
Abstract
Objectives
FLASH radiotherapy is garnering attention for its capacity to diminish skin toxicity without compromising tumoricidal efficacy, presenting a stark contrast to conventional (CONV) radiotherapy. Despite its promise, the underlying molecular mechanisms of FLASH irradiation (FLASH-IR) on skin are not yet fully elucidated.
Methods
This study investigated the transcriptomic responses of human foreskin fibroblast cells (HFF-1) via the FLASH-IR or CONV irradiation (CONV-IR), employing the next-generation RNA sequencing (RNA-seq) to capture the gene expression profiles. Our comparative analysis aimed to dissect the cellular and molecular pathways influenced by these two irradiation methods.
Results
We identified a spectrum of differentially expressed genes (DEGs), signaling pathways, and transcriptional networks that were either shared or divergent between FLASH-IR and CONV-IR. Particularly, transcription factor NR4A1 showed significant upregulation in response to FLASH-IR, while chromatin stability factor ELF3 was markedly downregulated following CONV-IR. The top 10 up-regulated DEGs were subjected to qPCR validation, confirming their differential expression in response to FLASH-IR and CONV-IR.
Conclusion
Collectively, our findings delineate unique regulatory landscapes of FLASH-IR and CONV-IR on skin cells, corroborating established effects and shedding new light on the molecular interplay within the context of ultra-high dose radiation.

Introduction
Radiotherapy stands as a cornerstone in oncology, utilized in more than half of all cancer cases to achieve cure or palliation.1-3 Despite its therapeutic importance, the treatment effectiveness is frequently mitigated by its toxicity on adjacent healthy tissues. 4 Skin tissue, with its high regenerative potential, is notably susceptible to radiation-induced damage. Clinically, a significant majority—approximately 90%—of patients receiving radiotherapy for tumors have been reported to burden moderate to severe radiation-induced skin injury (RSI). 5 These skin injuries not only diminish patients’ quality of life but also interrupt the completion of the intended radiotherapy course, thereby affecting treatment outcomes.
Radiation-induced skin injury (RSI) is broadly classified into acute and chronic periods. 5 Acute radiation dermatitis, recognized as the most common form of RSI, is graded on a severity scale from 1 to 4 by the National Cancer Institute, encompassing symptoms from mild erythema and dry desquamation to severe ulceration and necrosis. 6 Tissue reactions, trigger within hours to weeks after radiation exposure, resulting in substantial production of reactive oxygen species (ROS), irreversible DNA damage and inflammation. 7 The initial inflammatory phase is predominantly mediated by a complex interplay of proinflammatory cytokines (including IL-1, IL-3, IL-5, IL-6, IL-8, and TNF-α), chemokines (such as eotaxin), CCR receptors, adhesion molecules (E-selectin, ICAM-1, VCAM), and receptor tyrosine kinases. 5 This inflammatory milieu attracts eosinophils and neutrophils, exacerbating tissue damage and undermining the skin’s protective barrier. Chronic RSI encompasses conditions such as non-healing ulcers, wounds, fibrosis, and radiation-induced keratoses, which are typically resistant to spontaneous resolution. The chronic RSI is intricately linked to the TGF-β cytokine and the Wnt signaling pathway.6,8 TGF-β expression in skin tissue is induced in a manner that is contingent upon the radiation dose received. The elevated TGF-β levels have been detected in the fibrotic tissues of irradiated patients, contrasting with the non-irradiated counterparts. Mechanistically, radiation triggers a swift activation of TGF-β, which in turn stimulates the differentiation of fibroblasts into myofibroblasts and increases deposition of extracellular matrix (ECM), thereby disrupting the equilibrium between ECM synthesis and degradation. 9 Concurrently, recent research underscores the pivotal role of the Wnt/β-catenin signaling pathway in radiation-induced skin fibrosis. 10 Specifically, the microRNA MiR-17-5p has been reported to activate of the Wnt/β-catenin pathway and exacerbate radiation-induced liver fibrosis. 11 The study confirmed the critical role of the Wnt/β-catenin pathway in fibrosis development following radiation exposure. Distinguished from skin injuries caused by other etiologies, RSI is marked by its potential for latency, progression, and persistence. Consequently, endeavors aimed at preventing or attenuating radiation-induced skin and other normal tissue injuries during radiotherapy are likely to enhance the therapeutic index of radiation treatments.
