
Abstract
This study was performed to explore factors influencing the release of the proton pump inhibitor omeprazole from enteric-coated capsules in vitro and absorption in vivo in beagle dogs. Enteric-coated pellets with different enteric coating materials and coating levels were designed and prepared. All self-prepared formulations were characterized in vitro as well as in vivo and compared to the brand and generic commercial products. Evaluation of the corresponding release profiles suggested that coating material was the most critical factor. Enteric coating level determined the lag time before initiation of drug release, and subcoating level affected the drug release rate. Pharmacokinetic studies were performed in beagle dogs to further confirm the influence of formulation factors on drug absorption. Medium at pH 6.8 was a more biorelevant condition for in vitro drug release tests, although medium at pH 6.0 was better for discriminating release profiles of different formulations. A multiple level C in vitro/in vivo correlation was preliminarily established by which Tmax and Cmax of omeprazole formulations could be predicted with release parameters such as Tlag and T25. These results may facilitate quality evaluation and potentially improve the clinical efficacy of generic omeprazole products.
Introduction
Proton pump inhibitors (PPIs), efficacious agents for management of a variety of gastric acid-related disorders, are mostly designed as enteric-coated formulations to prevent degradation by gastric acid when administered orally. Omeprazole is the first PPI agent developed for the treatment of gastric/duodenal ulcers and other gastric acid–related disorders. The brand name drug consisting of omeprazole enteric-coated capsules is Losec (a product from AstraZeneca, London, UK), which was approved in 1989 by the US Food and Drug Administration (US FDA). A number of generic omeprazole enteric-coated capsules (or tablets) have become available since the patent for Losec expired. The use of generic products is encouraged in many countries over brand name versions of the same drug, as they are of the same performance and quality but have lower costs. Nevertheless, feedback from physicians and patients has shown that some generic omeprazole enteric-coated capsules are not as effective as the branded product. 1,2 Previous studies indicated that generic products showed remarkable delay in drug absorption compared to the branded product. 3,4 Similar results were also reported for other generic PPIs. 5,6 However, few studies have been performed to investigate the mechanisms underlying such delays.
A multiple-unit pellet system is commonly used to produce omeprazole enteric-coated capsules. An enteric-coated pellet consists of a drug core, a subcoating layer to prevent degradation of the drug due to contact with the acidic enteric coating material, and an enteric coating layer. Coating materials and the coating level of each functional layer can alter the drug release characteristics in vitro and in vivo. Absorption of most PPIs is speculated to occur in the proximal small intestine, where the enteric coating may lead to delayed drug release from the pellets in vivo, which may be responsible for the low drug absorption. 7 Therefore, an ideal enteric coating for omeprazole should be intact in the acidic gastric environment, and more importantly, it should dissolve rapidly in the duodenum after gastric emptying. 8,9 The branded excipient Eudragit L100-55 is a widely used enteric coating material containing a copolymer based on methacrylic acid and ethyl acrylate. An enteric coating of Eudragit L100-55 maintains the nonionized state, with poor permeability in the low pH environment of the stomach. Once it reaches an environment with a pH >5.5, the polymer ionizes and dissolves, resulting in release of the drug in the human duodenum. 10 Some analogs containing molecular functional groups similar to those used in Eudragit L100-55 are also employed for the same purpose in enteric-coated products. For example, polyacrylic resin emulsion fluid (PAEF) is a cheaper substitute for Eudragit L100-55 and is preferred by some manufacturers in China to produce generic omeprazole enteric-coated capsules. The longer ester chain and higher degree of esterification in PAEF increase the ionization and dissolution threshold of polymers to pH 6.5, which is likely to result in a greater delay in drug release. However, there have been no comparative studies regarding the use of PAEF and Eudragit L100-55 as enteric coating materials, including their respective influences on drug release in vitro and pharmacokinetics in vivo.
This study was performed to explore formulation factors that contribute to the performance of omeprazole enteric-coated capsules and to investigate the influences of different enteric coating materials. Five formulations of pellets coated with Eudragit L100-55 at various levels and 1 formulation coated with PAEF were designed and prepared. Use of a mixed polymer coating was reported to show improved performance of enteric-coated pellets, in which the ratio of polymers in the mixture was the determining factor for drug release rate. 11 -14 Thus, a formulation coated with a mixture of PAEF and Eudragit L100-55 was also designed and prepared in the present study. All formulations were characterized in terms of in vitro drug release profile in media at both pH 6.0 and pH 6.8 as well as in vivo pharmacokinetics in beagle dogs. One commercial generic omeprazole enteric-coated capsule product from the Chinese market and the brand product, Losec (AstraZeneca), were collected for comparative research with self-prepared formulations. Furthermore, an in vitro/in vivo correlation (IVIVC) was performed to allow prediction of the in vivo pharmacokinetic characteristics from the dissolution profile. The aims and related research of the present study are listed in Table 1.
Aims and Research Programs of the Present Study.

Abbreviations: Cmax , maximum plasma drug concentration; Tmax , the time to achieve Cmax ; AUC, areas under the curve; PAEF, polyacrylic resin emulsion fluid.
a Form.U: commercial generic product with unknown coating material and level.
