
Abstract
Introduction
Non-alcoholic fatty liver disease (NAFLD) has emerged as the most prevalent chronic liver condition globally, with its severe form, non-alcoholic steatohepatitis (NASH), leading to liver failure, hepatocellular carcinoma (HCC), and mortality. 1 Recent studies estimate the global prevalence of NAFLD at approximately 24%, although the epidemiological data on NASH remain inconsistent due to variations in diagnostic methods and criteria.2,3 Notably, the rising incidence of obesity has propelled NAFLD to become a leading cause of cirrhosis and HCC in several countries. 4
The pathogenesis of NASH and HCC is complex, involving multiple factors including mitochondrial dysfunction, insulin resistance, oxidative stress, and lipid metabolism disorders. 5 Accumulating evidence suggests that inflammatory responses play a pivotal role in the development and progression of NAFLD. 6 Recent observational studies have revealed associations among circulating inflammatory protein levels and NAFLD progression. For instance, Duan et al. found in 2022 that serum levels of C-reactive protein (CRP), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) positively correlated with NAFLD severity. 7 Additionally, experimental research has demonstrated that elevated fatty acid levels in the local hepatic microenvironment reduce CD4 + T lymphocyte numbers, potentially contributing to impaired immune regulation during NAFLD progression to cirrhosis and HCC. 8 However, these observational studies face limitations in establishing causal relationships among inflammatory proteins and NASH as well as HCC due to potential confounding factors and reverse causation. Understanding these causal relationships is crucial for developing effective prevention and treatment strategies.
Mendelian randomization (MR) is an emerging epidemiological method that utilizes genetic variants as instrumental variables to infer causal relationships between exposures and outcomes. 9 This approach is based on three key assumptions: (1) genetic variants are strongly associated with the exposure; (2) genetic variants are not associated with potential confounders; and (3) genetic variants influence the outcome only through the exposure. 10 Two-sample MR studies further enhance the power and generalizability of this method by utilizing exposure and outcome data from different populations.11–13 Compared to traditional observational studies, this approach effectively controls for confounding factors and reverse causation, providing a robust tool for exploring the etiology of complex diseases.
This study aims to systematically investigate the causal relationships among 91 circulating inflammatory proteins and NASH as well as HCC using a bidirectional two-sample MR approach. These inflammatory proteins include, but are not limited to, the IL family, TNF-α, CRP, and the CCL family, which have been previously implicated in NAFLD development.6,14 Through this method, we aim to: (1) identify specific inflammatory proteins that may play crucial roles in the pathogenesis of NASH and HCC, and (2) discover potential therapeutic targets or biomarkers, offering new perspectives for the prevention and treatment of NASH and HCC. The results of this study will contribute to a deeper understanding of the pathogenic mechanisms of NASH and HCC, particularly the role of inflammatory processes in cirrhosis progression. Moreover, by identifying inflammatory proteins with causal relationships, we may provide important clues for developing new diagnostic tools and therapeutic strategies, thereby improving the prognosis of NAFLD patients. For instance, if certain inflammatory proteins are proven to have causal relationships with the development of NASH and HCC, these proteins could become potential therapeutic targets, providing valuable evidence for risk assessment, prevention strategies, and treatment regimens in clinical practice.