FLASH radiotherapy (FLASH-RT) emerges as a groundbreaking modality in oncology, distinguished by its exceptional capacity to preserve normal tissues while exerting potent anti-tumor effects. 12 This novel approach starkly diverges from the CONV-IR with a dose rate of ≤0.03 Gy/s, and is widely recognized for the “FLASH effect” (dose rate ≥40 Gy/s). 13 In recent years, a series of preclinical researches have uncovered the potential protective effects of FLASH-RT on radiosensitive tissues, including lung, brain, intestine, hematopoietic systems, and skin, by leveraging diverse animal models. 14 A pivotal study by Vincent et al. demonstrated that FLASH-IR, at a dosage of 17 Gy, conferred a protective advantage against pneumonia and fibrosis in a validated murine model of lung fibrosis compared with CONV-IR. 15 The mitigating impact of FLASH-RT on skin toxicity has been corroborated by a series of preclinical investigations.16-18 Anastasia and colleagues delineated the protective mechanisms of FLASH-RT, highlighting its protective role in skin by reducing inflammation and the progression of lymphedema, alongside mitigating the atrophy of myofibers, hair follicles, and epidermal hyperplasia. 17 RNA sequencing (RNA-seq) analysis has further illuminated the notable upregulation of pathways implicated in apoptosis and keratinocyte signaling observed in CONV-RT-treated murine skin at 30 Gy, contrasting with the FLASH-RT group. In an assessment of the clinical FLASH proton beam scanning (PBS) system, Shannon et al. reported that FLASH-IR and CONV-IR exhibited comparable tumor control efficacy for head and neck cancers, yet FLASH-IR was associated with significantly reduced plasma and skin TGF-β1 levels, diminished skin toxicity, and lessened leg contracture in the murine models. 19 The promising outcomes of FLASH-IR have also been corroborated in higher mammals, exemplified by studies involving Göttingen mini-pigs and feline cancer patients, which affirmed the favorable therapeutic profile of FLASH-IR.20,21
Currently, the precise mechanisms by which FLASH-IR reduces skin toxicity remain unclear . A prevailing hypothesis posits that the rapid delivery of FLASH-IR rapidly depletes oxygen in normal tissues, consequently diminishes the generation of deleterious ROS and thus safeguards the structural and functional integrity of healthy cells.22,23 Additionally, FLASH-IR has been demonstrated to mitigate inflammatory responses and expedite the healing process of irradiated tissues, further enhance its protective effects on the skin. 24 It is imperative that additional research is conducted to elucidate the mechanisms by which FLASH-IR spares normal tissues while retaining its tumoricidal efficacy. Such insights are essential for the advancement of FLASH radiotherapy towards clinical adoption, particularly as technologies for precise and conformal delivery continue to evolve.
In our research, we sought to explore the impact of FLASH-IR compared to CONV-IR on normal skin cells. Utilizing RNA-seq on the irradiated skin cell model, we identified differentially expressed genes and associated signaling pathways (Figure 1). Specifically, the study revealed the transcription factor NR4A1 was markedly upregulated in response to FLASH-IR, while the chromatin stability factor ELF3 exhibited significantly downregulated following CONV-IR. Our findings offered novel insights in the shared or divergent molecular mechanisms of FLASH-IR and CONV-IR on skin cells. Schematic Representation of the Integrated Study Framework
Materials and Methods
Cell Culture
Human foreskin fibroblast (HFF-1) cells were purchased from American Type Culture Collection (ATCC) and cultured in the completed Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) to facilitate robust cell growth.25,26 In addition, 1% penicillin (100 U/mL)-streptomycin (100 μg/mL) were added to the completed liquid medium to avoid bacterial contamination. Cells were continuously nurtured in cell incubator set at a 37°C humidified atmosphere with 5% CO2. HFF-1 cells were passaged upon reaching 80%–90% and digested with 0.25% trypsin-EDTA solution for detachment.