Materials and Methods
Materials
The reference product, omeprazole enteric-coated capsules (10 mg, Lot NAAK; Losec) was obtained from the original manufacturer (AstraZeneca, London, UK). The active pharmaceutical ingredient of omeprazole used in this study was obtained from Jiangsu Aosaikang Pharmaceutical Ltd (Jiangsu, China). Sugar cores (0.90-1.12 mm in diameter) were purchased from Hangzhougaocheng Co, Ltd (Zhejiang, China). The commercial products, Opadry 03K19229 and Acryl-EZE 93O18508, were both gifts from Colorcon (Shanghai, China). Polyacrylic resin emulsion fluid was obtained from Wantai Co, Ltd (Jiangsu, China) and hydroxypropyl methylcellulose (HPMC, E6) from Dow Chemical Company (Midland, Michigan). Other excipients used for formulation optimization include
Preparation of Omeprazole Enteric-Coated Pellets
Seven formulations of enteric-coated pellets designed with different coating materials and coating levels were prepared as summarized in Table 2 (Form.1 to 5, Form.B, and Form.P). All formulations were prepared using a fluidized bed coater (Mini-DPL; Chongqing Jinggong Pharmaceutical Machinery Co, Ltd, Chongqing, China). Omeprazole was suspended in HPMC solution containing Tween-80 as a solubilizing agent and simethicone as a stabilizer. The suspension was then adjusted to pH 11 using
Enteric Materials and Coating Levels of Different Formulations Prepared in This Study.
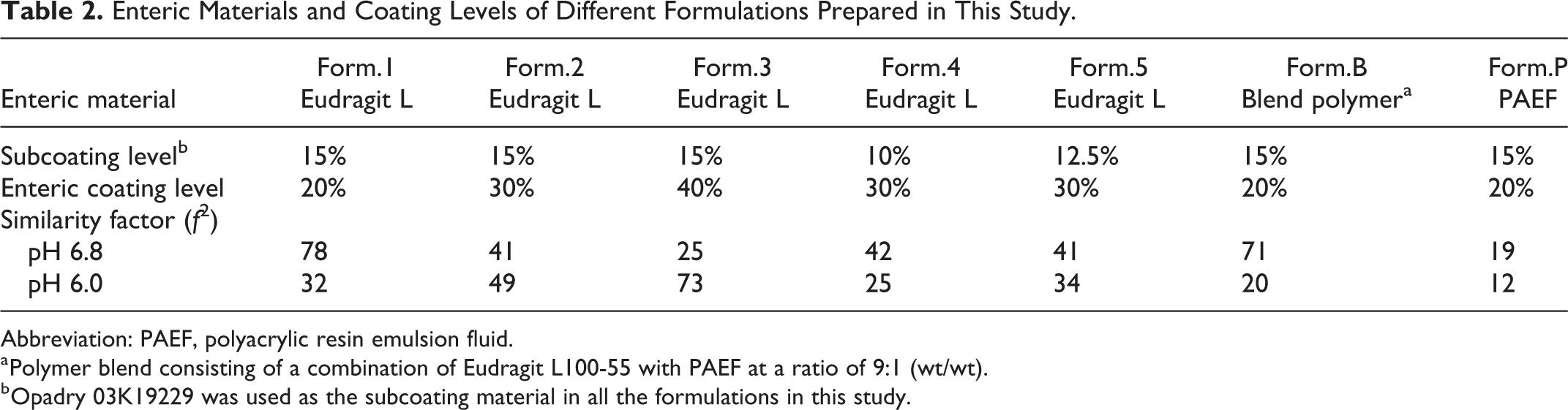
Abbreviation: PAEF, polyacrylic resin emulsion fluid.
a Polymer blend consisting of a combination of Eudragit L100-55 with PAEF at a ratio of 9:1 (wt/wt).
b Opadry 03K19229 was used as the subcoating material in all the formulations in this study.
Comparison of Two Types of Enteric Materials Used in the Design of the Polymer Blend.

Abbreviation: PAEF, polyacrylic resin emulsion fluid.
a MA, methacrylic acid; EA, ethyl methacrylate; BA, butyl methacrylate.
In Vitro Release Study
In vitro drug release experiments using self-prepared pellets and commercial products were carried out by the paddle method as described in Chinese Pharmacopoeia 2015. The media were maintained at a temperature of 37.0°C ± 0.5
Twelve capsules of each formulation and commercial products were tested to plot the drug release profile and calculate the similarity factor (f2 ). The f2 determined according to Equation 1, as suggested by the US FDA, 15 was used to evaluate the similarity of dissolution profiles between self-prepared formulations and the brand product, Losec.
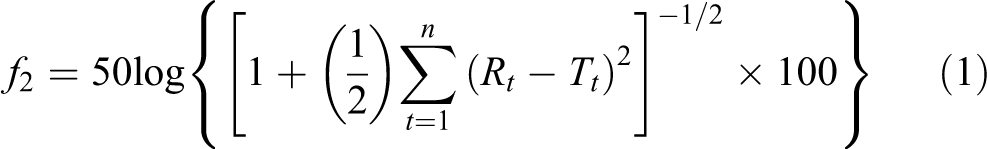
where Rt and Tt are the dissolution values of the brand product Losec and self-prepared formulation, respectively, at time t, and n is the number of time points selected to calculate the similarity factor f2 . Two drug release profiles were considered similar when f2 was not less than 50.