Methods
Figure 1 illustrates the comprehensive research workflow conducted from December 2023 to July 2024. Research workflow.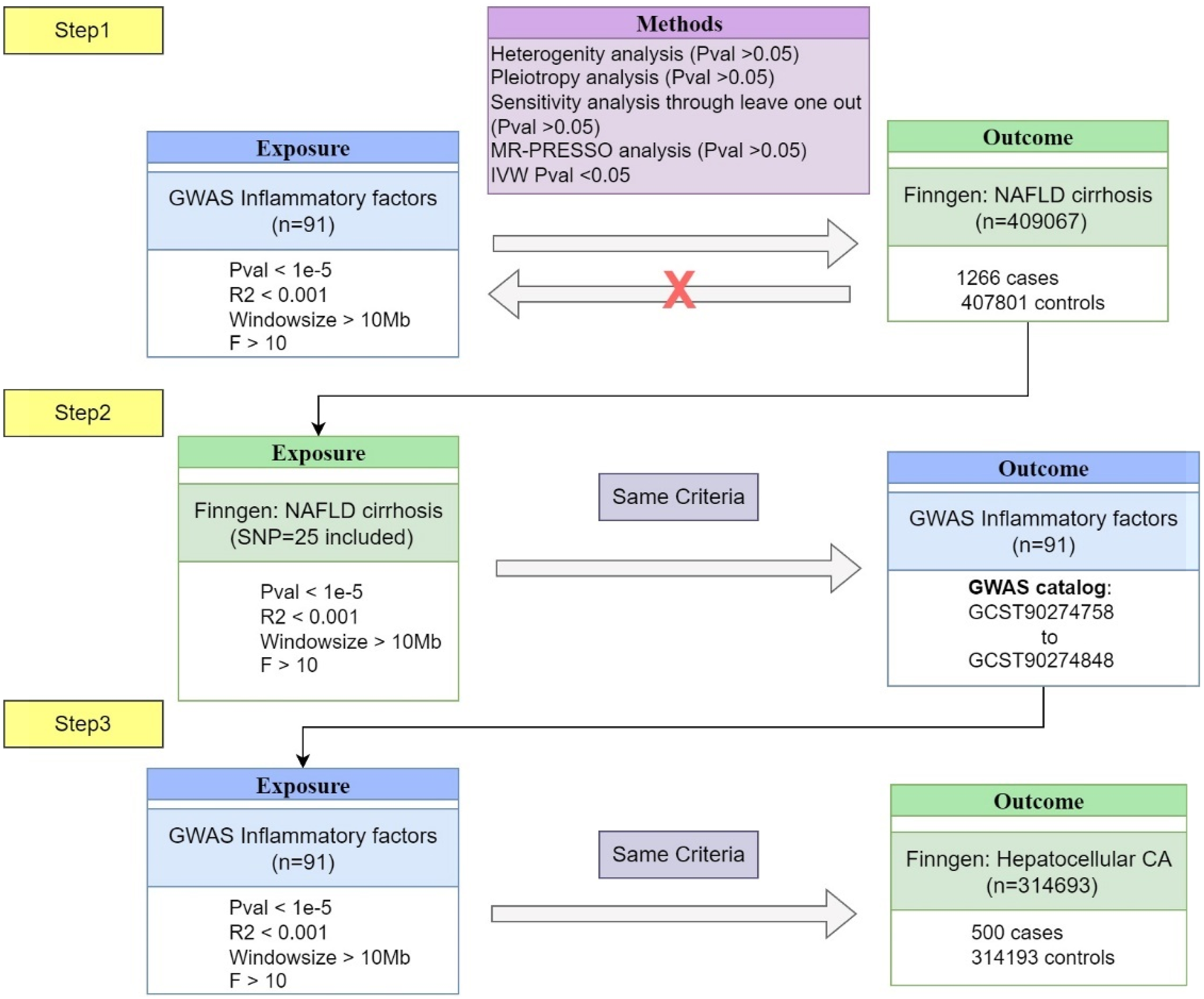
Data sources
For the selection of GWAS datasets on inflammatory factors, we utilized data from a large-scale proteomic GWAS conducted by the Department of Public Health and Primary Care at the University of Cambridge. 15 This study analyzed 91 circulating inflammatory proteins in 14,824 participants of European ancestry. We selected these datasets based on their comprehensive coverage of inflammatory factors, large sample size, and high-quality genotyping and imputation. The GWAS summary statistics for these 91 proteins were obtained from the GWAS Catalog (accession numbers GCST90274758 to GCST90274848).
For NASH and HCC data, we used the FinnGen biobank, which provides high-quality genetic and health data from the Finnish population. The CHIRHEP_NAS cohort comprised 1266 cases and 407,801 controls, and C3_HEPATOCELLU_CARC_EXALLC cohort comprised 500 cases and 314,193 controls with both datasets exclusively including individuals of European ancestry. They were selected due to their well-defined phenotypes, large sample sizes, and consistent data collection methods. These datasets were chosen to ensure robust genetic associations with our outcomes of interest.