Irradiation Treatment Strategy
Equal number of cells (1 × 106) were seeded in T25 flasks. The density of HFF-1 cells reached 70% after one day of culturing, and the cells were subsequently exposed to 10 Gy X-rays. FLASH-IR was conducted with the platform for advanced RT research (PARTER) at the Chengdu THz Free Electron Laser facility (CTFEL) in China in March 2024. The high-energy X-rays equipment can produce 6-8 MeV electrons with an adjustable mean current of up to 10 mA and a beam energy measured by root mean square is 8.2 MeV. For CONV-IR was performed by a biological research irradiator (Rad Source; RS-2000 Pro) at a dose rate of 2 Gy/min. Following irradiation, cells were continuously incubated in the completed medium for 24 h. Parallelly, control cells were cultured in the completed medium without any irradiation.
RNA Sequencing and Analysis
Three bottles of HFF-1 cells subjected to FLASH-IR, three bottles of HFF-1 cells subjected to CONV-IR, and two bottles of HFF-1 cells without IR were used for RNA sequencing (RNA-seq). For RNA-seq, 1 mL of cold TRIzol reagent was promptly added to HFF-1 cells and transferred to a 1.5 mL centrifuge tube. Total RNA was extracted following the protocols provided by OE company, in preparation for subsequent RNA-Seq analysis. The resultant clean reads were aligned to the human reference genome (GRCh38) utilizing the HISAT2 aligner. 27 Post-alignment, raw read counts were processed, entailing the exclusion of nonprotein-coding genes across the entire sample set. The bioinformatic analyses were executed with tools available on the OECloud platform. Additionally, the volcano plot and other graphical representations were generated using the R programming language, facilitated by the integrated environment on the OECloud platform at https://cloud.oebiotech.com/task/.
Differentially Expressed Genes (DEGs) Analysis
Differential gene expression analysis was performed using DESeq2 on the counts. 28 Prior to this analysis, a pruning step was implemented to refine the gene detection list from the sequencing results. Specifically, we filtered out protein-coding genes that expressed in less than 50% across the entire set of samples. Genes with an adjusted P-value <.05 and |log2FC|>1 were considered as significantly differentially expressed genes (DEGs).
Functional Enrichment Analysis
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis were performed on significantly DEGs using the clusterProfiler package. 29 GO analysis was used to elucidate the enriched gene sets associated with the biological processes (BP), molecular functions (MF), and cellular components (CC). Adjusted P-value <.05 were considered significantly enriched. In this study, Gene Set Enrichment Analysis (GSEA) was also used to identify biological pathways that were significantly enriched with all differential expressed genes between the experimental conditions. 30 The pre-ranked gene list was based on log2 fold change (log2FC). The collection of curated gene sets was compiled from Reactome. 31 The statistical significance of the enriched pathways was determined using an empirical phenotype-based permutation test approach, with 1000 permutations to estimate the null distribution of enrichment scores. The resulting P-value were adjusted for multiple hypothesis testing using the false discovery rate (FDR) method. Gene sets with an FDR less than 0.25 were considered significantly enriched, ensuring robustness against type I errors.
Prediction of Transcription Factors
To predict the regulatory relationships between human transcription factors (TFs) and their target genes in the FLASH-IR vs Control and CONV-IR vs Control groups, TFs were collected from the hTFtarget database (https://bioinfo.life.hust.edu.cn/hTFtarget/), which curated a comprehensive TF-target regulation from various ChIP-seq datasets of human TFs from NCBI Sequence Read Archive (SRA) and ENCODE databases. 32 We further screened the differentially expressed TFs and their target genes based on the above DEGs.