Pharmacokinetic Studies
Animals
Six male beagle dogs were used in 2 programs of pharmacokinetic studies. All animals in the experiments received care in compliance with the “Principles of Laboratory Animal Care” and “Guide for the Care and Use of Laboratory Animals” of Peking University. For program No. 1, a 3-period crossover single-dose design (Table 1, Program No. 1) was conducted with Form.1, Form.P, and the brand product to investigate the influence of different coating materials on product performance in vivo. Six beagle dogs (provided by Beijing Vital River Laboratory Animal Technology Co, Ltd, Beijing, China) weighing 10 to 11 kg was divided randomly into 3 groups. The dogs were fed standard laboratory chow and fasted overnight with water prior to drug administration. The enteric-coated pellets in capsules were orally administered to beagle dogs with water in a single dose of 40 mg. The washout period between administrations was 1 week. More formulations were compared in program No. 2, in which the animals were divided randomly into 2 groups. Investigations of Form.B versus Form.U and Form.3 versus Form.5 were performed in parallel experiments during 2 successive periods separated by a 1-week washout period. The dosage in program No. 2 was the same as that in program No. 1. In both programs, a blood sample of 2 mL was collected via the forelimb vein of each dog before dosing and at predetermined time intervals after dosing. The blood samples were centrifuged immediately at 3000 rpm for 10 minutes to separate the plasma. Aliquots of 0.3 mL of plasma were then sampled, and 0.1 mL of phosphate buffer (Na2HPO4, 0.25 mol/L) was added to improve the stability of omeprazole in plasma. The plasma samples were stored at −20
Determination of omeprazole concentration in plasma
Plasma concentrations of omeprazole in the beagle dogs were determined by HPLC using an internal standard method. Briefly, the plasma sample was mixed with 50 μL of internal standard solution (carbamazepine dissolved in methanol, 10 μg/mL) and then extracted with 3.0 mL of dichloromethane by vortexing for 2 minutes. Following centrifugation at 4000 rpm for 5 minutes, 2 mL of the organic phase was separated and evaporated under a gentle stream of nitrogen. The residue was reconstituted in 100 µL of mobile phase, and 30 µL was subjected to HPLC analysis.
Separation was performed on a C18 column (150 × 4.6 mm, 5 μm, Purospher STAR; Merck) at 30°C using acetate buffer solution (0.05 M CH3COONH4, adjusted to pH 7.0 with ammonium hydroxide)–acetonitrile–methanol (61:35:4, vol/vol/vol) as the mobile phase. The flow rate was 1.0 mL/min, and samples were monitored at 302 nm with an ultraviolet detector. Good linearity was obtained in the range from 0.02 µg/mL to 5 µg/mL with a limit of quantity of 0.005 µg/mL. The accuracy and precision of the method and sample stability were acceptable for quantitative analysis of omeprazole in plasma. 16
Pharmacokinetic parameters
WinNonlin software (version 6.3.0; Pharsight Corp, Mountain View, California) was used to calculate the pharmacokinetic parameters. The maximum plasma drug concentration (Cmax ) and the time to achieve Cmax (Tmax ) were obtained directly from the measured values. The areas under the plasma concentration–time curve (AUC0-t) and mean retention time were calculated by noncompartmental model analysis. The relative bioavailability (F) was determined using Equation 2:

where AUCT and AUCR refer to the AUC of the test and reference formulations, respectively. In Program No. 1, the ratios for lnCmax and ln AUC0-t of Form.1 and the reference were calculated, and the 90% confidence interval (CI) and probability of exceeding the limit of acceptance (80%-125%) were obtained by the 2-sided t test. 17 The formulations were considered bioequivalent if the ln-transformed ratios (test/reference) of Cmax and AUC0-t were within the predetermined equivalence range of 80% to 125%, and P < .05 for the 90% CI. 18
The pharmacokinetic parameters are presented as the means ± standard deviation in Table 4. The observed variation in pharmacokinetic parameters was tested with 1-way analysis of variance using SPSS software (SPSS Statistics version 19.0; IBM Corp, Armonk, New York). In all analyses, P < .05 was taken to indicate statistical significance.
Pharmacokinetic Parameters of the Omeprazole Enteric-Coated Pellets in the Beagle Dogs.a

Abbreviations: Abbreviations: Cmax , maximum plasma drug concentration; Tmax , the time to achieve Cmax ; AUC, areas under the curve; MRT, mean retention time; SD, standard deviation.
a Results are represented as mean ± SD.
b P < .05: significantly different from the reference.
