable of NSCLC status and the predictor variable of CNV frequencies to identify CNVs in association with NSCLC. Covariates in the statistical models were age, gender, smoking (pack-years). Furthermore, stratification analyses were performed to find CNVs associated with smoking status. In each group according to smoking status, CNVs associated with NSCLC were analyzed. Additionally, in each group of NSCLC and control, CNVs associated with smoking were analyzed. Significant correlation was defined as when the P value for the correlation test was < 0.05.
Real-time qPCR for CNV validation
We performed a direct qPCR in selected subjects by using the estimated copy-number from the visual examination for CNV validation. To run qPCR, we designed a specific amplification primer set (Assay_ID Hs00682250_cn, Hs04343438_cn, and Hs03873630_cn; Thermo Fisher Scientific, South San Francisco, CA, USA) to validate the CNV existence within chr6:8435314-8436141, chr8:144635308-144636026, and chr14:74004122-74004683, respectively. For copy-number determination analysis, we employed the ABI Prism 7900 sequence detection system. The RNase P gene was coamplified with the marker, which was then used as an internal standard. Amplification reactions (10 µL) were performed using 10 ng of template DNA, 2Ⅹ TaqMan® Universal Master Mix buffer (Thermo Fisher Scientific), 900 nM of each primer, and 250 nM of each fluorogenic probe. Moreover, we initiated thermal cycling by 50°C incubation for 2 min, followed by 95°C incubation for 10 min and 40 cycles of 95°C for 15 sec and 60°C for 1 min. The first three replicate reactions were performed for similar primer pairs, and each copy-number for an individual was calculated by CopyCaller v1.0 (Thermo Fisher Scientific) using the comparative CT method. Then, the extracted CNVs were examined for CNV validation with a P-value < 0.05.
The Gasdermin D (GSDMD) gene in chromosome 8 and the ACOT1 gene in chromosome 14 obtained the same result.
Results
Study characterization
Table 1 describes the baseline characteristics of the NSCLC and control groups. In all subjects, 86% were males. The mean age was 56.1 years in the NSCLC group and 55.6 years in the control group. In both groups, 48 current smokers, 30 past smokers, and 72 non-smokers were known. According to the histological classification of the NSCLC group, 108 and 42 patients had adenocarcinoma and squamous cell carcinoma, respectively. Among the 108 patients with adenocarcinoma, 70 had mutant EGFR (59.3%). In terms of cancer staging, 46 patients were diagnosed as stage I, 14 as stage II, 31 as stage III, and 59 as stage IV.
Baseline characteristics of patients with lung cancer and matched control subjects.
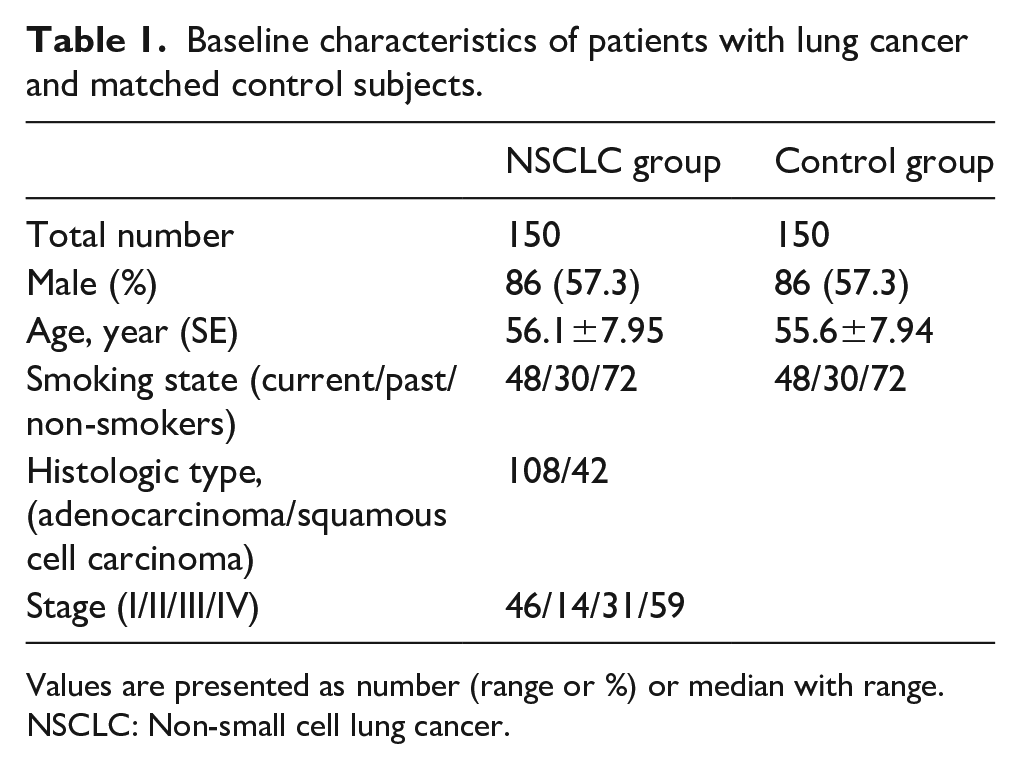
Values are presented as number (range or %) or median with range.
