
Abstract
The identification of driver mutations in epidermal growth factor receptor, anaplastic lymphoma kinase, the BRAF and ROS1 genes and subsequent successful clinical development of kinase inhibitors not only significantly improves clinical outcomes but also facilitates the discovery of other novel driver mutations in non-small cell lung cancer. These driver mutations can be categorized into mutations in or near the kinase domain, gene amplification or fusion. In this review, BRAF V600E, EGFR and HER-2 exon 20 mutation, FGFR1–4, K-RAS, MET, neuregulin-1, NRTK, PI3K/AKT/mTOR, RET and ROS1 gene aberration and their therapeutics will be discussed.
Introduction
Chemotherapy was the systemic therapy option in patients with treatment-naïve and pretreated, advanced or metastatic non-small cell lung (mNSCLC) until the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs), gefitinib, erlotinib and afatinib, were shown to improve overall response rate (ORR), median progression-free survival (mPFS), safety and tolerability when compared with platinum-based chemotherapy in treatment-naïve patients with mNSCLC who harboured activating EGFR mutations (deletion in exon 19 and L858R point mutation in exon 21).1–8 Afatinib and dacomitinib are second-generation, irreversible EGFR-TKIs that bind covalently to both wild-type (WT) and mutated (EGFRm+), and have shown improved mPFS when compared with gefitinib in patients who were treatment-naive, EGFRm+ mNSCLC. Furthermore, dacomitinib demonstrated an improvement in median overall survival proceeding (mOS) in the population that did not have brain metastases.9,10 Osimertinib, a third-generation EGFR-TKI, which selectively inhibits EGFR-activating and exon 20 T790M-resistant mutations, is also reported to have superior ORR, mPFS and tolerability over gefitinib or erlotinib, in the patient population with or without brain metastases. 11 In a press release from August 2019, osimertinib was reported to have a clinically meaningful improvement in mOS over gefitinib. The result was presented to the European Society of Medical Oncologists in September 2019.
Anaplastic lymphoma kinase translocation (ALK) was first identified in NSCLC by Soda and colleagues, 12 which led to rapid clinical development of a number of ALK inhibitors (ALKi). Crizotinib was the first ALKi to demonstrate improvement in mPFS, tolerability and mOS over chemotherapy and to receive regulatory approval in both treatment-naïve and pretreated mNSCLC with ALK translocation.13–15 To date, ceritinib,16,17 alectinib18–20 and brigatinib 21 have been shown to improve ORR and mPFS when compared with either chemotherapy or crizotinib in the ALKi-naïve or pretreated settings.
In addition, the combination of trametinib and dabrafenib in both treatment-naïve and previously treated patients with mutations in BRAF V600E and crizotinib in patients with ROS1 translocation have received regulatory approval throughout the world based on encouraging phase I–II data. Details of these studies are discussed below.
Advances in lung cancer therapeutics have led to the adaptation of comprehensive molecular profiling of known and novel driver mutations in mNSCLC, which may lead to the development of novel therapeutics that can further improve clinical outcomes.22–25 This review will provide an update on the clinical development of novel driver mutations, other than EGFR and ALK, in mNSCLC (Table 1, Figure 1).
Incidence, method of detection, and known secondary mutation in selected driver mutations.
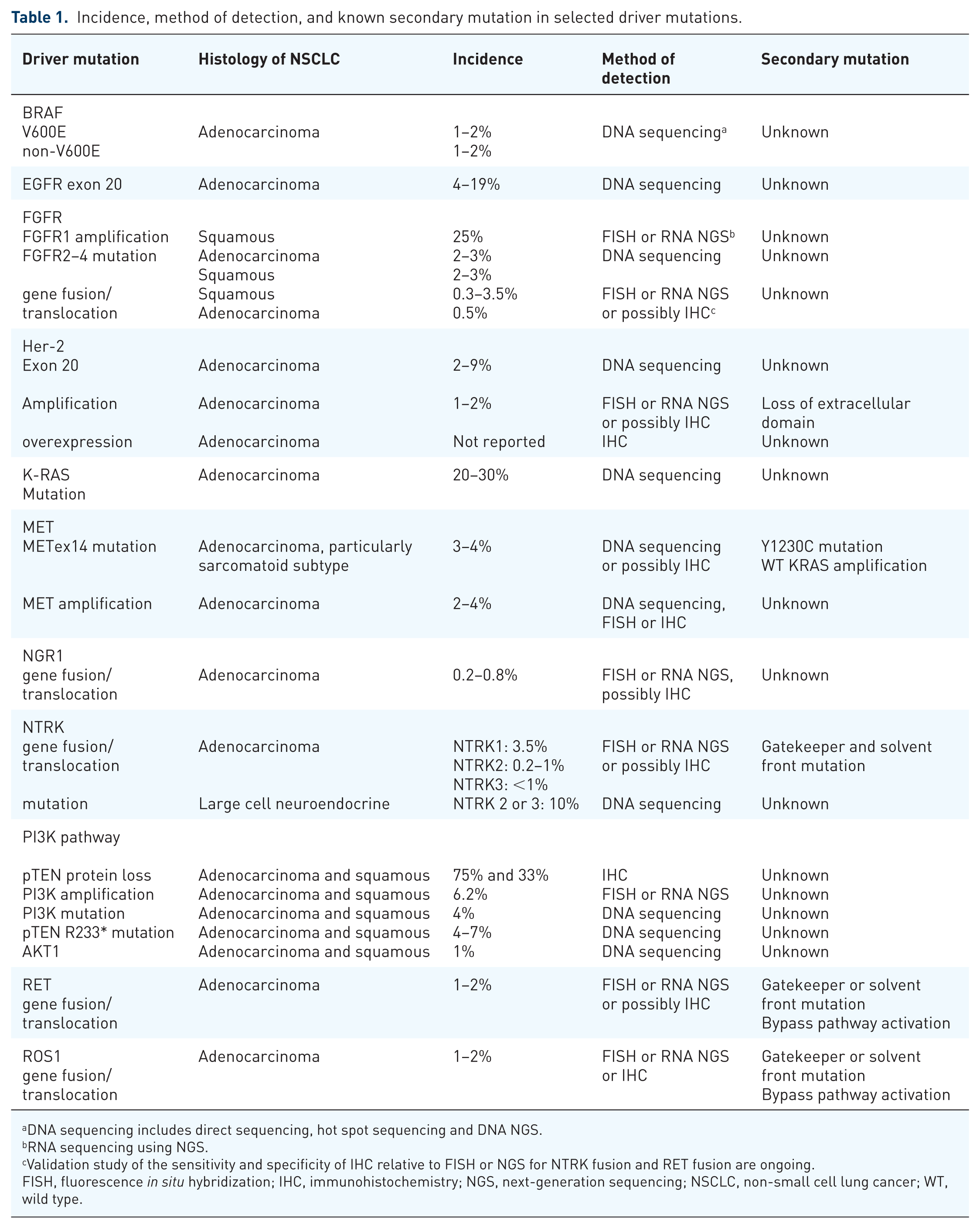
DNA sequencing includes direct sequencing, hot spot sequencing and DNA NGS.
RNA sequencing using NGS.
Validation study of the sensitivity and specificity of IHC relative to FISH or NGS for NTRK fusion and RET fusion are ongoing.
FISH, fluorescence in situ hybridization; IHC, immunohistochemistry; NGS, next-generation sequencing; NSCLC, non-small cell lung cancer; WT, wild type.