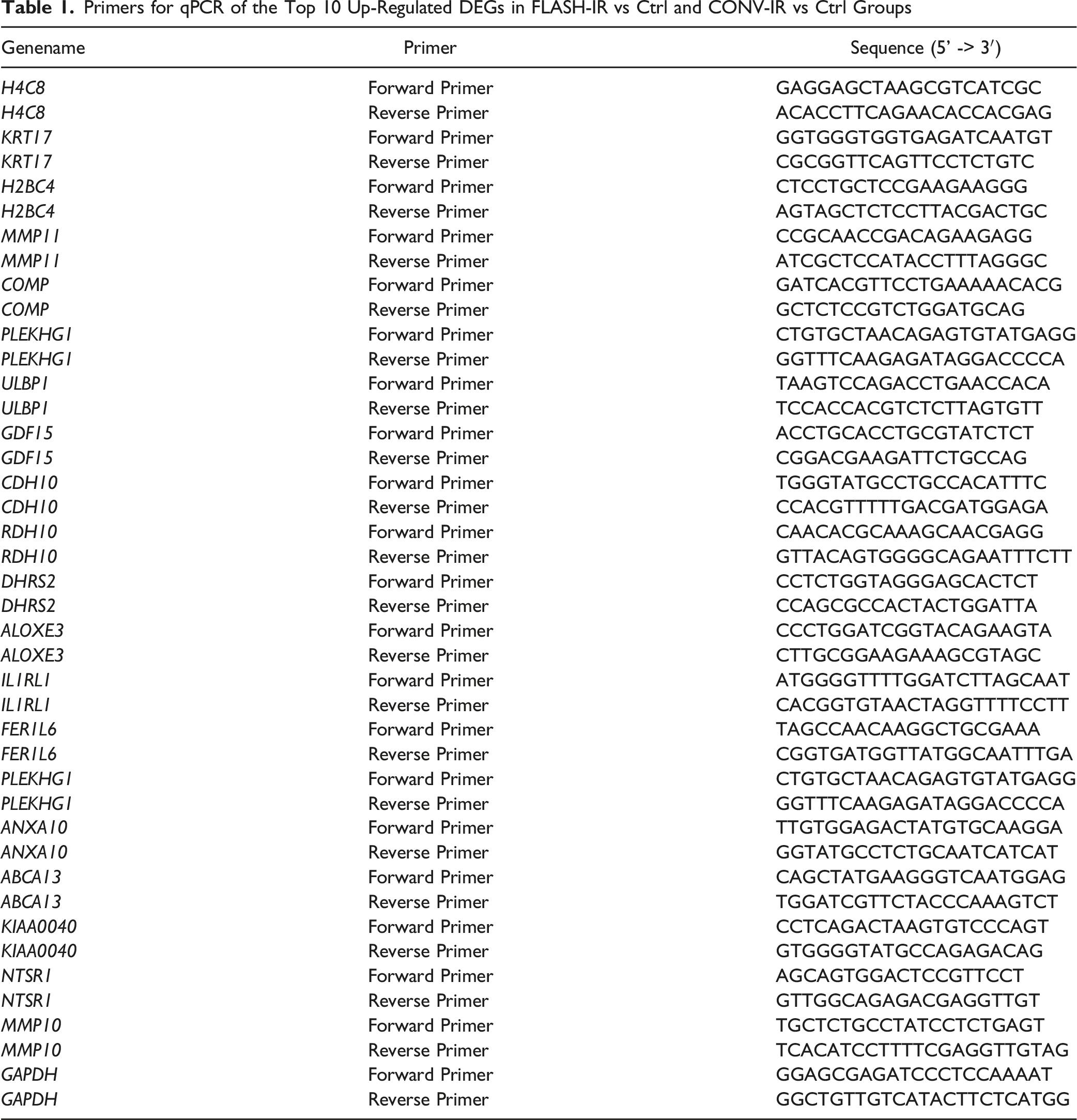
QPCR Analysis for Top Changed DEGs
Primers for qPCR of the Top 10 Up-Regulated DEGs in FLASH-IR vs Ctrl and CONV-IR vs Ctrl Groups
Statistical Analyses
Statistical analysis and visual representations were performed by GraphPad Prism version 7.0. Student’s t‐test analysis was used to determine the statistical significance between two groups. Kruskal–Wallis test was used to determine the statistical significance among 3 groups. The data are presented as the mean, complemented by the standard error of the mean (SEM). Adobe Illustrator (AI) software was employed to the edit and refine the figures.