NSCLC: Non-small cell lung cancer.
Genome-wide CNV analysis
We recorded 979 CNVs, with a copy-number gain of 582 and a copy-number loss of 967 (Figure 1). By sample, the average number of CNVs was 297 CNVs, with 92.3 for copy gain, and twice as many for copy loss. Figure 2 presents the average number of loss and gain per chromosome per sample.
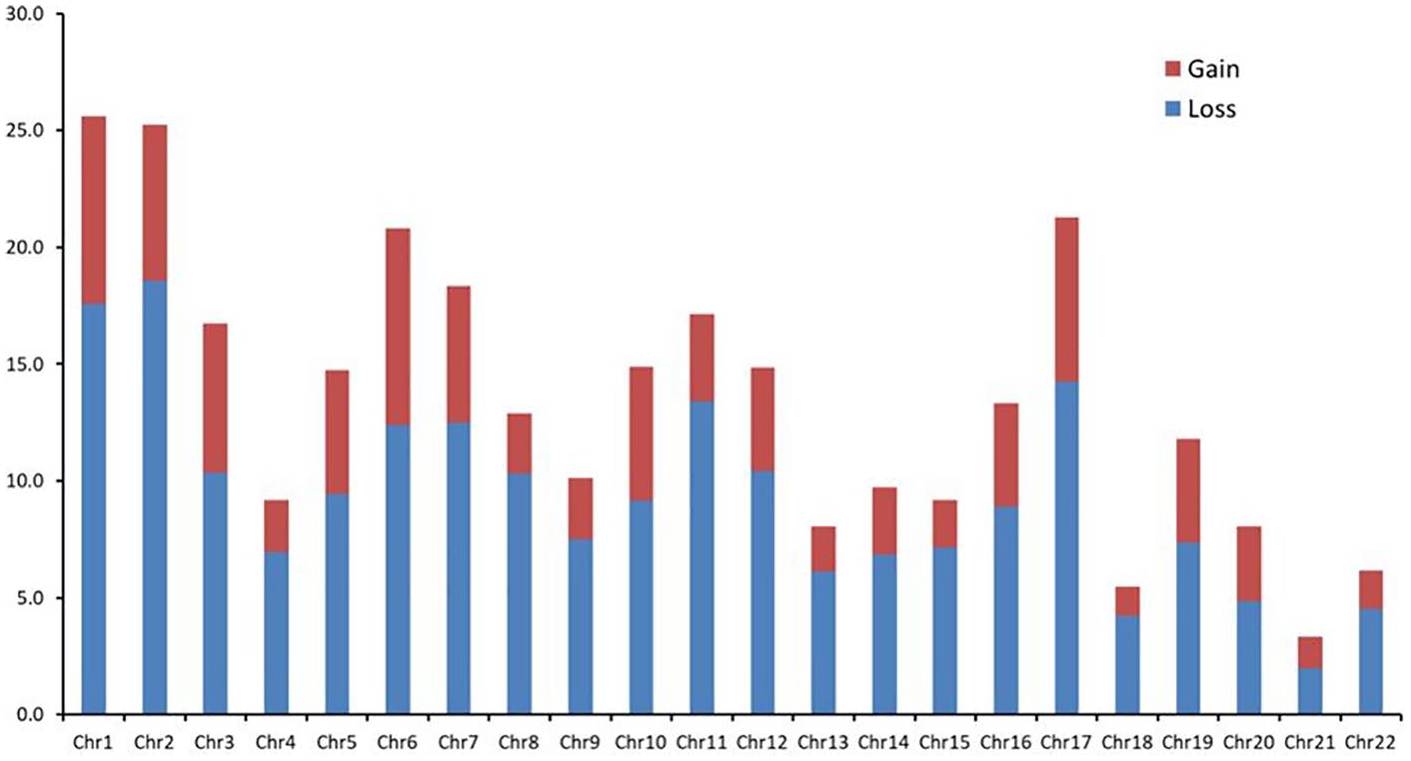
Copy number variation (CNV) discovery summary.