In Vitro/In Vivo Correlations
Release profiles in medium at pH 6.8 of all self-prepared and commercial products were first fitted by applying different models, including zero-order, first-order, Higuchi, Korsmeyer-Peppas, Hixson-Crowell, Weibull, and logistic equations. The best model (with a correlation coefficient of r > .99) was selected to estimate the parameters Tlag , T25 , T50 , and T90 , representing the start time of release and the times at which 25%, 50%, and 90% of drug had been released in vitro, respectively. To determine the relationship between the release curves and pharmacokinetic parameters, T25 , T50 , T90 , and Tlag from formulations with different release rates (fast, middle, and slow release) were determined using linear regression with different pharmacokinetic parameters (Tmax , Cmax , and AUC), and a multiple C level for IVIVC was developed.
The IVIVC was evaluated based on the correlation coefficient (r) and the internal/external predictability. Depending on the intended application of an IVIVC and the therapeutic index of the drug, evaluation of prediction error (PE) internally and/or externally may be appropriate. As described in the US FDA guidelines, 19 evaluation of internal predictability is based on the initial data used to define the IVIVC model. Evaluation of external predictability is based on additional test data sets, and the percent predicted error can be calculated by Equation 3:

The percentage PE (% PE) for each formulation should not exceed 15%.
Results and Discussion
In Vitro Drug Release
The dissolution profiles of the brand product Losec, commercial generic product, and 7 self-prepared formulations with different coating materials and coating levels in media at pH 6.8 and pH 6.0 were plotted. Under the experimental conditions described in Preparation of Omeprazole Enteric-Coated Pellet section, all formulations prepared had a high drug loading efficiency >90% and a coating efficiency >85% with rare adhesion pellets. The HPLC method (Section “In Vitro Release Study”) validation results demonstrated that the method met the requirements for concentration determination of omeprazole in the dissolution media. All products passed the gastroresistance test in media at pH 1.2 for 2 hours with degradation <10% (data not shown). The drug release of enteric-coated products is usually assessed in phosphate buffer at pH 6.8 according to the pharmacopoeia of China, Japan, and United States. On the other hand, as the pH of the luminal contents of the proximal duodenum shows more prolonged lowering in duodenal ulcers, 9 media with pH 6.8 and pH 6.0 were chosen to evaluate the drug release behavior. The release profiles are shown in Figure 1A and B. The f2 of each formulation compared to the reference brand product was calculated as listed in Table 2. Form.1 and Form.3 showed the greatest similarities in release profiles to the brand product in media at pH 6.8 and pH 6.0, respectively.

Dissolution profiles in media at pH 6.8 and pH 6.0. (A) pH 6.8; (B) pH 6.0; (C) pH 6.8 with different coating materials; (D) pH 6.0 with different coating materials. Form.1 to Form.5: formulations with different coating levels of enteric coating and subcoating using Eudragit L100-55 as enteric coating material; Form.P: formulation using PAEF as enteric coating material; Form.B: formulation using polymer blend consisting of Eudragit L100-55 and PAEF at a ratio of 9:1 as enteric coating material; Form.U: commercial generic product with unknown coating material and level; Ref.: Losec. n = 12. PAEF indicates polyacrylic resin emulsion fluid.
Effects of coating level on drug release
For most enteric coating formulations, coating level is a critical point that can markedly alter the dissolution profile. A prolonged release rate will usually be achieved with increased coating level due to increased diffusion pathways, according to Fick’s second law of diffusion. 20 The results of the present study clearly demonstrated the specific impact of variation in enteric coating levels and subcoating levels on omeprazole release. The dissolution profiles of all formulations showed a lag time until commencement of drug release (Figure 1A and B). The self-prepared formulations coated with Eudragit L100-55 showed that the thicker enteric coating caused a longer lag time. As the TWG% of coating material increased from 20% to 40% (Form.1, Form.2, and Form.3), the drug release curves obtained gradually extended the lag time but maintained almost the same slope in both media (Figure 2A and B), suggesting that formulations with different enteric coating levels had the same drug release rate once release had begun. On the other hand, increased subcoating thickness had the opposite effect as shown in Figure 2C and D. Formulations with subcoating levels from 10% to 15% (Form.4, Form.5, and Form.2) all released omeprazole after 10 minutes without marked differences with regard to lag time in medium at pH 6.0 (Figure 2D). The dissolution rate accelerated as the subcoating level decreased where Form.4, which had the minimal TWG% of subcoating, showed a burst release after the enteric coating dissolved (Figure 2D). Overall, the abovementioned results indicated that the enteric coating controlled the time until the formulation began to release the drug, whereas the subcoating obstructed release. Based on this, an optimized formulation of omeprazole enteric-coated pellets could be expected to exhibit the closet dissolution profile to that of the brand product. A better understanding of the factors impacting the release of omeprazole would help improve the quality of generic drugs.

Dissolution profiles of formulations with different coating levels: (A) pH 6.8, enteric coating level; (B) pH 6.0, enteric coating level; (C) pH 6.8, subcoating level; (D) pH 6.0, subcoating level. n = 12.