Results
FLASH-IR and CONV-IR Induced Distinct Gene Expression Patterns
Principal Component Analysis (PCA) was conducted to delineate the global patterns of gene expression variation across the samples. The resultant PCA plot revealed a clear demarcation of the samples into three discrete clusters, indicative of divergent gene expression profiles and underlying biological functions among the Control (Ctrl), FLASH-IR, and CONV-IR groups (Figure 2A). Samples within the same experimental cohort exhibited a propensity for co-clustering, underscoring the reliability and reproducibility of our experimental setup. Additionally, the volcano plot furnished a panoramic view of the differential gene expression landscape, highlighting the distinctions between the groups. 286 significantly changed DEGs, comprising 40 upregulated and 246 downregulated genes, were discerned in FLASH-IR group (Figure 2B). The CONV-IR group showcased 350 significantly changed DEGs, with 124 upregulated and 226 downregulated genes (Figure 2C). A comparative analysis identified 109 overlapped DEGs in FLASH-IR and CONV-IR groups, alongside 177 unique DEGs in the FLASH-IR group and 241 in the CONV-IR group (Figure 2D). Furthermore, a heatmap was employed to graphically represent the expression levels of the top 10 up- and down-regulated DEGs in FLASH-IR and CONV-IR groups (Figure 2E and Table 2). Collectively, these findings revealed the substantial differences in gene expression profiles between the FLASH-IR and CONV-IR groups. CONV-IR and FLASH-IR Exhibited Different Gene Expression Profiles. (A) PCA Diagram Showed the Global Difference of Gene Expression Pattern for Ctrl, FLASH-IR and CONV-IR Groups. (B) Volcano Plot Showed the DEGs of FLASH-IR. (C) Volcano Plot Showed the DEGs of CONV-IR Group. (D) Venn Plot Showed the Overlapped up- and Down-Regulated Genes Induced by FLASH-IR and CONV-IR Groups. (E) Heatmap Showed the Top DEGs in Ctrl, FLASH-IR and CONV-IR Groups (Ctrl, Control) Top 10 up- and Down-Regulated DEGs in FLASH-IR vs Ctrl and CONV-IR vs Ctrl Groups

FLASH-IR and CONV-IR Showed Different Regulatory Mechanisms on Skin Cells
To elucidate the functional implications of significant changed DEGs in response to FLASH-IR and CONV-IR, we conducted GO and KEGG pathway analyses. In the BP category of the overlapping DEGs, terms such as ‘nuclear division’, ‘organelle fission’ and ‘mitotic nuclear division’ were significantly enriched. The CC category highlighted terms like ‘chromosomal region’, ‘spindle’, ‘chromosome’, and ‘centromeric region’. The MF category revealed the enriched terms related to ‘microtubule binding’, ‘tubulin binding’, and ‘cyclin-dependent protein serine/threonine kinase regulator activity’ (Figure 3A). For distinct genes in FLASH-IR, the BP category showed enrichment in terms such as ‘nuclear division’, ‘organelle fission’, and ‘chromosome segregation’. The CC category identified terms like ‘chromosomal region’, ‘chromosome centromeric region’, and ‘condensed chromosome’. The MF category in FLASH-IR was enriched in terms associated with ‘ATP-dependent activity’, ‘acting on DNA’, and ‘catalytic activity acting on DNA’ (Figure 3B). In the case of distinct genes in CONV-IR, the BP category yielded terms such as ‘extracellular matrix organization’, ‘extracellular structure organization’, and ‘external encapsulating structure organization.’ The CC category pointed to terms related to ‘subcellular localization and structural organization’, ‘collagen-containing extracellular matrix’, and ‘complex of collagen trimers’. The MF category in CONV-IR was enriched in terms like ‘cytokine activity’, ‘chemokine activity’, and ‘glycosaminoglycan binding’ (Figure 3C). Furthermore, KEGG analysis indicated that the overlapping DEGs were enriched in pathways such as ‘Cell cycle’, ‘Oocyte meiosis’ and ‘Cellular senescence’ (Figure 3D). FLASH-IR was associated with pathways like ‘Cell cycle’, ‘DNA replication’, and ‘Nucleotide excision repair’ (Figure 3E). CONV-IR showed enrichment in pathways such as ‘Viral protein interaction with cytokine and cytokine receptor’, ‘Cytokine-cytokine receptor interaction’, and ‘TNF signaling pathway’ (Figure 3F). Collectively, these findings suggest that DEGs in the FLASH-IR group are closely associated with chromosomal and DNA stability, while CONV-IR group are related to chromosomal and DNA stability, extracellular matrix integrity, and inflammatory responses. FLASH-IR and CONV-IR Exhibited Different Functional Characteristics. (A) GO Functional Enrichment Analysis Showed the Top 5 Enriched Gene Sets in BP, CC, MF of the Overlapped DEGs in FLASH-IR and CONV-IR Groups. (B) GO Functional Enrichment Analysis Showed the Top 5 Enriched Gene Sets in BP, CC, MF of the Distinct DEGs in FLASH-IR Group. (C) GO Functional Enrichment Analysis Showed the Top 5 Enriched Gene Sets in BP, CC, MF of the Distinct DEGs in CONV-IR Group. (D) KEGG Functional Enrichment Analysis Showed the Top 5 Enriched Pathways of the Overlapped DEGs Between FLASH-IR and CONV-IR Groups. (E) KEGG Functional Enrichment Analysis Showed the Top 5 Enriched Pathways of the Distinct DEGs in FLASH-IR Group. (F) KEGG Functional Enrichment Analysis Showed the Top 5 Enriched Pathways of the Distinct DEGs in CONV-IR Group
GSEA Revealed the Functional Landscape of DEGs
We employed GSEA to reveal the enriched pathways from the ranked list of all genes and to provide a visual depiction of gene set activation across the entire transcriptome. Based on the Reactome database, 76 pathways were found to be commonly enriched in both FLASH-IR and CONV-IR groups, with 162 unique pathways identified in FLASH-IR and 14 in CONV-IR (FDR <0.25) (Figure 4A). Among the overlapping pathways, those related to different mitotic stages were notably enriched (Figure 4B), suggesting a downregulation of cell division and cell cycle responses in both irradiation methods. In FLASH-IR, 162 distinct pathways were enriched, including those involved in extracellular matrix organization, degradation, mRNA splicing, and elastic fiber formation, indicating a relative equilibrium in collagen formation and degradation (Figure 4C). While, in CONV-IR, pathways such as signaling by G alpha I and arachidonic acid metabolism were uniquely enriched, highlighting the significant activation of inflammatory mediator-related metabolic pathways (Figure 4D). FLASH-IR and CONV-IR Exhibited Different Functional Landscape of all the DEGs. (A) Venn Plot Showed the Overlapped and Distinct Pathways Induced by FLASH-IR and CONV-IR. (B) Classical GSEA Enrichment Graphic Showed the Top 5 Overlapped Enriched Pathways Between FLASH-IR and CONV-IR Groups. (C) Classical GSEA Enrichment Graphic Showed the Top 5 Distinct Enriched Pathways in FLASH-IR Group. (D) Classical GSEA Enrichment Graphic Showed the Top 5 Distinct Enriched Pathways in CONV-IR Group
Prediction of Transcription Factors
We performed an investigation into the network of transcription factor (TF)-target regulations to uncover the essential mechanisms underlying irradiation-specific gene expression patterns. In the FLASH-IR group, transcription factors NR4A1, GATA6, EHF, and EGR2 were upregulated, whereas MYBL2, LMNB1, LIN9, FOXM1, E2F2, and CENPA were downregulated (Figure 5A). In the CONV-IR group, KLF15 and ATF3 were identified as upregulated transcription factors, while LMO2, LMNB1, HES4, ELF3, E2F2, and CENPA were found to be downregulated (Figure 5B). FLASH-IR and CONV-IR Exhibited Different Exhibited Different Transcription Regulatory Networks. (A) Sankey Diagram Showed the Transcriptional Factors and Corresponding Targeted Genes in FLASH-IR Group. (B) Sankey Diagram Showed the Transcriptional Factors and Corresponding Targeted Genes in CONV-IR Group. (Red Font Denotes Up-Regulated, Blue Font Denotes Down-Regulated)
Differential Expression Analysis of Key Transcriptional Regulators