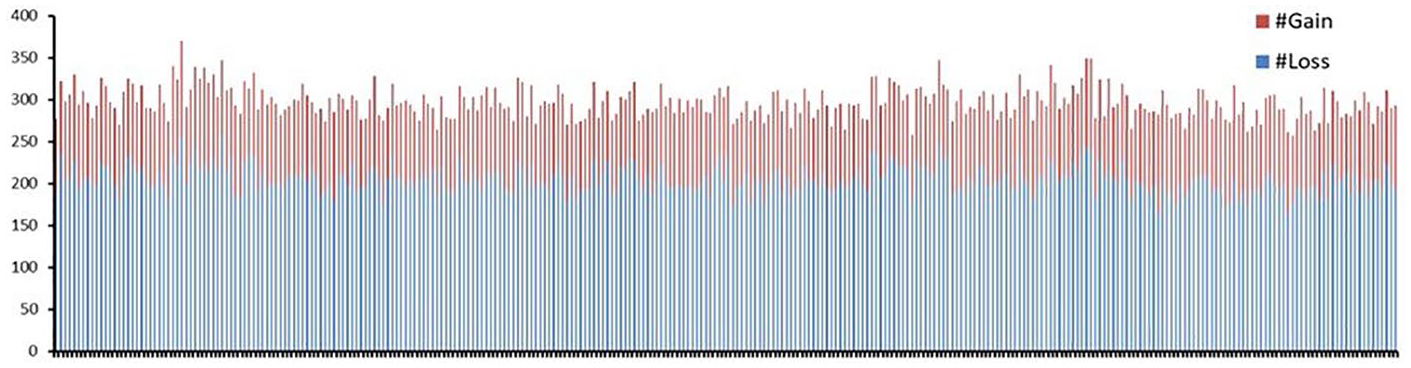
By sample, copy number variation (CNV) discovery summary.
The CNV frequency was not significantly different between the NSCLC and control groups. Among the current smokers (NSCLC group vs. control group = 48 vs. 48), CNV loss and gain of the NSCLC group were more commonly observed than those of the control group at the GSDMD gene in chromosome 8 (P=0.02) and at the ACOT1 gene in chromosome 14 (P=0.03), respectively. Among the non-smokers, the CNV loss of the NSCLC group was observed more frequently than that of the control group at the NAA60 gene in chromosome 16 (P=0.03).
In the NSCLC group (current smokers vs. non-smokers = 48 vs. 72), the CNV gain of the current-smoker group was observed more frequently at the ACOT1 gene in chromosome 14 (P= 0.003), whereas the CNV loss was observed more frequently at the HLA-DPA1 gene in chromosome 6 (P=0.02). Meanwhile, in the control group (current smokers vs. non-smokers = 48 vs. 72), the CNV loss of the current-smoker group was detected more frequently at the SLC35B3 gene (P=0.04).
Figure 3 shows the Manhattan plot of CNVs between the NSCLC and normal control groups. No significant difference of the CNV frequency was found between the all NSCLC and control groups (Bonferroni Correction, P-value < 5.1×10-5).

Manhattan plot of copy number variations (CNVs) between non-small cell lung cancer and control group.
Discussion
This study conducted an epigenome-wide association analysis using the peripheral blood of the NSCLC and normal control groups and analyzed the association between CNV and NSCLC. Using the 979 CNVs between the NSCLC and normal control groups from the epigenome-wide association analysis, we found five nominally significant associations in stratified analyses (P<0.05). Hence, CNVs may be associated with the mechanism of lung cancer development.
The association between lung cancer and CNVs has been identified in several studies. Qiu et al. 13 reported that a genome-wide CNV pattern was associated with a classification signature for NSCLC in tissue samples. They investigated the genome-wide CNV differences between two tumor types (adenocarcinoma and squamous cell carcinoma) and the corresponding nonmalignant tissues; they found that the CNV differences distinguished the two NSCLC subtypes. Thus, a CNV signature might be used as an adjunct test for the routine histopathologic classification of NSCLC. Given that associations between tumorigenesis and chromosomal instability have been widely reported, 14 Cai, et al. 15 found that methylome alterations are directly linked with tumor mutational burden (TMB) in NSCLCs. They checked CNVs and DNA methylation as markers for chromosomal instability in high- and low-TMB lung cancers and revealed that high-TMB NSCLCs had more DNA methylation aberrance and CNVs; thus, structural variant regions are associated with lung cancer. Considering that TMB is reportedly considered as a biomarker to check treatment effects for PD-1 blockade, 16 TMB-related DNA methylation changes or CNVs could also be expected as biomarkers for NSCLC. For example, a hypermethylated HOX gene was associated with NSCLC.17,18 However, the function of the HOX gene expression, which is related to lung cancer development, remains unknown. In addition, the genetic changes caused by CNV and DNA methylation still need a thorough investigation.