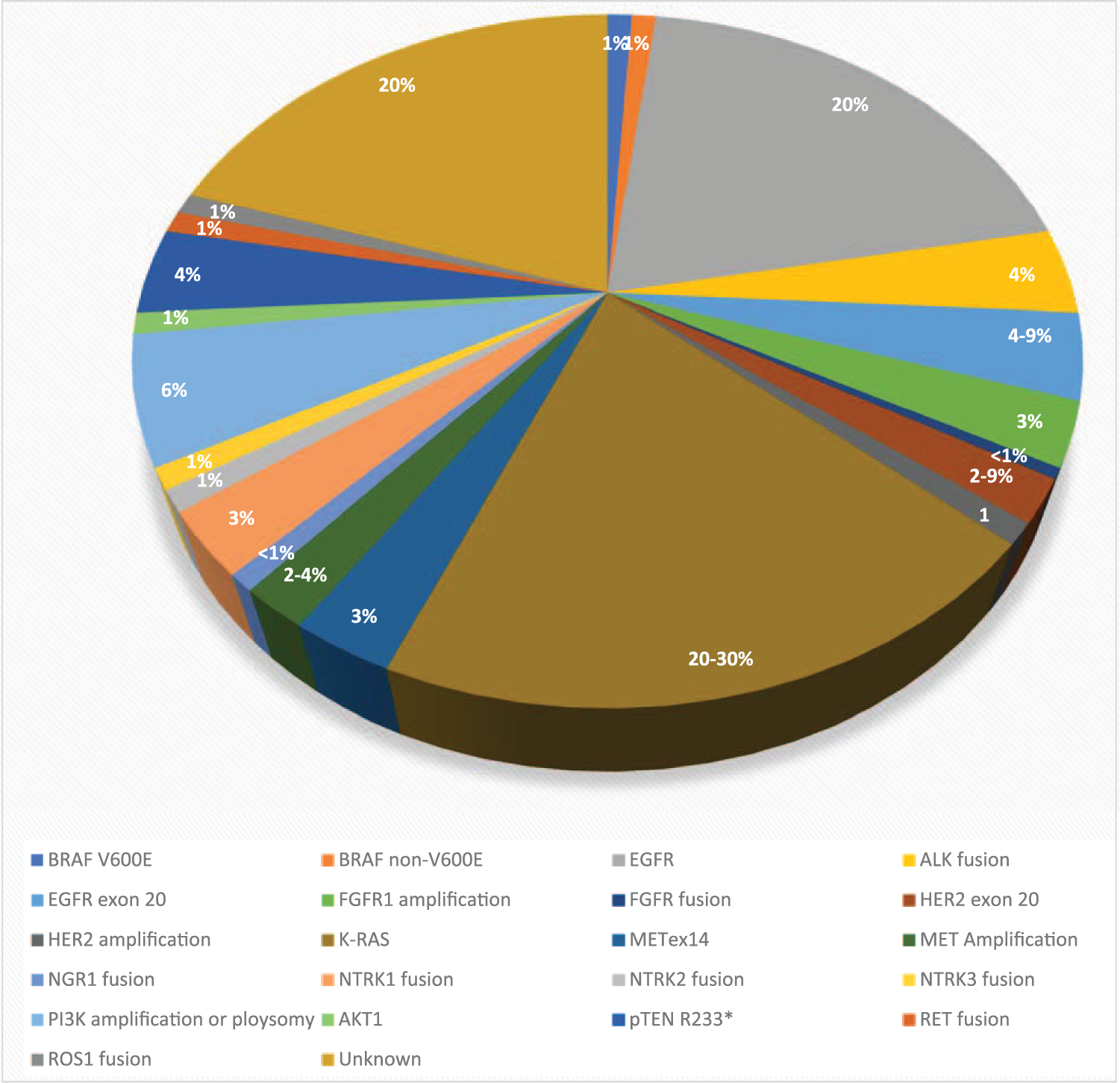
The distribution of various driver mutations in non-small cell lung cancer in Asian and White populations.
BRAF mutation
BRAF is an intracellular serine/threonine kinase that is activated by RAS, which, in turn, activates the downstream kinases, MEK and ERK (MAPK). BRAF mutation is identified in 50% of melanomas, 90% of which are of the subtype V600E. 26 BRAF mutation is detected in 2–5% of mNSCLC and can be classified into V600E and non-V600E subtypes. The former occurs in 1–2% of all mNSCLC. Multiple studies on clinical characteristics of BRAF-mutated NSCLC have been reported. Not only is there no distinguishing clinical characteristics, there is also no consistent information on the benefit of chemotherapy and prognosis of BRAF mutation, except that 20–30% of patients with the V600E subtype are nonsmokers and all patients with the non-V600E subtype are heavy smokers.27–34
Joshi and colleagues demonstrated that the treatment of a V600E NSCLC cell line with vemurafenib led to G1 arrest and an increase in Bcl-2-like protein 11 (BIM), followed by apoptosis. In addition, co-administration with trametinib abrogated the upregulation of AKT activity in both V600E and non-V600E BRAF-mutant lung cancer cell lines. Dual inhibition of BRAF and MEK was also shown to prevent paradoxical reactivation of MAPK, resulting in greater antitumour activity than each single agent alone. 35
Single-agent BRAF inhibition by either vemurafenib 36 and dabrafenib 37 demonstrated an ORR of 33–42% and mPFS of 5.5–7.3 months in previously treated BRAF-mutant NSCLC. Given the superior preclinical antitumour activity with concurrent BRAF and MEK inhibition, dabrafenib and trametinib were investigated in previously treated (n = 57) 38 and untreated (n = 36), 39 BRAF V600E mutation-positive mNSCLC. The respective ORRs determined by an independent review committee were 63.2% and 64%. The mPFS was 14.6 months in patients who were treatment-naïve and 9.7 months in those who were previously treated. The interim result of MyPathway study with vemurafenib in BRAF V600E and other patients who were BRAF positive, reported an ORR of 43% (n = 14) and 0% (n = 7), respectively. 40 A phase II study of dabrafenib and trametinib in V600E mNSCLC is currently underway in South Korea (ClinicalTrials.gov identifier: NCT03543306).
Novel paradox-breaking BRAF inhibitors are currently in early clinical development. These inhibitors may have improved efficacy and tolerability as they bind both BRAF and CRAF monomers, homodimers and heterodimers without activating the MAPK pathway.41–44 BGB-283 is a novel inhibitor to WT ARAF, BRAF, CRAF, BRAF V600E and EGFR. The recommended phase II dose (RDII) was 40 mg daily in patients with BRAF or KRAS/NRAS-mutated solid tumours. A partial response (PR) was observed in one patient who was BRAF or MEK inhibitor-naïve, KRAS-mutated mNSCLC. Dose-limiting toxicity (DLT) was caused by thrombocytopenia. The majority of patients experienced grade 1 or 2 fatigue (68%), anorexia (48%), constipation (42%), thrombocytopenia (39%), nausea (39%), vomiting (39%), acneiform rash (39%), hand–foot syndrome (35%), hypertension (35%), and dysphonia (32%). Grade 3 and 4 toxicities were thrombocytopenia (13%), fatigue (10%) and liver dysfunction (10%). 45 Janku and colleagues reported the phase I study of PLX8394 alone or in combination with cobistat, a CYP3A4 inhibitor, to increase the exposure of PLX8394, in refractory solid tumours. DLT for the combination was grade 3 elevation of aspartate transaminase (AST) and alanine transaminase (ALT). Other ⩾grade 3 treatment-related adverse events were diarrhoea (n = 1) and anorexia (n = 1). No BRAF inhibitor-related skin toxicity was reported. The combination of 900 mg of PLX8394 with 150 mg of cobistat was determined to be the RDII. PRs were reported in patients who were BRAF-inhibitor naïve, had V600E-positive gliomas or colorectal cancer. A phase II study in BRAF-mutated cancers is ongoing. 46 Thus far, there are no clinical data demonstrating antitumour activity of BRAF and MEK inhibitors in patients with non-V600E mutations despite preclinical efficacy.
EGFR exon 20 insertion
EGFR exon 20 insertion occurs in 4–10% of mNSCLC47,48 and in patients with adenocarcinoma who are nonsmokers, Asian and female. 49 This mutation involves the addition of 1–7 amino acids between residues 762 and 774 in the C-helix or regulatory domain of the kinase, leading to constitutive activation and dimerization of EGFR.50,51 Preclinical studies and clinical data demonstrated resistance of this heterogeneous mutation to gefitinib, erlotinib, afatinib and dacomatinib.50,52 Wu and colleagues reported a phase II trial of gefitinib in 16 evaluable patients with EGFR exon 20 mutation and only 4 patients had a response. 53 In a retrospective series of over 1000 patients with EGFR mutation, only 1 out of 25 patients with exon 20 mutation responded to EGFR-TKIs. 54 Naidoo and colleagues reported that the sensitivity of exon 20 mutation to EGFR-TKIs depended on the length and site of insertion. Those cases that harboured A703-Y704 insertion with FQEA amino acid residues were sensitive to EGFR-TKIs. 55 A phase II study of neratinib, a second-generation, irreversible, EGFR, HER2 and HER4 inhibitor, demonstrated an ORR of 3% and mPFS of <3 months. 56 In the post hoc analysis of LuxLung 2, 3 and 6 trials, response to afatinib was only observed in 8.7% of patients who harboured the exon 20 mutation. 57 The phase II study of osimertinib in patients who are positive for EGFR exon 20 mutation is ongoing (ClinicalTrials.gov identifier: NCT03414814).