Effects of coating materials on drug release
Form.1, Form.P, and Form.B were self-prepared formulations coated with Eudragit L100-55, PAEF, and mixed polymer materials of Eudragit L100-55 and PAEF at a ratio of 9:1, respectively. All were applied at the same coating level. The results showed that Form.P started to release the drug into the medium at pH 6.8 only after 40 minutes, which was approximately 35 minutes later than Form.1 and did not release the drug at all within 60 minutes in medium at pH 6.0 (Figure 1C and D). Interestingly, the commercial generic product, Form.U, also retained the drug in pellets at pH 6.0 and released the drug at 15 minutes in medium at pH 6.8, representing a delay of 10 minutes compared to Form.1 (Figure 1C and D). The results strongly suggested that the commercial generic product Form.U was coated with PAEF, which was used as the enteric coating material for Form.P, because both formulations showed the same obstruction for drug release in medium at pH 6.0. The shorter lag time of drug release from Form.U in medium at pH 6.8 may result from a lower enteric-coating level of Form.U than Form.P. Polyacrylic resin emulsion fluid is a widely used enteric coating substitute for Eudragit L100-55 in China because of its low cost and ease of manufacture. Although both possess the same methacrylic functional group, the ionization threshold of PAEF is at pH 6.5, whereas that of Eudragit L100-55 is pH 6.0, resulting from a more esterized structure in PAEF (Table 3). As illustrated in Figure 1D, the drug was hardly released due to the difficulty in dissolution of PAEF in media at pH 6.0. The marked difference in in vitro performance between Form.1 and Form.P suggested that polymer materials in enteric coatings with different chemical structures are the determining factors for the release profiles of enteric-coated formulations.
As Form.1 showed a faster release rate compared to the reference in medium at pH 6.0 (Figure 1D), 10% PAEF was added to Eudragit L100-55 to obtain a blend of polymers at a ratio of 9:1 to reduce the release rate of Form.B in medium at pH 6.0 and achieve similar release profiles to the reference brand product at both pH 6.0 and pH 6.8. Unexpectedly, Form.B showed even faster drug release than Form.1. As shown in Figure 1D, the drug was released abruptly from Form.B at 10 minutes, suggesting that the subcoating and enteric coating dissolved simultaneously. In the subcoating polymer, the chain of HPMC relaxes with decreasing polymer concentrations and increasing macromolecule mobility in water. 21,22 When pellets were coated with the blend of polymers, cracks may have appeared in the enteric coating film at the edges of areas of contact of the 2 materials due to the differences in pH sensitivity. In these regions, more water would diffuse into the enteric coating and accelerate dissolution of the subcoating layer in advance so that the collapse of the 2 coating layers would occur at the same time, resulting in abrupt drug release. As omeprazole is given intraduodenally and the pH of the duodenum ranges between 5.0 and 6.0, 23 the use of an enteric coating made of a blend of polymers may facilitate drug absorption and earlier onset of action.
Drug release in different dissolution media
The release profile of generic products should aim to match that of the brand formulation during the development of generic products. The release test results showed that none of our self-prepared formulations was similar to the reference product, Losec, in terms of release profile in media at both pH 6.8 and pH 6.0. For example, the release profile of Form.1, coated with Eudragit L 100-55 at 20% TWG%, was most similar to that of the reference in medium at pH 6.8 (f2 = 78; Figures 1C and 2A) but faster than that of the reference at pH 6.0 (f2 = 32; Figures 1D and 2B). To slow the release rate, Form.3 was coated with more enteric materials and matched the reference at pH 6.0 (f2 = 73; Figure 2B) but showed a more prolonged release profile than the reference at pH 6.8 (f2 = 25; Figure 2A). Form.B, coated with mixed polymers, showed similar in vitro performance to the reference at pH 6.8 (f2 = 71; Figure 1C) but the fastest release rate at pH 6.0 (f2 = 20; Figure 1D). Meanwhile, greater discrimination in the release profiles for different formulations was found at pH 6.0 (Figure 1B) than at pH 6.8 (Figure 1A), indicating that testing in medium at pH 6.0 would better reflect the formulation and process changes for omeprazole enteric-coated pellets than in medium at pH 6.8. However, investigation of the pharmacokinetics was still necessary to identify the formulation with the most similar in vivo performance to the reference product and biorelevant conditions for in vitro release tests.
In Vivo Pharmacokinetic Study
The mean plasma concentration–time curves with error bars of omeprazole for the self-prepared pellets and both commercial products are shown in Figure 3, and the main pharmacokinetic parameters determined by the noncompartmental approach are listed in Table 4. As there was a failure to administer the capsules to 1 beagle dog during the experiment in Program No. 2, in vivo data for Form.5 were only available from 2 dogs and were thus not included in the subsequent analyses.

Plasma concentration profiles in beagle dogs after oral administration. Each point represents the mean ± SD of 6 or 3 experiments. (A) Reference: Losec; (B) Form.1: formulation coated with Eudragit L100-55 at 20% TWG; (C) Form.B: formulation coated with blend of polymers; (D) Form.3: formulation coated with Eudragit L100-55 at 40% TWG; (E) Form.P: formulation coated with PAEF; (F) Form.U: commercial generic product; a, b, e: n = 6; c, d, f: n = 3. PAEF indicates polyacrylic resin emulsion fluid; SD, standard deviation; TWG, theoretical percentage of weight gained.