We further explored the expression the transcriptional regulators from different groups. To display differential expression of the screened TFs from different groups in all samples, we analyzed the expression of TFs in Ctrl, FLASH-IR and CONV-IR samples. The transcription factors such as LMO2, LMNB1, KLF15, HES4, NR4A1, LIN9, FOXM1, EGR2 exhibited significantly changed across all the samples (Figure 6A and B). Differential Expression Analysis of Key Transcription Regulators in FLASH-IR and CONV-IR Groups. (A) Transcription Factors Screened From FLASH-IR Group was Analyzed in all the Samples. (B) Transcriptional Factors Screened From CONV-IR Group was Analyzed in all the Samples. ( *P < .05, ns, No Significance)
Differential Gene Expression Detection by QPCR Analysis
We detected the expression levels of the top ten up-regulated DEGs in FLASH-IR and CONV-IR. KRT17, MMP11, COMP, ULBP1, GDF15, CDH10, and RDH10 were confirmed to up-regulate in FLASH-IR group by qPCR, while H4C8 and H2BC4 were down-regulated, and there was no significant change of PLEKHG1 in FLASH-IR vs Ctrl group (Figure 7A). DHRS2, ALOXE3, IL1RL1, FER1L6, PLEKHG1, ANXA10, ABCA13, KIAA0040, NTSR1, and MMP10 were confirmed to up-regulate by qPCR in CONV-IR group (Figure 7B). These results indicated that our transcriptome sequencing results had a high degree of agreement with the experimental results. Gene Expression was Detected by qPCR Analysis. (A) QPCR Analysis Showed the Top Up-Regulated Genes in FLASH-IR Group. (B) QPCR Analysis Showed the Top Up-Regulated Genes in CONV-IR Group. (*P < .05, **P < .01, ns, No Significance)
Discussion
Our study presents an in-depth comprehensive transcriptome profiling of HFF-1 skin fibroblasts subjected to FLASH-IR and CONV-IR irradiation. Our findings delineate distinct gene expression signatures associated with each irradiation method, indicating that FLASH-IR mediate therapeutic impact via distinctive molecular pathways compared to CONV-IR.
From the RNA-seq analysis of our study, a set of distinct genes including KRT17, COMP, ULBP1, GDF15, CDH10, and RDH10 were found to be significantly upregulated in the FLASH-IR group. KRT17, the largest fold change in FLASH-IR DEGs, is a member of the Keratin (KRT) family, which plays a crucial role in maintaining the structural stability and integrity of keratinocytes. 33 Its expression in cells is indicative of an activated and proliferative state under pathological conditions. KRT17 is also known to modulate the expression of multiple inflammatory cytokines, thereby influencing the development of adaptive T cell responses. 34 Previous studies on FLASH have predominantly focused on its capacity to minimize damage to surrounding tissues, our findings offer insights into the molecular targets affected by FLASH effects on tissue. This contributes to a theoretical framework for refining FLASH therapy. Consequently, KRT17 emerges as a promising candidate for therapeutic intervention in mitigating the skin effects of FLASH irradiation.
DHRS2, ALOXE3, IL1RL1, FER1L6, PLEKHG1, ANXA10, ABCA13, KIAA0040, NTSR1, MMP10 were top upregulated genes in CONV-IR group. Dehydrogenase/reductase member 2 (DHRS2), also known as Hep27, was initially isolated from the extract of hepatocelular carcinoma HepG2 cells and was identified as a specific cell cycle regulator. 35 DHRS2 has been reported inhibit the growth and metastasis of ovarian cancer (OC) cells by the interruption of choline metabolism in vitro and in vivo. 36 Wu et al have reported DHRS2 was repressed in prostate cancer (PCa) cells. Overexpression of DHRS2 suppressed PCa cell proliferation and promoted apoptosis. 37 The function of DHRS2 in inhibiting cell cycle in many urologic malignancies have widely reported, 38 while the role of DHRS2 in irradiation has not been reported. Our study thereby offering novel putative predictive biomarker in CONV-IR.
