
Abstract
In this study, a temperature responsive PGS/PLLA@PNIPAM core-shell nanofiber membrane was prepared by combining electrospinning with surface ATRP grafting polymer technology, in which the core layer of PGS/PLLA nanofiber was prepared by electrospinning, and then the shell layer of temperature responsive PNIPAM polymer was grafted on the nanofiber surface by ATRP reaction. In the experiment, a macromolecule-initiator PGS-Br was prepared and characterized by FTIR and 1H NMR. The surface morphology, composition, element of the core-shell nanofiber membrane were characterized by SEM, FTIR, and XPS. In addition, it is worth noting that the core-shell nanofiber membrane can respond to temperature in drug release tests.
Introduction
Electrospinning is a simple, fast, and efficient method for preparing nanofibers with the diameter from a few nanometers to tens of nanometers. 1 As a kind of one-dimensional nanofiber material, electrospun nanofibers have attracted more and more researchers' attention.2,3 Electrospun nanofibers have the advantages of length-to-diameter ratio, large specific surface area and high porosity. Electrospun nanofibers can be controlled by a variety of polymers and easy to be functionalized.4,5 Electrospun nanofibers have been shown to different applications in many fields, such as energy, 6 environment,7,8 tissue engineering, 9 medicine, 10 and sensors, 11 which excellent performances are due to their design tunable structure and composition. Among them, the core-shell nanofibers have novel structures and important applications due to combining the advantages of the inner and outer layer materials. The traditional methods for preparing core-shell nanofibers include coaxial electrospinning, 12 in situ growth 13 and the surface polymerization.14,15 Among them, the method of grafting on the nanofiber surface has a good prospect in the field of drug carrier control and release.16,17 In order to make the more intelligent carrier, a variety of conditional response drug carriers 18 were designed and prepared, such as temperature response carrier19,20 and pH response carrier. 21 Among them, surface-grafted electron transfer-atom-transfer radical polymerization (AGET-ATRP) is a well-known controlled polymerization method to adjust the degree of cross-linking of the polymer shell. 22
Poly (glycerol sebacate) (PGS), which has good biocompatibility, flexibility and degradability, is a kind of biomedical material with excellent performance, and is widely used in various organs and wound patches. However, its spinnability is low. It is not to form nanofiber morphology alone, which limit its application.23,24 Poly-L-lactic acid (PLLA), as a synthetic polymer that can be controlled to degrade, has been studied in clinical medicine for a long time and has good spinnability. It can be used as a spinning aid for PGS, so as to assist in the preparation of PGS nanofiber membrane.25,26 Yan et al. reported a core-shell poly(glycerol sebacate)-poly(l-lactic acid) electrospun membranes for oil spill remediation. 27 However, it does not further functionalize its surface. The polyn-isopropyl acrylamide (PNIPAM) macromolecular chain has hydrophilic acyl amino group and hydrophobic isopropyl group at the same time, which makes the linear PNIPAM aqueous solution and the cross-linked PNIPAM hydrogel show temperature sensitive characteristics. 28 Therefore, as an environmental response polymer has been widely used in drug controlled release and other fields.29–31 It would be very meaningful to combine PGS-PLLA nanofibers with PNIPAM polymer to realize the controlled drugs release of temperature-sensitive.
In this study, PGS/PGS-Br/PLLA nanofiber membrane was firstly prepared by coaxial electrospinning method, and PLLA was used as spinning aid. 32 Then, through ATRP grafting reaction, the surface of the nanofiber membrane was grafted with PNIPAM polymer, an temperature responsive polymer.33,34 Finally, the nanofiber membrane with PGS/PLLA as the core layer and PNIPAM polymer as the shell layer was obtained. It was used as a drug carrier to realize the temperature-sensitive controlled release of drugs.