Poziotinib is an orally available, quinazoline, irreversible inhibitor to EGFR and HER2. Exon 20 mutation leads to steric hinderance to binding of currently available EGFR-TKIs. Based on its small size, poziotinib slips into the ATP pocket and inhibits EGFR signalling in preclinical models. Poziotinib may be less potent to exon 20 mutation with addition of single amino acid than currently available EGFR-TKIs. 58 In 2018, the MD Anderson Cancer Center reported the preliminary result of a phase II study of poziotinib in 44 patients with EGFR exon 20 mutations. The ORR was 55% and the mPFS was 5.6 months. Grade 3 or higher toxicity occurred in 60% of patients, including skin rash (35%), diarrhoea (17.5%) and paronychia (9.5%). Overall, 45% of patients required dose reduction to 12 mg and 17.5% required further reduction to 8 mg. Response was reported irrespective of previous EGFR-TKI exposure. 59 Another phase II study of poziotinib in treatment-naïve and previously treated EGFR exon 20 or HER-2 exon 20 mutation is ongoing (ClinicalTrials.gov identifier: NCT03318933).
TAK788 (AP32788) is another orally available, irreversible EGFR and HER-2 tyrosine kinase inhibitor with broad spectrum preclinical antitumour activity to 14 EGFR and 6 HER2 mutant variants, including 8 variants of exon 20 mutation at an IC50 < 26 nM. 60 Doebele and colleagues presented the first report of the phase I study of TAK788 in advanced NSCLC. A total of 34 patients were treated at doses from 5–120 mg daily. Overall, 65% were female and 88% had two or more previous lines of systemic therapy. There were two patients that had dose-limiting pneumonitis at 80 mg and 120 mg. Common treatment-related toxicities included diarrhoea (47%), nausea (26%) and fatigue (21%). Grade 3 treatment-related toxicities were dyspnoea, anaemia, asthenia, dehydration, lung infection, pleural effusion, pneumonia and pneumonitis. All 3 PRs in the first 14 evaluable patients harboured EGFR exon 20 mutation. 61 Based on the encouraging preliminary antitumour activity, a phase III study of TAK788 or platinum/pemetrexed in treatment-naïve, EGFR exon 20 mutation mNSCLC is being planned. In addition, a phase I study of tarloxotinib in patients with EGFR or HER-2 exon 20 mutation is ongoing (ClinicalTrials.gov identifier: NCT038065841).
One of the most conceivable mechanisms of resistance to the current EGFR exon 20 TKIs is secondary mutation within the kinase or solvent front domain. It is highly possible that some patients who progress on one TKI will be sensitive to another. One of the important issues to investigate is the sequential use of these agents in this population.
FGFR pathway aberrations
The fibroblast growth factor (FGF) pathway consists of four receptors, FGFR1–4 and 18 ligands, including the hormone-like FGFs: FGF15, FGF19, FGF21 and FGF23, the canonical FGFs: FGF1, FGF4, FGF7, FGF8 and FGF9, and intracellular FGF11. Binding of the ligand to the corresponding receptor leads to activation of RAS/RAF/MAPK, PI3K/AKT/mTOR, STAT and PLC γ, which in turn leads to cell growth, proliferation, differentiation, migration and survival.62,63 Dysregulation of the FGF pathway can be a result of overexpression of the ligand or receptor, gene alteration from alternative splicing, amplification, point mutation or chromosomal rearrangement and loss of downregulation or degradation of the activated FGF pathway. 64 In squamous cell carcinoma of the lung, the FGFR pathway aberration includes FGFR1 amplification65–67 (25%), point mutation in FGFR2 (3%), FGFR3 (3%), FGFR4 (2%) 68 and chromosomal translocation such as FGFR3-TACC3 (3.5%), BAG4-FGFR1 (<1%) and FGFR2-KIAA1967 (0.3%). 63 FGFR2 or FGFR3 point mutation in either the extracellular domain or the kinase domain leads to constitutive dimerization and thus ligand-independent activation of the receptor. 68 FGF pathway dysregulation is less common in nonsquamous NSCLC, which includes FGFR1 amplification in 2–3% and FGFR3-TACC3 translocation in 0.5% of cases. 63 Chandrani and colleagues showed that S249C mutations in the extracellular domain and G691R mutations in the kinase domain of FGFR3 were sensitive to FGFR inhibition in preclinical models. 69
In the phase I study of BGJ398, a dose-expansion cohort of 36 patients with NSCLC with FGFR1 amplification were enrolled. Only 11.1% patients achieved a PR and 50% patients had disease controlled, 70 whereas there was no response reported with erdafitinib (JNJ-42756493) in this population. 71 Both studies did not explicitly define the criterion for FGFR1 amplification. In two phase II studies of AZD4547, only one PR was reported in an FGFR1-amplified mNSCLC, which was defined as FGFR1:CEP8 ⩾ 2.8.72,73
The LUNG-MAP SWOG S1400D was a randomized phase II study of AZD4547 or docetaxel in previously treated, squamous cell carcinoma patients with FGFR pathway activation. A total of 43 patients with FGFR1 amplification (56%), FGFR3 S249C mutation (9%), FGFR3 amplification (7%) and FGFR3 fusion (5%) were enrolled. The median age was 66.3 years and 30% were female. Out of the 25 patients with an evaluable response treated with AZD4547, only 1 with an S249C FGFR3 mutation had an unconfirmed PR (uPR) for a duration of 1.5 months. The mPFS for patients treated with AZD4547 was 2.7 months. Grade 3 or higher treatment-related toxicity was reported in five patients, including dyspnoea, fatigue, hyponatremia, lung infection and retinopathy. 74
Although the initial studies in this population are disappointing, additional studies are ongoing using different criteria for FGFR amplification. For example, the study of rogaratinib (BAY1163877, ClinicalTrials.gov identifier: NCT03762122) uses elevated FGFR1–3 mRNA levels, which may be more reflective of FGFR pathway activation. Other agents in clinical development include TAS-120 (ClinicalTrials.gov identifier: NCT02052778), pemigatinib (ClinicalTrials.gov identifier: NCT02393248) and others in malignancies with FGF/FGFR aberrations, including mNSCLC, are ongoing. It is hoped that these ongoing studies will help confirm whether FGFR gene aberration is a true driver mutation for NSCLC.
HER-2 exon 20 mutation and amplification
HER-2 exon 20 insertion or deletion occurs in 2–9% of adenocarcinomas of the lung, which accounts for 90% all HER-2 gene aberrations. The rest harbour HER-2 amplifications.75–80 Comparable with EGFR exon 20 mutation, the length of insertion or deletion is highly variable,78,79 which leads to constitutive kinase activation. 58 Most of the insertion occurs between codons 775 and 781, with either duplication or insertion of four amino acid residues (YVMA) or three base pair insertions with complex insertions or substitutions at codon 775. L755S and G776V/C point mutations have also been reported.78,80
HER-2 is first identified to be a potential target for mNSCLC based on a superior ORR (83% versus 41%) and mPFS (8.5 versus 7.0 months) to trastuzumab in combination with cisplatin/gemcitabine in patients with HER-2 2+ or 3+ by immunohistochemistry (IHC). 81
A phase II study of dacomitinib in mNSCLC with either HER-2 exon 20 mutation or amplification, reported an ORR of 12% and 0%, respectively. 82 The two phase II studies of afatinib in patients with exon 20 mutation reported disappointing results of disease control rate (DCR) at 71% including one out of seven uPRs, and a progression-free rate at 12-weeks of 54%, respectively.83,84 In a multinational, retrospective study of afatinib, 3 out of 27 patients with HER-2 exon 20 mutations responded. Overall, two of the responders harboured YVA or VAG insertions and had received previous trastuzumab and pertuzumab. 85 Heymach and colleagues reported a preliminary report of poziotinib with an ORR of 50% in 12 patients with HER-2 exon 20 mutation. 59
HER-2 antibodies, including trastuzumab, ado-trastuzumab emtansine and DS-8201a have been investigated in this population. Li and colleagues reported an ORR of 44% and mPFS of 5 months with ado-trastuzumab emtansine in 18 patients with either exon 20 insertion or point mutation at the extracellular or transmembrane domain. 86 Responses were independent of HER-2 mutation subtype. Another phase II study with ado-trastuzumab emtansine in patients who had HER-2 2+ or 3+ by IHC reported an ORR of 0% and 25% respectively. 87 However, Hotta and colleagues reported no patients with Her-2 2+ or 3+ by IHC and only one patient with an exon 20 mutation responded to aldo-trastuzumab emtansine. 88 The interim result of the MyPathway study with trastuzumab and pertuzumab, targeting HER-2 dimerization with other HER family receptors, reported an ORR of 13% and 19% in 16 patients with HER-2 amplification and 12 patients with mutated mNSCLC, respectively. 40
DS-8201a is a HER-2 targeted antibody drug conjugate with a topoisomerase I payload. An update of the phase I study in HER-2 IHC 1+ or higher or mutated mNSCLC was presented at the World Conference for Lung cancer in 2018. The median age was 58 years and patients had a median of three previous regimens. A total of 18 patients were evaluable for response: 8 out of 11 patients who were HER-2 mutated and 10 out of 17 patients with either HER-2 overexpression or mutation responded. Only 3 of 12 patients experienced ⩾grade 3 toxicity. The common toxicity included grade 1 or 2 anorexia (66.7%), nausea (58.3%), alopecia (41.7%) and fatigue (41.7%). 89
The EUHER2 study reported an ORR of 45.3% and mPFS of 6 months in HER-2 exon 20 mNSCLC treated with platinum-based chemotherapy. Patients treated with ado-trastuzumab emtansine or trastuzumab had an ORR of 51% and mPFS of 4.8 months. No response was observed in patients treated with TKIs. The mOS of the entire cohort was 24 months, which was comparable with that reported in prospective phase II studies. 90
To date, targeting the HER-2 exon 20 mutation has been more successful with HER-2 antibodies than TKIs, except for poziotinib. The benefit to the HER-2 antibody or antibody drug conjugate confined to either patients who were HER-2 over-expressed or amplified was modest, whereas efficacy with TKIs was a disappointment. Only with further elucidation of the biology of HER-2 amplification or overexpression can we improve the therapeutic benefit of HER-2 targeting agents. Comparable with EGFR exon 20, poziotinib is active in mNSCLC with exon 20 mutations, which is probably due to its small size and flexible structure, allowing it to bind to the kinase domain, while other HER-2 TKIs are too bulky to fit into the ATP pocket. 58 Tarloxotinib is currently being investigated in patients with exon 20 mutations in EGFR or HER-2 (ClinicalTrials.gov identifier: NCT03805841). Given the heterogeneous nature of exon 20 mutation, translational study is needed to understand which subgroup will not benefit from the TKIs, poziotinib or tarloxotinib, and HER-2 targeted antibodies. Resistance mechanisms have yet to be reported in NSCLC; however, it is conceivable that the loss of the extracellular domain in patients treated with HER-2 targeted antibodies can be a potential mechanism, as observed in breast cancer. Other mechanisms may include concurrent HER-2 amplification and bypass pathways, including EGFR amplification, activation or mutation.