In Program No. 1, drug plasma concentrations of the reference product increased quickly and reached Cmax at 1.277 ± 0.327 hours after oral administration (Figure 3A and Table 4), suggesting that omeprazole was released rapidly and absorbed in vivo. Form.1, with a coating of Eudragit L100-55, showed a very similar in vivo profile to the reference product, with no significant differences in the Cmax , Tmax , or AUC0-8 h values (P > .05; Figure 3B and Table 4). The 90% CI of the ratio of the AUC0-8 h of Form.1 to the reference was 95.6% to 119.8%, falling within the bioequivalence limit range of 80% to 125% according to the guidelines of the US FDA. 24 The ratio for Cmax was 88.0% to 145.1%, which was beyond the bioequivalence limit, and this may have been due to the small number of test cases and high variation in omeprazole. 25 However, Form.1 was still expected to show bioequivalence with the reference product. Form.P, coated with PAEF, showed much slower drug release in vitro (Figure 1C and D) and exhibited significantly delayed absorption in vivo (Figure 3E). The Tmax of Form.P was nearly 3 times greater than that of the reference product as well as that of Form.1. Cmax and AUC0-8 h were also significantly decreased in comparison to the reference product, from 1.904 ± 0.320 to 0.981 ± 0.453 and from 3.569 ± 0.798 to 2.496 ± 0.729 (Table 4), respectively. The results outlined earlier indicated that enteric coating material plays a key role in drug release in vitro and absorption in vivo.
The pharmacokinetic behaviors of Form.3, Form.B, and Form.U were examined in Program No. 2. Form.3, with the highest coating level of Eudragit L100-55, showed delayed absorption with Tmax at 2.667 ± 0.577 hours, which was almost double that of the reference product. However, the AUC0-8 h (4.006 ± 0.982 μgh/mL) and Cmax (1.856 ± 0.464 μg/mL) of Form.3 were not significantly different from those of the reference product (P > .05; Table 4), indicating that increasing the enteric coating level within a certain range may affect the rate but not the extent of drug absorption in vivo. For Form.B (Figure 3C), formulated with a blend of polymers, no significant differences related to the fastest drug release in vitro in medium at pH 6.0 were observed in the pharmacokinetic parameters of Tmax , Cmax , and AUC0-8 h in comparison to the reference (Figure 1D and Table 4). Form.U exhibited a similar pattern to Form.P, with no significant differences in terms of Cmax , Tmax , and AUC0-8 h (P > .05). Although Form.U showed faster drug release than Form.P in vitro in medium at pH 6.8 (Figure 1C), both formulations showed decreased absorption to the same degree compared to the reference (Figure 3F and E). These observations suggest that for any formulation, if drug release is delayed for over 30 minutes in vitro, bioavailability may be severely reduced. The inhibition of drug absorption may be related to the decreased water availability as the drug moves distally from the small intestine and increased fluid viscosity in the colon. 26 The poor solubility also makes omeprazole sensitive to such factors and influences drug absorption. 27 The results of our pharmacokinetic study in dogs indicated that it was highly possible Form.U was not bioequivalent to the brand product, Losec, in humans, despite its status as an approved generic product. It is necessary to optimize the formulation and process and reevaluate commercial products such as Form.U in vitro and in vivo.
For all tested formulations and products, the trend in Tmax values increased in the order: reference product ≈ Form.1 ≈ Form.B < Form.3 < Form.P ≈ generic product (Form.U), which showed better agreement with the in vitro drug release delay in medium at pH 6.8 than at pH 6.0. The trends of Cmax and AUC0-8 h better matched the change in the release profile at pH 6.8 than at pH 6.0. These observations indicated that pH 6.8 better reflected the performance in vivo, which was inconsistent with the inferences from in vitro release tests, where medium at pH 6.0 was better for discriminating the different formulations.
In Vitro and In Vivo Correlation
As outlined earlier, the self-prepared formulations and 2 commercial products showed differences in both drug release rate in vitro and extent of absorption in vivo, making it possible to determine the vitro and in vivo correlation. The release profiles in medium at pH 6.8 were used to establish IVIVC because of the high degree of biorelevance of these conditions. Form.1, Form.3, and Form.U, representing fast, middle, and slow release, respectively, were used along with the reference to explore and establish the IVIVC.
Fitting of drug release equation
The mechanism of omeprazole release at pH 6.8 from enteric-coated pellets was investigated by fitting the in vitro data into zero order, first order, Higuchi, Korsmeyer-Peppas, Weibull, and logistic models. The correlation coefficient (r), release rate constant (k), and release exponent (n) values obtained after linear regression in various kinetic models are shown in Table 5. The correlation coefficient (r) was used to determine the best model (with r > .99). The release rate data showed the best fit to the Korsmeyer-Peppas model, with r > .99 for all formulations. The release exponents (n) ranged between 0.088 and 0.200 except for Form.U, confirming that diffusion was the principal mechanism of drug release (n < 0.5). 28 Moreover, the processes of polymer erosion and drug dissolution resulted in S-shaped release profiles for enteric capsules; thus, release data also showed a good fit to the Weibull and logistic models (r > .99) that can describe S-shaped/sigmoidal release profiles. 29 As a simpler fitting equation is preferable, the equation of the Korsmeyer-Peppas model was selected to describe the release curve for developing the IVIVC.