Selection of instrumental variables
To assess the causal relationship between inflammatory factors, NASH and HCC, we employed a two-sample MR approach individually. This method relies on three key assumptions: (1) Relevance: The selected instrumental variables (genetic variants) must be strongly associated with the exposure (blood inflammatory factors). To identify robust instrumental variables satisfying this assumption, we conducted association analyses on the GWAS summary statistics. Given that the conventional genome-wide significance threshold (p < 5 × 10^-8) yielded a limited number of single nucleotide polymorphisms (SNPs), potentially insufficient for further analysis, we relaxed the threshold to p < 1 × 10^-5. Furthermore, to validate the strength of the instrumental variables, we calculated F-statistics and excluded SNPs with F-values below 10. (2) Independence: The genetic variants must be independent of any potential confounding factors that influence both the exposure and the outcome. While this assumption cannot be directly tested, we mitigated potential violations by performing linkage disequilibrium (LD) pruning using PLINK software, applying a physical distance threshold of 10,000 kb and an R^2 threshold of 0.001 to ensure independence among the selected SNPs. (3) Exclusion restriction: The genetic variants must influence NASH or HCC solely through the inflammatory factors, without any direct causal pathways. To address this, we conducted sensitivity analyses including MR-Egger regression to detect and account for potential pleiotropy.
Failure to meet assumption (1) precludes MR analysis, while violations of assumptions (2) and/or (3) may lead to false-positive results. To further validate these assumptions and enhance the robustness of our causal inference, we performed additional analyses: • Heterogeneity tests (Cochran’s Q) to assess the consistency of causal estimates across different SNPs. • MR-PRESSO tests to detect and correct for horizontal pleiotropy. • Leave-one-out analyses to identify influential outliers that might violate the MR assumptions.
These steps collectively address potential violations of the core MR assumptions and strengthen the validity of our causal inferences in the two-sample Mendelian randomization approach.
Mendelian randomization analysis and sensitivity analyses
To comprehensively evaluate the causal relationships among blood inflammatory factors, NASH, and HCC, we employed multiple MR methods: 1. Inverse-Variance Weighted (IVW): Selected as our primary approach for estimating causal effects due to its robustness in causal inference.
16
2. MR-Egger regression: Used to detect and account for potential pleiotropy. 3. Weighted Median (WM): Provides consistent estimates even when up to 50% of the information comes from invalid instrumental variables. 4. Simple Mode and Weighted Mode: Offer alternatives that are robust to outliers.
To assess the robustness of our MR results, we conducted several sensitivity analyses: 1. Leave-one-out cross-validation: To evaluate the influence of individual SNPs on the overall causal effect estimate. 2. MR-PRESSO (Pleiotropy Residual Sum and Outlier): To detect and correct for horizontal pleiotropy. 3. Cochran’s Q test: To assess heterogeneity in causal effect estimates across different SNPs. 4. MR-Egger regression intercept: To examine directional pleiotropy bias.
17
Bidirectional MR analysis
To explore potential reverse causation, we conducted a bidirectional MR analysis. For the reverse MR analysis, with NASH as the exposure, we selected SNPs significantly associated with NASH from the FinnGen biobank database as instrumental variables. The selection criteria were consistent with those previously described, including p-value thresholds, LD pruning, and F-statistic evaluation.
In the bidirectional MR analysis, we employed the same set of MR methods and sensitivity analyses as in the forward analysis to ensure consistency and robustness of our results.
Statistical analysis
All MR analyses were performed using R software (version 4.4.1) and relevant packages, including “TwoSampleMR” and “MRInstruments”. This comprehensive analytical approach enhances the validity and reliability of our causal inference, providing a robust framework for investigating the complex relationship between inflammatory factors, NASH and HCC.
Results
Causal relationship assessment between circulating inflammatory factors and NASH
We employed a two-sample MR approach to evaluate the causal relationships between circulating inflammatory factors and NASH. Preliminary results (Figure 2) indicated potential causal associations between NASH risk and the levels of five inflammatory factors: C-C motif chemokine 25 (CCL25), C-X-C motif chemokine 11 (CXCL11), Protein S100-A12 (S100A12), Interleukin-18 (IL18), and Leukemia inhibitory factor receptor (LIFR). Forest map of MR results of significant statistical inflammatory protein levels and nonalcoholic fatty cirrhosis.