K-RAS mutation
RAS is a guanosine triphosphatase gene superfamily, including K-RAS, H-RAS and N-RAS. It can be activated by either upstream tyrosine kinase or by point mutation at codons 12, 13, 14, and 60/61 of RAS. In turn, RAS activates the RAF–MAPK and PI3K pathways for cell proliferation, growth and anti-apoptosis.
In NSCLC, K-RAS is mutated in 20–30% of patients.22,91 Mutation at codon 12 is the most common: G12C (G→T, 43%), G12V (G→T, 18%) transversion and G12D (G→A, 11%) transition.91–93 K-RAS mutation occurs predominantly in patients who have adenocarcinoma, are non-Asian and smokers. The incidence seems to correlate with the number of cigarettes smoked, particularly for transversion.22,91 The meta-analysis by Meng and colleagues showed that early-stage K-RAS-mutated adenocarcinoma had a poorer outcome,94,95 especially in those patients with a codon 12 mutation 95 or transversion. 96 Therefore, K-RAS mutation is heterogeneous in biology and different therapeutic approaches will be required.
One of the most logical strategies to target K-RAS is to inhibit downstream MEK, which, in turn, inhibits ERK. The phase III study of docetaxel with or without selumetinib, an allosteric MEK1/2 inhibitor, in previously treated, advanced K-RAS mutation-positive NSCLC failed to improve both mPFS [3.9 versus 2.8 months, hazard ratio (HR) = 0.93] and mOS (8.7 versus 7.9 months, HR = 1.05) despite an improvement in mPFS in the randomized phase II study (HR = 1.14).97,98
RAS activation leads to G1/S cell cycle progression via cyclin-dependent kinase 2 and 4 (CDK2/4), which induces cyclin D1 and downregulates p27KIP. Cyclin D1 activates CDK4/6, which phosphorylates retinoblastoma protein, leading to G1/S transition. 99 Preclinical studies demonstrated that K-RAS-mutant NSCLC cell lines were sensitive to CDK4/6 inhibition.100,101 Based on the synthetic lethal relationship between K-RAS and CDK4/6, inhibition of CDK4/6 alone or in combination with inhibition of the RAF/MEK/ERK pathway may be a potential therapeutic strategy.
JUNIPER was a randomized phase III study of abemaciclib 200 mg twice daily, a CDK4/6 inhibitor, or erlotinib in patients with mNSCLC with K-RAS codon 12 or 13 mutations who had no more than two previous lines of therapy, including platinum-based chemotherapy. Despite an improvement in mPFS (3.6 versus 1.9 months, HR = 0.58, p < 0.001) and ORR (8.9% versus 2.7%, p = 0.01), mOS was not improved (7.4 versus 7.8 months, HR = 0.97, p = 0.77) with abemaciclib. Molecular subgroup analysis has yet to be presented. It will be of interest if there is any difference in benefit between codon 12 and 13, transversion and transition and cyclin D overexpression or amplification status. 102
Small molecular inhibitors to K-RAS G12C mutations have been shown to have significant antitumour activity in animal models. Phase I–II studies of AMG 510 (ClinicalTrials.gov identifier: NCT03600883) and MRTX849 (ClinicalTrials.gov identifier: NCT03785249) in this population have just commenced and the results are highly anticipated.
Sullivan and colleagues reported on the phase Ib study of trametinib and palbociclib in patients with solid tumours without selecting for K-RAS mutation. The RDII was once daily of 2 mg of trametinib and 75 mg of palbociclib or 1 mg of trametinib and 100 mg of palbociclib for 3 out of 4 weeks. DLTs observed at the RDII were grade 3 mucositis (1 in 7 patients) and grade 3 decrease in left ventricular ejection fraction (1 in 6 patients), respectively. Common adverse events were diarrhoea (67.9%), acneiform rash (64.3%) and fatigue (53.6%). Overall, two patients with WT BRAF or RAS melanoma and one patient with colorectal cancer and NRAS Q61K transversion responded. 103 Preliminary result of the phase Ib study of palbociclib and PD0325901 (MEKi) in RAS-mutant tumours reported the maximum administered dose to be 125 mg of palbociclib daily and 8 mg of PD-0325901 twice daily. A dose-limiting pneumonitis occurred at the 100 mg palbociclib and 4 mg PD-0325901 dose level. The most frequent drug-related toxicities were leukopenia (72%), anaemia (72%), thrombocytopenia (72%), neutropenia (64%), acneiform rash (64%), diarrhoea (52%), fatigue (44%), lower extremity oedema (32%), vomiting (28%), nausea (28%), oral mucositis (24%), increased AST (20%), increased creatinine (12%), epistaxis (12%) and blurred vision (12%). One PR and five cases of stable disease (SD) for more than 6 months were observed in K-RAS mutant NSCLC. K-RAS mutation with or without concomitant p53 or CDKN2A/B loss was predictive for response. 104 Other MEK and CDK4/6 inhibitor combinations are currently in early clinical development (ClinicalTrials.gov identifiers: NCT03170206 and NCT02703571). Based on the significant toxicity and modest clinical activity, these combinations are not ready for clinical use. Further optimization of the schedule and the dose of these combinations is needed to reduce toxicity and improve efficacy.
Even though K-RAS G12C-specific inhibitors may have significant clinical benefit, there is still an unmet need for other K-RAS mutation subtypes. Only through continual preclinical and translational studies of non-G12C mutants, can successful clinical strategies be developed.
MET EX14 point mutation and amplification
MET and its ligand hepatocyte growth factor play a role in cell growth and development and epithelial mesenchymal transition in cancer cells, through the activation of the downstream RAS/RAF/MAPK, Pi3K/AKT/mTOR, WNT/β-catenin and STAT pathways (Table 2).
Comparative toxicity and ORR of selected MET inhibitor.