Release Kinetic Parameters and Correlation Coefficients of Each Equation for Different Formulations.

Development and evaluation of multiple level C correlation
The parameters Tlag , T25 , T50 , and T90 , representing the lag time before the start of drug release, and early, middle, and late time points with 25%, 50%, and 90% of drug released, respectively, were used to describe the drug release in vitro. Tlag was read directly from the release profiles, whereas T25 , T50 , and T90 were calculated by fitting to the equation of the Korsmeyer-Peppas model (Table 6). T25 , T50 , T90 , and Tlag were subjected to linear regression analyses with different pharmacokinetic parameters (Tmax , Cmax , and AUC) for the different formulations, that is, the reference product, Form.1, Form.3, and Form.U (Table 7).
In Vitro and in Vivo Parameters Used in Establishment of IVIVC.

Abbreviations: Cmax , maximum plasma drug concentration; Tmax , the time to achieve Cmax ; AUC, areas under the curve; IVIVC, in vitro in vivo correlation.
a In vitro parameters were calculated by equation of Korsmeyer-Peppas with Tlag .
b In vivo parameters were pharmacokinetic parameters presented as mean values.
Linearity of In Vitro and In Vivo Parameters Presented in Multiple Level C Correlation.

Abbreviations: Cmax , maximum plasma drug concentration; Tmax , the time to achieve Cmax ; AUC, areas under the curve.
a A definite linearity (r >.95) was presented.
Definite linear correlations (r > .95) were established between Cmax and T25 , T50 , and T90 , with r > .97 for all (Table 7; Figure 4A). The internal predictability test indicated that the correlation equation between Cmax and T25 yielded the most accurate predicted result, although all 3 equations gave predicted values within the US FDA criteria (Table 8). 19 A more rigorous linear correlation between Tlag in vitro and Tmax in vivo was established, with r = .999 (Table 7; Figure 4B). This result indicated that Tmax in vivo could be predicted by Tlag in vitro. The dissolution of enteric polymers reflected by Tlag and Tmax had a direct influence on drug release and absorption. For carboxylic polymers, polymer dissolution in aqueous solution was proposed to consist of 5 steps, 29 with diffusion of water and hydroxyl ions into the polymer matrix and disentanglement of polymer chains out of the gel layer to the polymer–solution interface being the most important steps. Variations in the pH, concentration, and acidity of proton carriers is due primarily to changes in concentration of hydrogen ions at the polymer–solution interface. Based on the abovementioned analysis, the linear correlation between Tlag in vitro and Tmax in vivo observed in this study indicated that the in vitro dissolution medium (pH 6.8) reflected the dissolution process of the enteric layer in vivo. The pharmacological activity of omeprazole was mostly reflected by the AUC, whereas linear correlations (with r = .8-.95) observed between in vitro parameters with AUC values were weaker than those of Cmax and Tmax . This was mainly due to Form.3, for which Tmax increased with increasing Tlag , but AUC did not significantly change (Table 7; Figure 4C and D).
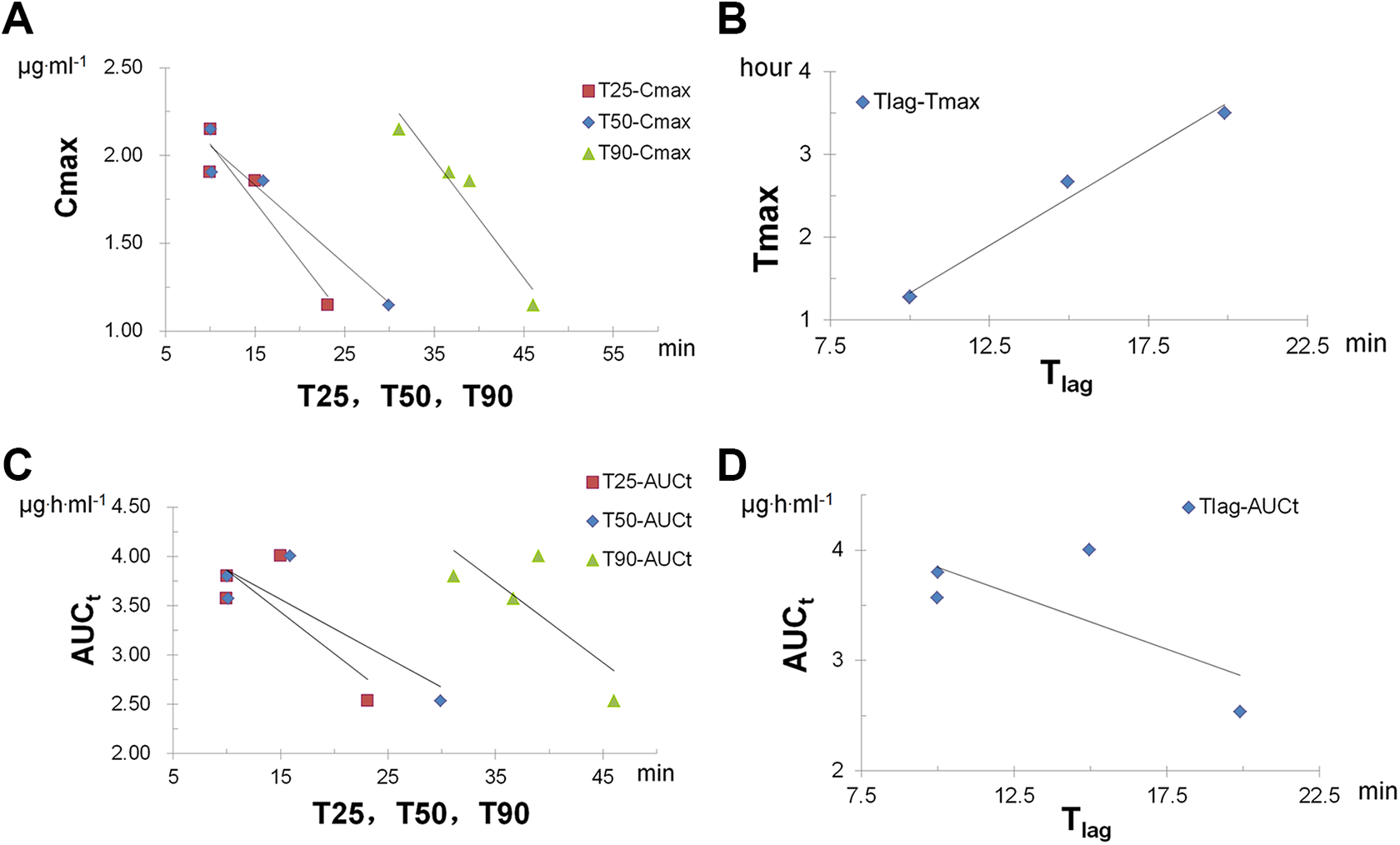
Multiple level C in vitro/in vivo correlation (IVIVC) regressions between in vitro parameters and in vivo parameters. (A) T25, T50, T90 vs Cmax; (B) Tlag vs Tmax; (C) T25, T50, T90 vs AUCt; (D) Tlag vs AUCt. AUC indicates area under the curve.
Evaluation of the Predictability of the Multiple Level C Correlation of Cmax.