Specifically, our analysis revealed the following associations, CCL25 showed a negative correlation with NASH risk (OR = 0.8703, 95% CI: 0.7774-0.9743, p = .0159), S100A12 demonstrated a negative correlation with NASH risk (OR = 0.7582, 95% CI: 0.6001-0.9580, p = .0204), IL18 exhibited a significant negative correlation with NASH risk (OR = 0.7412, 95% CI: 0.6099-0.9007, p = .0026), LIFR displayed a significant negative correlation with NASH risk (OR = 0.6804, 95% CI: 0.5654-0.8187, p < .001), and CXCL11 was the only factor showing a positive correlation with NASH risk (OR = 1.2625, 95% CI: 1.0161-1.5687, p = .0354).
Heterogeneity, pleiotropy, and MR-PRESSO test results.

Causal effect assessment of NASH on circulating inflammatory factors
To investigate potential bidirectional relationships between NASH and circulating inflammatory factors, we conducted reverse MR analyses, treating NASH as the exposure and the 91 circulating inflammatory factors as outcomes. This approach aimed to validate the absence of reverse causality for previously identified significant inflammatory factors and explore potential causal effects of NASH on a broader spectrum of circulating inflammatory factors.
The reverse MR analysis revealed statistically significant causal associations between NASH and levels of 10 circulating inflammatory factors (Figure 3). Notably, the five inflammatory factors identified in Section 3.1 (CCL25, CXCL11, S100A12, IL18, and LIFR) did not show significant reverse causal relationships (Supplemental files 2), further supporting their unidirectional causal influence on NASH. Forest plot of reverse MR analysis: Effects of NASH on circulating inflammatory factor levels.
Our analysis identified significant effects of NASH on the following inflammatory factors, Beta-nerve growth factor levels: OR = 1.0286 (95% CI: 1.0036–1.0541, p = .0245),C-C motif chemokine 20 levels: OR = 1.0382 (95% CI: 1.0150–1.0619, p = .0012),T-cell surface glycoprotein CD5 levels: OR = 1.0268 (95% CI: 1.0004–1.0539, p = .0466), CUB domain-containing protein 1 levels: OR = 1.0371 (95% CI: 1.0096–1.0653, p = .0078), Macrophage colony-stimulating factor 1 levels: OR = 1.0231 (95% CI: 1.0004–1.0463, p = .0458), Interleukin-12 subunit beta levels: OR = 1.0261 (95% CI: 1.0014–1.0515, p = .0381), Interleukin-22 receptor subunit alpha-1 levels: OR = 0.9722 (95% CI: 0.9476–0.9974, p = .0311), Macrophage inflammatory protein 1a levels: OR = 1.0372 (95% CI: 1.0143–1.0605, p = .0013), Osteoprotegerin levels: OR = 1.0436 (95% CI: 1.0101–1.0782, p = .0103), and Urokinase-type plasminogen activator levels: OR = 1.0320 (95% CI: 1.0093–1.0551, p = .0054).
These results suggest that NASH may lead to slight increases in multiple inflammatory factor levels, with only Interleukin-22 receptor subunit alpha-1 showing a minor decreasing trend. Notably, C-C motif chemokine 20 and Macrophage inflammatory protein 1a levels demonstrated more significant changes (p < .01), potentially indicating a more direct or strong influence of NASH on these factors.
Heterogeneity, pleiotropy, and MR-PRESSO test results.

In conclusion, these findings reveal complex interactions between NASH and the circulating inflammatory network. While certain inflammatory factors may be potential causes of NASH, the condition itself appears to induce changes in levels of other inflammatory factors. This bidirectional analysis approach provides a more comprehensive understanding of disease mechanisms, potentially informing future therapeutic strategies.
Causal effect assessment of circulating inflammatory factors and HCC
To further investigate the potential impact of cirrhosis-induced changes in inflammatory factor levels on HCC, we conducted an additional MR analysis to evaluate the causal relationships between circulating inflammatory factors and HCC risk. Our analysis revealed statistically significant associations between two inflammatory factors and HCC risk (Figure 4). Specifically, Interleukin-1-alpha (IL-1α) levels showed a positive correlation with HCC risk (OR = 1.5335, 95% CI: 1.0246-2.2951, p = .0377), and Sulfotransferase 1A1 (SULT1A1) levels demonstrated a negative correlation with HCC risk (OR = 0.7190, 95% CI: 0.5445-0.9493, p = .0200). Forest plot of MR results showing significant associations between inflammatory protein levels and HCC risk.