ALT, alanine transaminase; AST, aspartate transaminase; CNS, central nervous system; GCN, gene copy number; GGT,
MET point mutation occurs in 3–4% of mNSCLC, with the most common being exon 14 skip mutations (METex14), which occurs in 2–3% NSCLC. Smokers, who are older than 70 years and with a sarcomatoid histology more commonly harbour this mutation. Other reports found METex14 mutation in acinar or solid subtypes of adenocarcinoma. Furthermore, stage I–IIIA METex14-positive NSCLC is more likely to recur. For those patients who had recurrence, the OS was comparable with that in ALK-positive mNSCLC. 111 MET exon14 is located in the juxtamembrane domain of the receptor and is involved in degradation by ubiquitin ligase, Cbl. Thus, METex14 leads to an increase in MET expression and activity.111–115 METex14 may have concurrent MET amplification, defined as a MET:CEP7 ratio ⩾5.0 and MET overexpression.112,114,115 Both METex14 (HR = 2.156, p = 0.026) and amplification (HR = 3.444, p = 0.007) are independent negative prognostic factors in treatment-naïve mNSCLC. 116
Crizotinib, an ALK, ROS1, MET inhibitor, was the first TKI to demonstrate clinical activity in METex14 mNSCLC. 117 Drilon and colleagues then reported 5 PRs and 5 uPRs for crizotinib in 15 patients who were evaluable for response with METex14 mutation. The mPFS was not reached in the 2016 update. 105 Responses to cabozantinib, a multikinase inhibitor to MET, VEGFR2 and RET, were reported in patients who were naïve to MET inhibitors or pretreated with crizotinib.118–120 Ou and colleagues reported that Y1230C mutations conferred resistance to crizotinib but sensitivity to other type II MET inhibitors, such as cabozantinib. 119 About 4% of METex14 was found to have concurrent K-RAS G12 mutation irrespective of previous crizotinib exposure. K-RAS mutation not only led to activation of the RAS/MEK/ERK pathway, but also enhanced MET expression, leading to resistance to MET inhibition. Dual inhibition of MET with either EGR/HER-2 or MEK inhibited growth in cell lines and xenograft models more significantly than MET inhibition alone. 121
The phase II study of capmatinib, a selective type I MET inhibitor, in patients with METex14 mNSCLC reported an ORR of 39%. Over 70% of patients had received 1–2 previous lines of therapy. Toxicity included peripheral oedema (49%), nausea (43%), vomiting (43%), an increase in creatinine (25%), dyspnoea (24%), anorexia (21%) and fatigue (21%), with the majority being grade 1–2. 108 Preliminary results from the phase II study of tepotinib, another selective type I MET inhibitor, reported an ORR of 60% in the first 13 evaluable patients. Tepotinib was well tolerated with mostly grade 1–2 treatment-related oedema and diarrhoea. Grade 3 toxicities were increased amylase, Gamma-glutamyl transpeptidase (GGT) and interstitial pneumonia. 109
An interim report of a phase II study of savolitinib in 41 mNSCLC patients with either sarcomatoid (n = 11) or other histology (n = 20) and METex14 mutation was presented at the American Association of Cancer Research Meeting in 2019. Of the 31 patients evaluable for response, the ORR was 54.8% for both cohorts and the mPFS and median duration of response (mDOR) were not reached at the time of the presentation. The treatment-related adverse events that occurred in ⩾10% patients were grade 1–2 nausea (41.5%), peripheral oedema (36.8%), elevation of AST/ALT (26.8%), vomiting (24.4%), anorexia (14.6%) and pyrexia (10%). Grade 3 toxicities observed were peripheral oedema (5%), elevation of AST/ALT (7.2%) and pyrexia (2.4%). Overall, five patients discontinued due to toxicity. 110 Many other MET inhibitors, such as sitravatinib, AMG337 and tivantinib, are in clinical development for this patient population.
MET amplification is not only a primary driver mutation but also a resistance mechanism to EGFR-TKIs. In this review, only de novo MET amplification will be discussed, which occurs in 2–4% of newly diagnosed mNSCLC.122–126 MET amplification is associated with poorer survival in both resected122,127 and metastatic NSCLC. 126 The definition of MET amplification remains to be clearly defined. The ORRs to crizotinib were 0%, 20% and 50% in patients with a low (⩾1.8 to ⩽2.2), intermediate (>2.2 to <5) and high (⩾5) MET:CEP7 ratio. 106 The responses were more common in females and ex-smokers. Patients with high MET gene copy number (GCNs) did not respond. This study was recently updated with a change in the cutoff for intermediate and high-level amplification. The ORRs were 33.3%, 14.3% and 40% for low (⩾1.8 to ⩽2.2), intermediate (>2.2 to <4) and high (⩾4) MET amplification. The corresponding mPFS rates were 1.8, 1.9 and 6.7 months. 107
Capmatinib is also being investigating in patients with MET amplification, defined as a MET:CEP7 ratio ⩾2, GCN ⩾ 4 to <6 or ⩾6 or IHC 2+ and 3+. In the preliminary report, 5 of 17 patients with IHC 3+, 2 of 7 with GCN ⩾ 4 and <6 and 9 of 12 with GCN ⩾ 6 had a PR. 128 Other MET inhibitors (METi) are being investigated in the MET-amplified setting, including tepotinib (ClinicalTrials.gov identifier: NCT028664992), cabozantinib (ClinicalTrials.gov identifier: NCT03911193) and sitravatinib (ClinicalTrials.gov identifier: NCT02219711).
To date, the definition of MET amplification is still unclear. Hopefully, this will be confirmed with ongoing trials using various definitions of MET amplification. The selective MET inhibitors seem to have an improved efficacy and toxicity profile over multitargeted or type II kinase inhibitors. Mechanisms of resistance, including Y1230C kinase domain mutation and K-RAS mutation, and hence corresponding therapeutic strategies will be elucidated from ongoing trials.
Neuregulin-1 fusion
The first neuregulin-1 (NRG1) fusion with CD74 in lung adenocarcinoma was reported in 2014. 129 Liu and colleagues reported NRG1 fusion in 0.2–0.8% of multiple tumour types, including cholangiocarcinoma, thyroid, ovarian, pancreas and breast cancers, NSCLC and sarcoma. In NSCLC, multiple 5′ fusion partners have been identified and the break point occurs either at exon 2 or 5 of NRG1. NRG1 fusion NSCLC is more commonly found in mucinous adenocarcinoma, females and nonsmokers with a median age of 70 years. Tp53 (50%) and pTEN loss are two common co-mutations.130,131 NRG1 is a natural ligand for HER-3, which leads to dimerization and activation of HER-2 and HER-3 followed by PI3K/AKT129,131. These findings support targeting NRG1 fusion with a pan-HER inhibitor. There are case reports of prolonged responses to anti-HER3 antibodies and afatinib.132–135 Tirunagaru and colleagues reported a novel, irreversible EGFR and HER-2 inhibitor, tarloxotinib, with potent antitumour activity in NRG fusion-positive cell lines. Responses are more durable than with afatinib. 136 Trials with pan-HER or EGFR/HER-2 TKIs, such as tarloxotinib and HER2 or 3-directed antibodies will be of interest in this population.
NTRK mutation and chromosomal fusion
There are three members in the neurotrophic tyrosine kinase (NTRK) family, which are involved in the development of different neuronal tissues. In adults, NTRK1 and NTRK3 are responsible for sensory function, while NTRK2 controls cortical function. Neurotrophin-3 (NT-3) and nerve growth factor are ligands for NTRK1, NT-3–5 and brain-derived neurotrophic factor for NTRK2 and NT-3 for NTRK3. Binding of the ligands to the respective NTRK activates downstream PI3K/AKT/mTOR, RAS/RAF/MAPK and PLC-γ, leading to proliferation, growth and survival of neurons in the peripheral and central nervous system. NTRK dysregulation can result from chromosomal fusion, point mutation, overexpression and alternative splicing.137–139 To date, alternative splicing of NTRK has not been identified in NSCLC. Gene fusion of NTRK1, NTRK2 and NTRK3 occurs in 3.5%, 140 0.2–1% 141 and <1% 142 of NSCLC, respectively. Marchetti and colleagues also reported point mutations in NTRK2 and NTRK3 in 10% of large-cell neuroendocrine carcinoma of the lung and none was documented in other NSCLC histological subtypes. All the NTRK somatic mutations are in the activation or catalytic domain, which lead to constitutive activation. Furthermore, NTRK mutations are only found in the neuroendocrine portion of large-cell neuroendocrine tumours. These findings suggest somatic NTRK2 or NTRK3 mutation may be an oncogenic driver for large-cell neuroendocrine tumour (Table 3). 143
Comparative toxicity and ORR of selected NTRK and ROS1 inhibitors.

ALT, alanine transaminase; AST, aspartate transaminase; CNS, central nervous system; GI, gastrointestinal; ORR, overall response rate.