a Criteria of the US Food and Drug Administration.
The predictive IVIVC was established and used to examine internal and external validation. As defined in the US FDA guidelines, a multiple level C correlation relates one or several pharmacokinetic parameters of interest to the amount of drug dissolved at several time points in the dissolution profile. Multiple level C correlation can be as useful as level A correlation, especially in the early stages of formulation development during the pilot formulation selection process. Considering the different medicinal properties of generic drugs on the market, 30 the establishment of an in vitro discriminative method and application of multiple level C correlations could help to predict drug absorption in vivo and facilitate quality evaluation of generic products.
It should be noted that to investigate more influential factors, IVIVC in the present study was established with formulations used in 2 pharmacokinetic programs; thus, the interindividual and interperiod variabilities were neglected. In addition, the pharmacokinetic parameters were collected in dogs that have higher and more variable gastric pH than humans. 31,32 If the dogs had been pretreated with pentagastrin before the pharmacokinetic studies, the environment of the gastrointestinal tract would have been closer to that of humans, with a lower pH and decreased variation both between and within individuals. 9 The results and extrapolation would have been more representative of the situation in humans.
Conclusions
This study was performed to investigate the dissolution profiles of enteric-coated pellets prepared with different materials and various coating levels, together with pharmacokinetic studies in beagle dogs. The results indicated that the coating material was the most critical determinant of drug release and absorption. Modified enteric coating with a blend of polymers has the potential to accelerate drug release and result in earlier absorption, as well as earlier onset, of medication. In addition, this study showed that coating level plays a key role in drug release rate and that excessive coating may inhibit drug release and absorption. Enteric coating level determined the lag time before the start of drug release, and subcoating level affected drug release rate. Drug release tests in medium at pH 6.0 better reflected the formulation and process changes for omeprazole enteric-coated pellets than those in medium at pH 6.8. However, pH 6.8 is a more biorelevant condition for drug release tests in vitro than pH 6.0. The multiple level C correlation established in preliminary experiments not only indicated the impact of formulation factors on drug release but also helps to predict drug absorption in vivo through its release.
In conclusion, the results of the present study will help to clarify strategies for developing omeprazole enteric-coated capsule products, including formulation design, process modification, and quality evaluation. The system developed here may be applied to other PPIs due to the highly conserved physicochemical characteristics and mechanisms of action among these drugs.
Footnotes
Authors’ Note
Cheng Cui and Jiabei Sun contributed equally to this work.
Acknowledgments
The authors would like to thank Prof Qiang Zhang, from the School of Pharmaceutical Sciences, Peking University, for his very helpful advice to the present study, especially to the formulation design and of pharmacokinetic study.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.