These findings suggest that increased levels of IL-1α may be causally associated with an elevated risk of HCC, while higher levels of SULT1A1 may have a protective effect against HCC development.
Further analysis of these two factors revealed no significant heterogeneity or horizontal pleiotropy. The MR-PRESSO test did not detect any significant bias in the causal relationships, with all relevant test results yielding p-values >0.05 (Supplemental files 4).
Discussion
This study employed a bidirectional MR approach to systematically investigate the causal relationships among 91 circulating inflammatory proteins, NASH, and HCC, revealing several important findings. In the forward analysis, we identified five inflammatory factors (CXCL11, CCL25, S100A12, IL18, and LIFR) significantly associated with NASH risk: CCL25, S100A12, IL18, and LIFR levels showed negative correlations with NASH risk, while CXCL11 levels exhibited a positive correlation. Specifically, decreased levels of CCL25, S100A12, IL18, and LIFR, along with increased levels of CXCL11, were associated with an elevated risk of NASH. These results suggest that these inflammatory factors may play crucial roles in the pathogenesis of NASH. Notably, further analysis of these five factors showed no significant heterogeneity or horizontal pleiotropy, and the MR-PRESSO test did not detect significant bias in the causal relationships, further supporting the robustness of these associations.
The reverse analysis revealed statistically significant causal associations between NASH and levels of 10 circulating inflammatory factors. Our findings suggest that NASH may lead to slight increases in multiple inflammatory factor levels, including Beta-nerve growth factor, C-C motif chemokine 20, T-cell surface glycoprotein CD5, CUB domain-containing protein 1, Macrophage colony-stimulating factor 1, Interleukin-12 subunit beta, Macrophage inflammatory protein 1a, Osteoprotegerin, and Urokinase-type plasminogen activator. Only Interleukin-22 receptor subunit alpha-1 showed a minor decreasing trend. Importantly, the five inflammatory factors identified in the forward analysis (CCL25, CXCL11, S100A12, IL18, and LIFR) did not show significant reverse causal relationships, further supporting their unidirectional causal influence on NASH.
To further explore the potential impact of cirrhosis-induced changes in inflammatory factor levels on HCC, we conducted an additional MR analysis. This analysis revealed statistically significant associations between two inflammatory factors and HCC risk. IL-1α levels showed a positive correlation with HCC risk, while SULT1A1 levels demonstrated a negative correlation. These findings suggest that increased levels of IL-1α may be causally associated with an elevated risk of HCC, while higher levels of SULT1A1 may have a protective effect against HCC development. Further analysis of these two factors revealed no significant heterogeneity or horizontal pleiotropy, and the MR-PRESSO test did not detect any significant bias in the causal relationships.
With regard to heterogeneity, pleiotropy, and MR-PRESSO tests, the majority of inflammatory factors demonstrated no significant heterogeneity or pleiotropy in their associations with NASH. However, several notable exceptions were observed. Osteoprotegerin levels, in particular, exhibited significant heterogeneity and yielded a global MR-PRESSO test p-value <.001, suggesting that its association with NASH might be influenced by horizontal pleiotropy or potential outliers. Furthermore, the global MR-PRESSO test p-value for CUB domain-containing protein 1 levels was <0.05, despite showing no significant heterogeneity, indicating possible horizontal pleiotropy or outliers that could affect causal effect estimation. These findings may reflect the multifaceted roles of OPG and CUBD1 in hepatic pathophysiology, including their involvement in inflammatory regulation and cell survival.