Larotrectinib and entrectinib are the first NTRK inhibitors (NTRKi) in clinical development. Larotrectinib is a highly selective NRTKi. Drilon and colleagues reported the RDII to be 100 mg twice daily and an ORR of 75% by independent radiological review in the first 55 NTRK-positive solid tumours. Response was observed irrespective of age, histology, NTRK1–3 and 5′ fusion partner. 144 This study was recently updated to include a total of 122 patients. The ORR was 81% with a 17% complete response (CR) across all tumour types. Preliminary intracranial antitumour responses were observed. Both the mDOR and mPFS have not been reached after a median follow up of 7.4 months. 145 A total of 11 mNSCLC with NTRK fusions were enrolled. The median age was 52 years. Overall, eight patients had NTRK1 fusion and the reminder had NTRK3 fusion. In total, seven patients with mNSCLC were evaluable for response. Overall, one CR and four PRs with mDOR ⩾ 7 months were reported and no patients had primary progressive disease. 146 Larotrectinib was generally well tolerated with mostly grade 1 and 2 toxicity, including fatigue (36%), dizziness (29%), nausea (29%), constipation (27%), anaemia (27%), elevation of AST/ALT (26%), cough (26%), diarrhoea (23%), vomiting (23%), pyrexia (18%), dyspnoea (18%), headache (16%), myalgia (16%) and peripheral oedema (15%). The most common grade 3 or higher toxicity was anaemia, which was reported in 10% of patients. Only 9% of patients required a dose reduction and <1% terminated treatment, secondary to toxicity. 145
Entrectinib is a pan-TRK, ROS and ALK inhibitor. The combined results of the phase I studies, ALKA-372-001 and STARTRK1, determined 600 mg daily to be the RDII. Overall, three patients with NTRK fusions responded. Furthermore, five out of eight patients with NTRK fusion with brain metastases in the phase II study, STARTRK2, had a documented intracranial response. 147 Demetri and colleagues reported the combined analysis of 54 patients with refractory NTRK-fusion-positive solid tumours from the ALKA-372-001, STARTRK-I and STARTRK-2 trials. The ORR was 57.4% with CR at 27.3%. Comparable with larotrectinib, responses were observed across all histology and fusion partners. The mPFS and mDOR were 11.2 and 10.4 months respectively, and the mOS was 20.9 months. The enrolled patients with NTRK fusion and mNSCLC had a median age of 62.5 years and 60% had documented brain metastases. Males and females were equally likely to harbour an NTRK fusion. The ORR was 70% (7/10) and four out of six patients had intracranial responses. 148 Majority of the patients experienced grade 1 or 2 toxicity including dysgeusia (41.4%), fatigue (27.9%), dizziness (25.4%), constipation (23.7%), diarrhoea (22.8%), nausea (20.8%), weight gain (19.5%), paraesthesia (18.9%), increase in creatinine (15.2%), myalgia (15.2%), peripheral oedema (14.1%), vomiting (13.5%), arthralgia (12.4%), anaemia (12.1%), AST elevation (10.7%) and muscular weakness (7%). The most common grade 3 toxicity was weight gain (5.1%), anaemia (4.5%) and fatigue (2.8%). A total of 27% and 25% of patients required dose reduction and interruption, respectively, but only 4% of patients discontinued due to adverse events. 149
Both larotrectinib and entrectinib reported on-target neurological toxicity, such as dizziness, paraesthesia and myalgia. The pathophysiology of anaemia and peripheral oedema remain to be elucidated.
Unfortunately, resistance will occur in patients treated with larotrectinib and entrectinib due to mutation in the solvent front region of the kinase (for example G595R and G623R in NTRK1 and NTRK3) or the xDFG motif (for example G667C in NTRK1), leading to steric hinderance to current NTRKi binding in the kinase domain. 154 LOXO-195 and repotrectinib (TPX-0005) are currently in early clinical development to overcome these NTRK resistance mechanisms.
LOXO-195 is a low molecular weight macrocyclic inhibitor that binds to the kinase domain without steric hinderance from the bulky amino acid moiety, resulting from secondary NTRK mutation. Preliminary antitumour activity has been reported in a patient with colorectal cancer and infantile fibrosarcoma with a G595R mutation in the NTRK fusion. 154 The phase I/II study is currently ongoing (ClinicalTrials.gov identifier: NCT03215511). Repotrectinib is a novel ALK/ROS1/NTRK inhibitor designed to reside within the ATP binding pocket and avoid steric hindrance from solvent front substitutions. 151 Dose-limiting dizziness and dyspnoea occurred at doses ⩾160 mg daily. Other common toxicity, mostly grade 1 and 2, were dizziness (49%), dysgeusia (48%), paraesthesia (28%) and constipation (20%). A total of seven patients who were NTRK fusion-positive were evaluable for response and two patients who were NTRKi-pretreated responded. Overall, one of the responders had mammary analogue secretory carcinoma and G623E solvent front mutation in the NTRK3 fusion. 155 The phase II study of reprotectinib is currently ongoing (ClinicalTrials.gov identifier: NCT03093116). Thus far, proof-of-principle responses have been observed. The ongoing studies will confirm the clinical efficacy and will help to elucidate the resistance mechanism to these second-generation NTRKi.
PIK3CA/AKT/PTEN/MTOR pathway gene aberrations
Activation of the PI3K/AKT/mTOR pathway is central to cellular proliferation, growth and apoptosis. In normal cells, following activation of PIK3CA and downstream AKT/TORC1/p70S6K, activated p70S6K exerts a negative feedback on PIK3CA. In addition, pTEN acts as a negative regulator of this pathway via inhibition of AKT. Overall, three classes of PI3K have been identified. PIK3CA is a heterodimer of a p85 regulatory subunit and one of the four isoforms of p110 catalytic subunits (α, β, γ and δ). Each p110 subunit is preferentially expressed in both normal and malignant tissue. There are also three isoforms of AKT. Malignant transformation from this pathway results from activation or mutation of upstream pathways, such as tyrosine kinase or RAS and from aberration in PI3K, AKT, pTEN or RICTOR, which leads to tumour proliferation, growth and survival.156–158
In NSCLC, activating mutations in exon 9 helical (E542K, E545A/G/K/Q) and exon 20 kinase (H1047L/R/Y) domains of PIK3CA are detected in up to 4% of patients with mNSCLC.159–161 These mutations are more common in patients with chronic obstructive pulmonary disease (10.4% versus 1.7%, p = 0.015). 162 Amplification or polysomy of PIK3CA is the predominant molecular aberration in squamous cell carcinoma (33.1% versus 6.2% in adenocarcinoma). 161 All in all, PIK3CA pathway aberration is more commonly found in squamous cell carcinoma, which is also prognostic (mOS of 8.5 versus 10.1 months, p < 0.0001) and predictive of higher disease burden, intracranial metastases and genomic heterogeneity. 163 Activating E17K AKT1 mutations in exon 4 kinase domains occurs in 1% of all NSCLC, with the majority being squamous in histology.164–166 Loss of pTEN protein expression can result from allelic loss167,168 or promotor methylation,167,169 which occurs in up to 75% of NSCLC. Overall, 4–7% of NSCLC have activating R233* mutations in exon 7 or the C2 domain. This mutation controls the attachment of pTEN to the cell membrane, leading to activation of AKT.168,170,171 Loss of pTEN function leads to PIK3CAβ activation, which may predict sensitivity to PIK3Caβ, AKT 172 or mTOR 173 inhibition.
Buparlisib (BKM120), a pan-PI3K inhibitor, was tested in 30 patients with squamous and 30 patients with nonsquamous mNSCLC with PI3K/AKT/mTOR pathway activation. Approximately 50% of patients had previous chemotherapy. The progression-free rates at 12 weeks were 23.3% and 20%, respectively. The corresponding ORRs were 3.3% and 3.0% and mOS were 7.98 months and 7.20 months. PIK3CA mutation in exons 1, 5, 7, 9 and 20 were documented in 20.7% of squamous and nonsquamous NSCLC. Almost 28% of squamous and 4% of nonsquamous mNSCLC had pTEN mutations in exons 1–9. pTEN protein losses by IHC were found in 62.1% of squamous and 24% of nonsquamous mNSCLC. None of these aberrations correlated with outcome. The most common adverse events in ⩾25% of patients, regardless of relationship to study drug treatment, were nausea, vomiting, and diarrhoea (61.9%), asthenia/fatigue (49.2%), hyperglycaemia (39.7%), liver toxicity (31.7%) and hypersensitivity or rash (28.6%). Treatment-related grade 3 or 4 events were hyperglycaemia (23%), asthenia (6.7%) and fatigue (6.7%) in the squamous cohort, whereas elevation of AST/ALT (15.2%), hyperglycaemia (12%), asthenia (6%) and rash (6%) were reported in the nonsquamous cohort. Grade 3 psychiatric toxicity, described as confusion, anxiety or hallucination was reported in 10% of patients with squamous histology, whereas grade 3 depression or altered mood was reported in 6% of adenocarcinoma patients. A total of 13% patients with squamous NSCLC discontinued due to hyperglycaemia, whereas 21% of nonsquamous NSCLC discontinued due to dyspnoea, elevation in ALT or depression. The most common reason for discontinuation was toxicity in the squamous cohort as compared with disease progression in the nonsquamous cohort. 174 There seemed to be some difference in the toxicity profile between the two histologies, which may be related to the fact that patients with squamous NSCLC might have more comorbidity.