Functional enrichment analyses including GO, KEGG and GSEA have demonstrated that both FLASH-IR and CONV-IR irradiation methods compromised chromosomal and DNA stability, impacting key processes such as cell proliferation and apoptosis.39-41 Additionally, CONV-IR group exhibited the enriched pathways associated with extracellular matrix functions and inflammation, which were the activated factors of skin fibrosis formation. In contrast, these effects were subdued in the FLASH-IR group. These findings support the hypothesis that FLASH-IR may elicit a diminished inflammatory response and reduced skin fibrosis, potentially accounting for the decreased adverse effects noted. 42 FLASH-IR has reported to reduce the inflammatory response in several preclinical studies.43,44
Furthermore, our data indicated an altered expression of several key transcriptional regulators of chromosomal stability. For instance, the expression levels of NR4A1 was up-regulated by FLASH-IR. NR4A1is critical to maintain genomic stability, and its deletion causes chromosomal defects compromising proliferation and survival. 45 NR4A1 was previously reported to regulate mesenchymal cell function and dampen fibrogenic signaling, and the activation of NR4A1 in human myofibroblasts reduced TGF-β1-induced collagen deposition and fibrosis-related gene expression. 46 By leveraging NR4A1 as a biomarker, clinicians can enhance their ability to predict treatment responses, assess skin injury risks, and explore new therapeutic strategies, ultimately optimizing radiotherapy management and improving overall patient prognosis. In addition, NR4A1 targeted gene known as matrix metallopeptidase 11 (MMP11) was slightly upregulated in FLASH treatment group. MMP11, also known as stromelysin-3, which plays a crucial role in the breakdown of extracellular matrix. 47 MMP11 also played important role in inflammatory response by modulating the activity of cytokines and chemokines, thus influenced the infiltration and activity of immune cells in tissues. 48 The transcription factor ELF3 was downregulated by CONV-IR. ELF3 was expressed in various types of cells and regulated a variety of biological behaviors, such as cell proliferation, differentiation, apoptosis, migration, and invasion. In recent years, studies have shown that ELF3 played an important role in the occurrence and development of many tumors and inflammation and immune related diseases. 49
The clinical implications of our findings are significant, by analyzing the impact of two distinct irradiation modalities on the skin cell transcriptome. Our comprehensive study reveals both commonalities and divergences in their regulatory effects on skin cells. While both irradiation types share certain regulatory mechanisms on chromosomal and DNA stability, meanwhile they exhibit unique molecular expression profiles and distinct pathways of action. However, this study involves a small sample size, which may lead to results being biased due to insufficient sample size. Such bias could affect the generalizability of the results and their association with larger populations, thereby reducing the reliability and validity of the study’s conclusions. To address this issue, future research should consider increasing the sample size to more comprehensively assess the relationships between study variables and ensure the accuracy and reproducibility of the findings. In addition, this study only examined fibroblasts in the foreskin 24 hours after radiation exposure. While these cells play an important role in the radiation response, the significance of this research is limited due to the diverse cell populations that comprise human skin. The limitation lead the failure to comprehensively assess the dynamic changes and interactions of these cell populations. To make more holistic and fundamental statements regarding the different mechanisms of action following FLASH-IR and CONV-IR exposure, advanced techniques such as single-cell RNA sequencing are actually needed. This method allows for in-depth analysis of the gene expression profiles of various cell populations at different time points, thereby revealing the complex signaling pathways and interactions among cell types in response to FLASH-IR and CONV-IR exposure. Future clinical trials, design based on these molecular insights could help to optimize FLASH radiotherapy protocols and potentially lead to better patient outcomes.
Conclusion
In summary, our study offers significant insights into the molecular underpinnings that distinguish the effects of FLASH-IR from those of CONV-IR. By revealing the precise molecular alterations triggered by FLASH irradiation in skin fibroblasts, our work not only lays a robust groundwork but also enriches the scientific discourse. This contribution is poised to inform future investigations, potentially leveraging these insights to bolster the safety and effectiveness of radiotherapy in oncological applications.
Footnotes
Acknowledgements
We thank the National Natural Science Foundation of China (12275192), the project from State Key Laboratory of Radiation Medicine and Protection (GZK1202308, GZK12024046), the Nuclear Technology Application Excellent Experts Program of the Second Affiliated Hospital of Soochow University (XKTJ-HRC2021002), Medical Key Discipline Funding Plan of Suzhou City (SZFCXK202137), Discipline Priority of Suzhou City, Discipline Construction Support Project (XKTJ-XK202410) for financially supporting the research.
Author Contributions
XM: Study design, data acquisition, data analysis, and data interpretation and manuscript drafting. PQ: Data collection and analysis. ZJ: Data acquisition, and analysis. XZ: Data collection. CX: Data collection. CZ: Study design, manuscript review, and study supervision. ZY: Study design, manuscript review, and study supervision. All authors have read and approved the final manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (12275192), the project from State Key Laboratory of Radiation Medicine and Protection (GZK1202308, GZK12024046), the Nuclear Technology Application Excellent Experts Program of the Second Affiliated Hospital of Soochow University (XKTJ-HRC2021002), Medical Key Discipline Funding Plan of Suzhou City (SZFCXK202137), Discipline Priority of Suzhou City, Discipline Construction Support Project (XKTJ-XK202410).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.