Our finding that CCL25 levels negatively correlate with NASH risk seems to contradict its known functions. CCL25 is typically considered a pro-inflammatory factor, attracting CCR9 + T cells to participate in hepatic inflammatory processes.18,19 However, recent research suggests that the CCL25/CCR9 axis may have dual roles in the liver. 20 In 2021, Spinnen et al. reported that high concentrations of CCL25 might lead to strong inflammatory responses, including increased secretion of various inflammatory factors, but may exert protective effects under certain conditions. This protective action might be related to CCL25 concentration, release method, or local microenvironment. For instance, a sustained-release system of CCL25 encapsulated in poly(lactic-co-glycolic acid) (PLGA) microparticles might induce a milder immune response, avoiding the intense inflammatory reaction potentially caused by high CCL25 concentrations. 21 Furthermore, CCL25’s role in maintaining intestinal immune homeostasis may indirectly influence NAFLD progression, considering the importance of the gut-liver axis in NAFLD pathogenesis. 22
Our study revealed a negative correlation between S100A12 levels and NASH risk, potentially reflecting S100A12’s complex role in NAFLD progression. This complexity is further elucidated in a review by Delangre et al. in 2022, which highlights S100 proteins’ involvement in various key cellular functions, including proliferation, apoptosis, migration, and inflammatory responses, with effects often dependent on specific cell types and microenvironments. 23 S100A12 may influence cellular behavior through interactions with p53, Wnt/β-catenin, and MAPK signaling pathways. It may also regulate cytoskeletal dynamics, cell-cell junctions, matrix metalloproteinase activity, and extracellular matrix degradation, thus affecting tissue remodeling. Moreover, S100A12 participates in regulating intracellular reactive oxygen species (ROS) levels and vascular remodeling, functions that may play crucial roles in NAFLD development. 24
Our observation of a significant negative correlation between IL18 levels and NASH risk aligns with some existing studies. For instance, in 2016, Yamanishi et al. found that IL18-deficient mice were more susceptible to developing steatohepatitis. 25 IL18 may exert protective effects through various mechanisms, including inhibition of lipogenesis, promotion of fatty acid oxidation, and regulation of hepatic immune responses. 26 Furthermore, IL18 might indirectly influence NAFLD progression by affecting intestinal barrier function and gut microbiota composition. 27
LIFR (Leukemia Inhibitory Factor Receptor) is a cytokine receptor involved in multiple biological processes. Our study is the first to report a significant negative correlation between LIFR levels and NASH risk. Although LIFR’s role in NAFLD is not extensively studied, evidence suggests that the LIF/LIFR signaling pathway plays a crucial role in liver regeneration and protection. 28 Yuan et al. found that LIF can protect hepatocytes from lipotoxicity by activating the STAT3 signaling pathway, suggesting that decreased LIFR levels might weaken this protective mechanism, thereby increasing the risk of NAFLD progression to cirrhosis. 29
Our study found a positive correlation between CXCL11 levels and NASH risk, consistent with CXCL11’s pro-inflammatory role. 30 CXCL11 can recruit and activate T cells, particularly Th1 cells, exacerbating hepatic inflammation and fibrosis. Activation of the CXCL11/CXCR3 axis positively correlates with the degree of liver fibrosis. 31 Additionally, CXCL11 can serve as a biomarker for liver fibrosis. 32
The subsequent reverse MR analysis revealed significant effects of NASH on the levels of 10 circulating inflammatory factors, providing deeper insights into the complex role of inflammatory networks in disease progression. We focused on factors showing the most significant changes, such as CCL20 and CCL3, which were markedly elevated in NASH patients. CCL20 may exacerbate hepatic inflammation by recruiting CCR6 + immune cells while promoting liver fibrosis, 33 whereas CCL3 might accelerate fibrosis progression by activating hepatic stellate cells. 34 Other affected factors include β-NGF, IL-12β, and OPG, which are involved in neuro-immune regulation, Th1 immune responses, and bone metabolism disorders, respectively.35–37
Most importantly, although no direct effect of NASH on inflammatory factors that directly influence HCC was found, the changes in other inflammatory factors caused by cirrhosis may indirectly affect the expression or activity of IL-1α. For example, cirrhosis significantly increased the level of CCL20, which recruits CCR6 + immune cells and Th17 cells. 38 Th17 cells can produce IL-1α, so the increase in CCL20 may indirectly lead to an increase in IL-1α levels, thereby promoting the development of HCC. Moreover, cirrhosis increased the level of IL-12β. Macrophages can secrete both IL-12β and IL-1α to affect hematopoietic stem cells, which are the main cells involved in liver regeneration. 39 Therefore, the increase in IL-12β may indirectly influence IL-1α, potentially weakening its protective effect against HCC. In terms of the microenvironment, the increased levels of CCL3 and CSF1 caused by cirrhosis can also significantly affect IL-1α through macrophages, promoting the development of HCC. Specifically, CCL3 can recruit and activate tumor-associated macrophages (TAMs), while CSF1 is crucial for macrophage proliferation and differentiation, leading to increased TAM infiltration. 40