Preliminary results of taselisib, a selective PI3Kα inhibitor, in the SWOG 1400B study in previously treated, squamous NSCLC that harboured PIK3CA exon 9 or exon 20 mutations reported an ORR of 4%, mPFS of 2.8 months and mOS of 5.9 months. Further enrollment was halted after this interim analysis. Overall, five patients each experienced grade 3 hyperglycaemia or diarrhoea and three had grade 3 lymphopenia. 175
The initial results from the trials targeting the PI3K pathway have been disappointing from both efficacy and toxicity perspectives. Hyperglycaemia and gastrointestinal toxicity limit the dose intensity which may, in turn, limit continuous inhibition of the pathway and thus, antitumour activity. LoRusso suggested inhibition to specific PIK3CA or AKT isoforms may lead to compensatory activation of other isoforms, such as chronic lymphocytic leukaemia treated with PI3Kδ leading to activation of PI3Kα.176,177 Furthermore, inhibition of the PI3K/AKT pathway also leads to loss of AKT inhibition by p70S6K and paradoxical activation of the pathway.172,177
Until a better understanding of whether aberration in the PI3K/AKT/pTEN pathway is a driver mutation in mNSCLC and novel agents have improved toxicity profiles, successful clinical development of agents targeting this pathway is doubtful.
RET chromosomal fusion
Giant cell-derived neurotrophic factor is the ligand for RET, a tyrosine kinase receptor, and its activation leads to downstream activation of RAS/RAF/MAPK, PI3K/AKT/mTOR and PLC-γ, followed by cellular proliferation, migration and differentiation. In normal tissue, RET is responsible for renal and enteric nervous system development.178,179 RET is expressed in cells of neural crest origin, such as neurons, sympathetic and parasympathetic ganglia, testicular germ cells, urogenital tract, adrenal medulla and C cells in the thyroid (Table 4). 180
Comparative toxicity and ORR in selected RET inhibitors.

ALT, alanine transaminase; AST, aspartate transaminase; CNS, central nervous system; GI, gastrointestinal; ORR, overall response rate; PR, partial response; uPR, unconfirmed PR.
RET fusion occurs in 1–2% of patients with NSCLC who are younger than 60 years, have never smoked or are minimal smokers and is equally common in men and women. Multiple 5′ fusion partners have been identified with KIF5B being the most common. Most of RET fusion mNSCLC cases have either mixed or solid subtype adenocarcinoma and 30% demonstrate signet ring morphology.187–190
Currently, the multitarget kinase inhibitors, cabozantinib, vandetanib, lenvatinib, sitravatinib, alectinib and ponatinib, and the specific RET inhibitors, RXDX-105, BLU-667, LOXO-292, BOS172738, are in clinical development.
A phase II study of cabozantinib at 60 mg daily in 25 patients with previously untreated (25%) and treated (75%) mNSCLC with RET fusion had an ORR of 28%, mPFS of 5.5 months and mOS of almost 10 months. Grade 3 treatment-related adverse events were elevation of lipase in four patients and elevation of AST/ALT or thrombocytopenia and hypophosphatemia in two patients each. 181 Cabozantinib-treated patients in the Global RET Registry (GLORY) reported an ORR of 33%, mPFS of 3.6 months and mOS of 4.9 months. 191
The two phase II studies of vandetanib in heavily pretreated RET fusion NSCLC reported an ORR of 18–47%, mPFS of 4.5–4.7 months and mOS of 11.6 months. The grade 3 toxicities reported were hypertension, due to vascular endothelial growth factor receptor (VEGFR) inhibition, diarrhoea, rash, dry skin, prolongation of QTc interval and elevation of AST/ALT.192,193 The GLORY registry reported an ORR of 18%, mPFS of 2.9 months and mOS at 10.2 months in 11 patients treated with vandetainib. 191
Lenvatinib is a multitargeted kinase inhibitor to VEGFR1–3, FGFR1–4, PDGFRα, c-KIT and RET. It has received United States Food and Drug Administration and other regulatory agency approval for radioactive iodine-refractory, well-differentiated thyroid cancer, anti-angiogenic inhibitor pretreated renal cell carcinoma and untreated hepatocellular carcinoma. The ORR of lenvatinib at 14 mg daily in 25 patients with heavily pretreated, RET fusion-positive mNSCLC was 16% with mDOR of 16 weeks. The mPFS was 7.3 months and the mOS was not reached at the time of the report. Treatment-related adverse events occurred in 92% of patients with three being fatal. In addition, adverse event that required dose reduction, dose interruption and termination occurred in 64%, 76% and 20% patients, respectively. Common toxicities included hypertension, nausea, anorexia, diarrhoea, proteinuria and vomiting. 182 The efficacy of lenvatinib may not be justified by the toxicity.
Alectinib is an ALK and RET kinase inhibitor and has been found to have activity to V804L/M gatekeeper mutations in a KIF5B-RET fusion NSCLC mouse model. Lin and colleagues reported that two out of four patients with RET fusion who had previous RET inhibitors responded to alectinib. 194 A phase I/II study determined 450 mg twice daily to be the RDII. DLTs observed in 3 of 6 patients treated with 600 mg twice daily were grade 3 rash, elevation of AST, erythema multiforme, thromboembolic disease and elevation of creatine phosphokinase (CPK). No efficacy data have been presented to date. The phase II study at 450 mg twice daily is currently ongoing, which is higher than the 300 mg twice daily dose approved in Japan for ALK-positive mNSCLC.18,195 The B-FAST study is also evaluating alectinib in patients with RET fusion detected by blood-based next-generation sequencing (ClinicalTrials.gov identifier: NCT03178552).
RXDX-105 is a novel VEGFR1/2-sparing, multikinase inhibitor with antitumour activity in WT RET, KIF5B-, CCDC6- and NCOA4-RET fusion, WT or V600E mutated BRAF tumour xenografts. Drilon and colleagues reported a dose-escalating phase I study followed by dose expansion in RET fusion-positive mNSCLC, colorectal cancer, hepatocellular carcinoma, medullary thyroid cancer and other solid tumours. Only 11% of patients had no previous systemic therapy. The RDII was 275 mg daily in the fed state. A total of 39 patients who were RET fusion-positive with mNSCLC and 31 who were RET inhibitor-naïve were enrolled in the phase Ib portion. The most common fusion partners were KIF5B (65%), followed by CCDC6 (20%). The ORR in RET fusion-positive mNSCLC who were RET inhibitor-naïve was 19%. All the responses were reported in the patients who were non-KI5FB-RET-positive. A total of four DLTs were observed during the dose-escalating portion of the study: grade 3 rash at 200 mg daily, grade 3 fatigue and grade 3 diarrhoea at 275 mg and grade 3 hyperbilirubinaemia at 350 mg. Dose reduction was required in 28% of all patients enrolled with 31% at 275 mg daily. The most common adverse events that led to dose reduction were elevation of AST/ALT (9%) and cutaneous toxicity (6%). A total of 25 patients discontinued due to toxicity. The most common RXDX-105-related toxicity, included fatigue (26%), diarrhoea (24%), rash, maculopapular and nonmaculopapular, (25%), hypophosphatemia (18%), nausea (15%), elevated AST/ALT (14%), muscle spasms (13%), anorexia (11%) and vomiting (11%). Grade 3 or higher toxicities were nausea, diarrhoea, hypophosphatemia, rash and elevated AST/ALT in <10% of patients. Interestingly, three patients experienced cutaneous hypersensitivity consistent with drug rash with eosinophilia and systemic symptoms, which has also been observed in vemurafenib. 183 It is postulated that the rash may be related to BRAF inhibition. It is intriguing about the discrepancy of response in xenograft models and in patients with KIF5B-RET fusion NSCLC. Further exploration of the biology of the various fusion partners of RET will help to understand this observation. Overall, the antitumour activity was comparable with other multikinase RET inhibitors, such as cabozantinib, vandetanib and lenvatinib.