Experimental section
Materials
Sebacic acid (99%, MW = 202.25, C10H18O4) and glycerol (A.R., MW = 92.09, C3H8O3) were purchased from Aladdin (China), and Sigma (USA), respectively. The poly-L-lactic acid (PLLA, Mn = 200 kDa) were purchased from the Jinan Daigang Biomaterial Co. Ltd (China). N-isopropylacrylamide (A.R., MW = 113.16, C6H11NO) was obtained from Aladdin (China) and was recrystallized by water. 2-Bromoisobutyryl bromide (BiBB, 99%, MW = 229.91, C4H6Br2O) was purchased from Aladdin (China). Triethylamine (TEA, MW = 101.19, C6H15N) was purchased from Sigma (USA). CuBr was purchased from Macklin (China) and was reduced by glacial acetic acid (98%, Aladdin, China). Pentamethyldiethylenetriamine (PMDETA, MW = 173.3, C9H23N3) and Ascorbic Acid (AA, MW = 176.12, C6H8O6) were obtained from Aladdin (China). Dichloromethane (DCM), ethyl acetate (EtOAc), methanol (MeOH), and N,N-dimethylformamide (DMF) were of reagent grade and purchased from Sigma (USA). Deionized water was prepared by water purification machine (UTP-1-10T). Diluted hydrochloric acid (0.01 mol/L) was prepared by diluting the concentrated hydrochloric acid (37.5% HCl) from Beijing Chemical Work (China). All solvents were used without further purification.
Fabrication of the macro-initiator pre-PGS-Br
First, sebacic acid was recrystallized using absolute ethyl alcohol. Then, equimolar ratios of glycerol and sebacic acid were mixed in the flack under 140°C oil bath to melt the sebacic acid, then heat up to 160°C for 8 h to form the PGS pre-polymer. And the nitrogen flow was blown at a gas flow of 0.2 m³ h−1 during the reaction. 35
About 10 g pre-PGS was dissolved in dichloromethane (DCM) in the 50 mL flask, then 900 μL BiBB was added to solution drop by drop. 200 μL TEA was as acid-binding agent in order to promote the reactivity. The solution was stirred by magnetic stirrers at room temperature for 24 h. DCM of the solution was evaporated by rotatory evaporator after reaction ending. EtOAc was added to the above-mentioned product due to remove the reactive triethylamine hydrochloride. And 0.01 mol/L diluted hydrochloric acid was used to remove the remaining TEA by separating funnel. At the end, EtOAc was removed by rotatory evaporator (Yarong RE-52AA, Shanghai, China).
Fabrication of the PGS/PGS-Br/PLLA nanofiber membrane
During fabrication of the PGS/PGS-Br/PLLA nanofiber membrane, 0.8 g pre-PGS, 0.2 g pre-PGS-Br and 1.0 g PLLA were mixed and dissolved in the mixed solvent of DCM and DMF (v:v = 3:1. The concentration of PLLA in the mixed solvent was 10 wt%. The as-prepared solutions were loaded into two 5 mL syringe. The electrospinning equipment is QINGZI NANO E02 (China). The inner diameter of needle is 0.6 mm. And the flow rate of accesses was 0.6 mL h−1. In addition, the electrospinning voltage was 18 kV and the distance between the needle and collector (aluminum foil) was set at 15 cm. The obtained PGS/PGS-Br@PLLA nanofiber membrane was exposed on room temperature to evaporate any residual solvent. Furthermore, the membrane was thermo-cured in 120°C under vacuum drying oven (101-2A, Tianjin, China) for 72 h to cure PGS nanofibers.
Fabrication of the PGS/PLLA@PNIPAM core-shell nanofiber membrane by surface ATRP grafting
The above-mentioned nanofiber membrane containing the macromolecule initiator was added to the three-mouth flask containing methanol or water, and nitrogen was rinsed under the ice water bath for 20 min. The ligand pentamethyldiethylenetriamine (PMDETA) and the monomer NIPAM were added to the flask and degassed with nitrogen for 10 min. Next, the reducing agent AA and catalyst CuBr were added to the flask in a nitrogen atmosphere, and then changed to an oil bath (dimethyl silicone oil), and then reacted at 60°C for 60 min. Finally, the reaction is terminated by removing nitrogen atmosphere and touching the air. After the reaction is completed, the nanofiber membrane is washed with deionized water for three times and then put into a vacuum drying oven (101-2A, Tianjin, China) for drying 24 h.