Overall, in the progression of NAFLD, various immune cells collectively shape a complex and dynamic immune landscape. Regulatory T cells (Tregs) play a crucial role in maintaining immune tolerance and controlling inflammatory responses, potentially serving as novel immunotherapeutic targets. 41 Effector T cells, particularly Th1 and Th17 cells, exacerbate liver inflammation and fibrosis through the secretion of pro-inflammatory cytokines.42,43 CD8 + T cells and Natural Killer (NK) cells are important in anti-tumor immunity, but their function may be suppressed in chronic inflammatory environments. 44 Macrophages, including liver-resident Kupffer cells and infiltrating macrophages, play a central role in regulating inflammation, fibrosis, and tissue repair processes. 45 Unconventional immune cells such as γδ T cells and innate lymphoid cells (ILCs) may have unique roles in innate immune responses and tissue repair in the liver. 46 The interactions and balance among these immune cells determine the progression of NAFLD from simple steatosis to NASH, and further to cirrhosis and HCC, constituting a dynamic immune network that evolves with disease stages.
Which means, these alterations in inflammatory factor levels may lead to a cascade of effects, including amplification of inflammatory responses, immune imbalance, increased risk of complications, and elevated liver cancer risk. These effects not only accelerate NASH progression but may also increase the risk of systemic complications in patients. Simultaneously, these findings provide important clues for developing new diagnostic markers and therapeutic targets. However, given the heterogeneity and complexity of NAFLD, these results still require further clinical validation and mechanistic studies.
Limitations
However, this study also has several limitations: Firstly, although the MR method can effectively control for confounding factors and reverse causality, it cannot completely rule out the influence of pleiotropic effects. Secondly, this study is based on data from European populations, and the results may not be directly generalizable to other ethnicities. Thirdly, our analysis primarily focused on known inflammatory factors, potentially overlooking some unknown but potentially important factors. Lastly, while we have identified several significant associations, the specific biological mechanisms still require further experimental validation. Future research should combine functional experiments and clinical studies to more comprehensively elucidate the role of these inflammatory factors in NAFLD progression and explore their potential as therapeutic targets.
Conclusion
In conclusion, our study systematically investigated the causal relationships among circulating inflammatory proteins, NASH, and HCC using a bidirectional MR approach, revealing several important findings. We found that levels of CCL25, S100A12, IL18, and LIFR were negatively correlated with NASH risk, while CXCL11 levels showed a positive correlation. Reverse analysis indicated that NASH might lead to slight increases in various inflammatory factor levels, including β-NGF, CCL20, CD5, CSF1, IL-12β, CCL3, and uPA, along with a decrease in IL-22Rα1 levels. Furthermore, these changes in the inflammatory microenvironment may contribute to an increased risk of HCC by elevating IL-1α levels. These findings provide new insights into the complex interplay between inflammatory factors, NASH, and HCC, offering potential targets for future diagnostic and therapeutic strategies.
Supplemental Material
Supplemental Material - Systemic inflammatory factors in non-alcoholic steatohepatitis-induced hepatocellular carcinoma: A bidirectional Mendelian randomization analysis
Supplemental Material for Systemic inflammatory factors in non-alcoholic steatohepatitis-induced hepatocellular carcinoma: A bidirectional Mendelian randomization analysis by Kang Fu, Yiming Zhou, Zhongyi Guo, Xiaotong Chen, Lin Sun, and Xiao Hu in European Journal of Inflammation.
Footnotes
Author contributions
Kang Fu, Yiming Zhou, and Xiao Hu designed the study; Kang Fu, Lin Sun, and Zhongyi Guo collected the data and carried out the computation and generated figures; Kang Fu and Xiaotong Chen analyzed the data and wrote the manuscript, Kang Fu and Xiao Hu revised the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical statement
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.