BLU-667 is a next-generation highly selective RET inhibitor with preclinical activity to the most common RET fusion, KIF5B-RET and CCDC6-RET, and activating mutation, C643W, M918T and V804L/M, and WT RET at sub-nanomolar concentrations. With VEGFR2 sparing, BLU-667 is anticipated not to have toxicities such as hypertension, thrombosis and haemorrhage. Proof of concept antitumour activity from the ongoing phase I study (ClinicalTrials.gov identifier: NCT03037385) has been reported in two patients with medullary thyroid cancer with RET mutation. In addition, 4 PRs and 1 uPR were reported in 11 patients with NSCLC. Most toxicity was grade 1, including constipation (23%), elevated AST/ALT (16%), diarrhoea (14%), fatigue, elevated creatinine, neutrophilia and hypertension (12% each). Overall, three patients experienced grade 3 toxicity: elevation in ALT, hypertension and tumour lysis syndrome.184,185
LOXO-292 is another next-generation RET inhibitor with preclinical antitumour activity to various RET 5′ fusion partners, including KIF5B and CCDC6. It demonstrated better response and survival than cabozantinib and ponatinib in xenograft or orthotopic models. LIBERETTO-1 is a dose-escalation phase I study of LOXO-292 in patients with solid tumours who have failed or were not candidates for standard therapy. A total of 82 patients were enrolled at doses from 20 mg to 240 mg once or twice daily. Patients had a median of three previous lines of therapy. A total of 45% of patients had no previous RET inhibitor. The most commonly fusion partners were KIF5B (60.6%), followed by CCDC6 (26.3%). DLTs were observed in four patients at 240 mg twice daily: grade 3 diarrhoea, elevation of AST/ALT, thrombocytopenia and tumour lysis syndrome. The RDII was determined to be 160 mg twice daily. The majority of the treatment-related toxicity were grade 1, including diarrhoea (23%), fatigue (22%), dry mouth (21%), constipation (20%), hypomagnesaemia (13%), cough (12%), headache (12%) and nausea (12%). The grade 3 treatment-related toxicities reported were diarrhoea (1%) and headache (1%). A total of 68% of the 38 patients with RET fusion-positive mNSCLC enrolled had a PR. A response for ⩾6 months was reported in 92% of the responders. Primary progression was only observed in two patients. Furthermore, all four patients with measurable intracranial metastases responded. Over 90% of patients had a decrease in circulating tumour RET fusion DNA. The study is currently enrolling patients with RET fusion-positive solid tumours with ⩾1 previous standard therapy, treatment-naïve RET fusion-positive solid tumours, RET-mutated medullary thyroid carcinoma with ⩾1 previous therapy, treatment-naïve RET-mutated medullary thyroid cancer and RET-altered solid tumours without measurable lesions or with RET fusion detected in circulating DNA (ClinicalTrials.gov identifier: NCT03157128). 186
To date, the antitumour activity of RET-specific inhibitors, LOXO292 and BLU-667, is encouraging with manageable toxicity. Tumour and plasma biopsy collected will help to understand the secondary resistance, which will likely include both gatekeeper and solvent mutations as well as bypass pathway activation such as EGFR and RAS.
ROS1 fusion
ROS1 is a kinase receptor that has significant structural homology to ALK. ROS1 fusion activates downstream RAS/RAF/MAPK, PI3K/AKT/mTOR and STAT-3, and leads to cell growth, proliferation and survival. Multiple 5′ fusion partners have been identified in NSCLC and other cancers, which may impact on the therapeutic benefit to ROS1 inhibitors. 196 ROS1 fusion is found in 1–2% of lung adenocarcinoma and these patients are usually younger with a median age of 48.9 years, nonsmokers, female, Asian and in the advanced setting.197–199 A total of 36% of patients with ROS1 fusions presented with brain metastases, which was similar to that reported in ALK and other driver-mutation-positive mNSCLC. 200 Ng and colleagues also reported a higher incidence of thromboembolic disease in patients with ROS1 (34.7%) than patients with ALK (22.3%), EGFR (13.7%) or K-RAS (18.4%) mutations. 201 Overall, patients with ROS1 may have a better outcome with mOS of 3 years in those who had received only standard chemotherapy and over 5 years in those with both chemotherapy and crizotinib (Table 3). 202
Crizotinib is the first TKI reported to have significant activity in ROS1 fusion mNSCLC. In this phase Ib/II study, the ORR was 72%, mPFS was 19.2 months and 1-year OS rate was 85% in the 50 patients with ROS1 fusion. A total of seven 5′ fusion partners were identified with two being novel, LIMA1 and MSN, which had no differential benefit to crizotinib. 203 Unfortunately, the brain was the first site of treatment failure in patients with ROS1 treated with crizotinib at even higher incidence than crizotinib-treated ALK+ mNSCLC (47% versus 33%). 200 The Korean investigators also reported an impressive ORR of 84%, mPFS of 19.3 months and mOS of 24 months for ceritinib in ROS1 inhibitor-naïve mNSCLC. Overall, five out of nine patients with documented intracranial metastases responded. Toxicity was not different from that in the patients who were ALK positive. A total of 15 patients underwent FISH, IHC and next-generation sequencing (NGS) for ROS1. A patient who was FISH+ with negative IHC and NGS progressed after 1 month, whereas three additional patients who were FISH+ who were negative by NGS or IHC had at least SD. 152 Thus far, there is no clear evidence whether FISH, NGS or IHC is the superior companion diagnostic or a combination of these diagnostic tests is required.
Resistance mechanism to patients with ROS1 treated with crizotinib includes solvent front mutation,204–209 G2032R, D2033N, and gatekeeper mutation, G2026N, and L2155S, 208 c-KIT mutation 210 and upregulation of RAS 211 or the EGFR pathway. 197 Next-generation ROS1 inhibitors to the secondary mutation are in clinical development, including for example, lorlatinib, entrectinib and repotrectinib.
Lorlatinib is a selective ALK/ROS1 inhibitor that has preclinical antitumour activity in G2032R and G2026M mutations. Lorlatinib is also shown to penetrate the blood–brain barrier. 206 Shaw and colleagues reported the phase I study of lorlatinib in ALK or ROS1-positive mNSCLC, with the RDII at 100 mg twice daily. Only one dose-limiting grade 2 neurocognitive change was observed. The most common toxicities were hypercholesterolaemia (72%), hypertriglyceridaemia (39%), peripheral neuropathy (39%), peripheral oedema (39%), speech changes (19%), cognitive disturbances (17%), elevation of lipase (17%), weight gain (17%), fatigue (15%), mood changes (15%), increase in amylase (13%), elevation of AST (13%), constipation (13%), tinnitus (13%), visual changes (13%) oedema (11%) and nausea (11%). Grade 3 or higher toxicities were hypercholesterolaemia, hypertriglyceridaemia, cognitive changes and elevation of amylase or AST, which each occurred in <10% of patients. A total of 12 patients with ROS1 were enrolled, out of which 7 had previous crizotinib. The preliminary ORR was 50% and the mDOR and mPFS were 12 and 7 months, respectively. Overall, 60% (3 of 5) patients had a response in their brain metastases. 153
A total of 53 patients with ROS1 fusion were treated with entrectinib in the STARTRK-2, STARTRK-1 and ALKA372-001 studies, with an ORR of 77.4% and mPFS of 19 months. The mPFS in patients with or without brain metastases was 13.6 and 26.3 months, respectively. Intracranial responses to entrectinib was observed in 55% of the 23 patients with brain metastases. 150
Repotrectinib has antitumour activity in G2032R and D2033N-resistant mutation ROS1 cell lines. Preliminary results of repotrectinib in 29 patients with ROS1 fusion mNSCLC reported an ORR of 11% and 71% in those with and without previous ROS1 inhibitors, respectively. A proof-of-principle response was reported in two patients with G2032R mutations and two additional patients had SD. A patient experienced an intracranial response. 155 Enrolment in patients with ROS1 is ongoing (ClinicalTrials.gov identifier: NCT03093116).
In additional to the evaluation of resistance mechanism to second-generation ROS1 inhibitors, translational research to evaluate the differential benefit of various fusion partners to ROS1 inhibitors will be important. Clinical investigation with concurrent inhibition of ROS1 and bypass resistance mechanisms, such as EGFR and RAS, is warranted. 212
Conclusion
Many novel driver mutations are or have been investigated in mNSCLC, with some successes and some failures. With better understanding of the biology of various subtypes of each driver mutation will help not only to choose the right inhibitor for the right patient, but also to elucidate the respective resistance mechanisms and their therapeutic strategies. These goals can only be achieved through serial tumour or plasma biopsy and multiplex molecular testing. In addition, the optimal sequence of the inhibitors for each driver mutation remains to be determined. It is not inconceivable that different molecular subtype of each driver mutation will require a different sequence of inhibitors. It is still not clear whether selective or multitargeted kinase inhibitors have superior antitumour activity; however, selective inhibitors seem to have more favourable toxicity profiles. Activation of bypass pathways represents an important resistance mechanism to driver mutation inhibitors. Further preclinical and clinical evaluation of concurrent inhibition of the bypass pathways and driver mutation is warranted. These efforts may both delay the emergence of resistance or further improve therapeutic benefits from an existing treatment strategy and overcome some secondary resistance.