In the stratified analyses, five nominally significant associations were identified (ACOT1, NAA60, GSDMD, HLA-DPA1, and SLC35B3 genes). In the current-smoker group (NSCLC group vs. control group = 48 vs. 48), the NSCLC group had more CNV losses at the GSDMD gene in chromosome 8 than the control group (P=0.02). The GSDMD gene is a human homologation of Gasdermin, which is primarily associated with cancer of the upper gastrointestinal tract. 19 The GSDM family, which is composed of GSDMB, GSDMC, and GSDMD, is involved in regulating epithelial apoptosis. 20 GSDMD genes are related to the cancer of the upper gastrointestinal tract by epithelial cell proliferation and/or differentiation including the apoptosis process, which can affect tumorigenesis in many organs, including the lungs. 21 Moreover, in the NSCLC group (current smokers vs. non-smokers = 48 vs. 72), the current-smoker group had more CNV losses at the HLA-DPA1 gene in chromosome 6 than the non-smoker group (P=0.02). The expression of the HLA-DPA1 gene is deregulated in the Myc/VEGF mouse tumor metastatic signature and in the lung metastasis signature from primary human breast cancer. 22 In addition, this gene is downregulated in brain metastasis site from lung cancer 23 ; thus, the HLA-DPA1 gene is associated with lung cancer and migrates to other organs. Meanwhile, in the control group (current smoker vs. non-smoker = 48 vs. 72), the current-smoker group had more CNV losses at the SLC35B3 gene than the non-smoker group (P=0.04). The SLC35B3 gene encodes 3′-phosphoadenosine 5′-sulfate transporter protein, which is related to the synthesis of sulfated proteoglycan and glycoproteins. 24 The SLC35 gene family is related to nucleotide sugar transporters, which are associated with neoplasm metastasis, cellular immunity, and morphological characteristics. 25 For instance, downregulation of the SLC35F2 expression can attenuate the proliferation, migration, and invasion capacities of H1299 cells, suggesting that SLC35F2 may be a potential oncogene of lung cancer. 26 The SLC35B3 gene is a member of SLC35 gene; therefore, this gene might also be a potential oncogene of lung cancer. The GSDMD, HLA-DPA1, and SLC35B3 genes have been associated with lung cancer and other cancers; thus, changes in these genes may be related to lung cancer development.
Compared with other studies, which mostly used the CNV of tissue samples,13 -15 our study used the CNV of the peripheral blood in evaluating its association with NSCLC, implying the study’s particular point of attention. Using blood samples is more advantageous than tissue samples because they are less invasive and more cost effective, and could have potential for the development of useful biomarkers.
However, this study has several limitations. First, it has a limited number of subjects. We only tested 150 people with NSCLC and 150 normal controls. Further study with a large sample size could provide more meaningful results. Nonetheless, our study could become more useful for future validation study. Second, the ability to assess CNV has been limited by the lack of techniques for accurately measuring the copy-number level of each CNV in each individual. Exploring CNV is limited by low sensitivity and accuracy. 27 Further research with advanced techniques for detecting CNV and related genes may help to determine the biomarker for lung cancer development.
In conclusion, we identified five nominally significant associations between NSCLC and CNVs from an epigenome-wide analysis on peripheral blood samples. This study shows that CNVs may be associated with the mechanism of lung cancer development. However, further research is needed to determine the role of CNV in this cancer development.
Footnotes
Author contributions
Conceptualization: Y Heo, WJ Kim, Y Hong
Methodology: Y Heo, HS Cheong, WJ Kim, Y Hong
Provision of study materials or patients: WJ Kim, HS Cheong, Y Hong
Collection and assembly of data: HS Cheong, WJ Kim, Y Hong
Data analysis and interpretation: Y Heo, HS Cheong, WJ Kim, Y Hong
Manuscript writing: Y Heo, Y Hong
Final approval of manuscript: Y Heo, J Heo, SS Han, HS Cheong, WJ Kim, Y Hong
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by 2018 Research Grant from Kangwon National University and 2018 Research Grant from Kangwon National University Hospital.
Notation of prior abstract publication/presentation
The abstract of this paper was presented at the 2019 European Respiratory Society (ERS) Congress, which was held 28 September to 2 October at the IFEMA Exhibition Centre, Madrid, Spain.