Characterizations of pre-PGS-Br and the PGS/PLLA@PNIPAM nanofiber membrane
The pre-PGS and pre-PGS-Br were smeared on the potassium bromide (KBr) tablet and characterized by Fourier-transform infrared spectrometer from 4000 to 500 cm−1(FTIR, Nicolet 5700, Thermo Company, USA). The structure of pre-PGS and pre-PGS-Br were characterized by hydrogen nuclear magnetic resonance (1H NMR, AV400 NMR, Bruker Company, Germany). It was tested at 600 MHz using DCl3 as solvent. The morphology of the PGS/PLLA@PNIPAM nanofiber membrane was examined by scanning electron microscopy (SEM, FEI Quanta 250, The Netherlands). At the same time, SEM could also be used with electronic energy spectrum, qualitative test sample elements. The dry membrane sample ensuring they are completely free of moisture was pasted on the sample table and then were sputter coated with gold (Model 550; Electron Microscope Sciences) in preparation for SEM. 35 X-ray photoelectric spectroscopy (XPS, ESCALAB 250, Thermo VG Scientific, USA.) was used to detect the N elements, the corresponding content and to analyze the chemical state within 10 nm of the sample surface.
Characterization of physical properties
The wettability of membranes was characterized by water contact angle measurements (WCA, Samsung FA-CED camera, Korea). To illustrate the thermoresponsive property of PGS/PLLA@PNIPAM membranes.
Drug release test in different temperature
In order to test the temperature responsive effect of drug release, the characteristic absorption peak of gatifloxacin (model drug) at 285.5 nm was measured by ultraviolet spectrophotometry (UV, u-3900, Hitachi, Japan). The quantitative PGS/PLLA@PNIPAM nanofibers loading gatifloxacin were immersed into 50 mL deionized water under different temperature (20°C and 50°C), respectively in oscillation incubator (BS-2F, Dingfeng, China). 1 mL solution was taken out at different times (0, 5, 15, 30, 60, 90, 120, 180, 240, 360, 480, and 600 min) respectively, then tested by ultraviolet absorption three times to calculate the drug release concentration.
Results and discussions
As shown in Scheme 1, the preparation process of the PGS/PLLA@PNIPAM nanofiber membrane consists of two main steps: synthesis of the macro-initiator PGS-Br and grafting PNIPAM polymer by AGET-ATRP on the surface of electrospun PGS/PGS-Br/PLLA nanofiber membrane.

(a) The material synthesis process and (b) the preparation process of the PGS/PLLA@PNIPAM.
Preparation of the pre-PGS-Br
As depicted in 1H NMR spectrum of Figure 1(a), it showed –CH2 groups in the backbone supported by sebacic acid at 1.3, 1.6, and 2.34 ppm, and –CH2, –CH– at 4.18 and 3.7 ppm from glycerol, which were influenced by the distance from ester groups. As shown in Figure 1(b), macro-initiator pre-PGS-Br was evidenced by –CH3 group from BiBB at 2.0 ppm, which means BiBB was successfully grafted on pre-PGS. The pre-PGS was synthesized by the condensation reaction, the FTIR spectrum of pre-PGS in Figure 1(c) shows the peaks for –C=O at 1740 cm−1 and the peaks for –OH at 3438 cm−1, which is completely consistent with the previous report. 35 And in Figure 1(d), the peak of C–Br was showed at 737 cm−1. It also confirmed the formation of pre-PGS-Br. X-ray photoelectron spectroscopy (XPS) results show the co-presence of C, O, N, and Br in the as-prepared samples (Figure 2(b)) compared to the PGS/PLLA membrane (Figure 2(a)), suggesting the successful reacting of BiBB. The high resolution Br 3d spectrum provided in Figure 2(e).
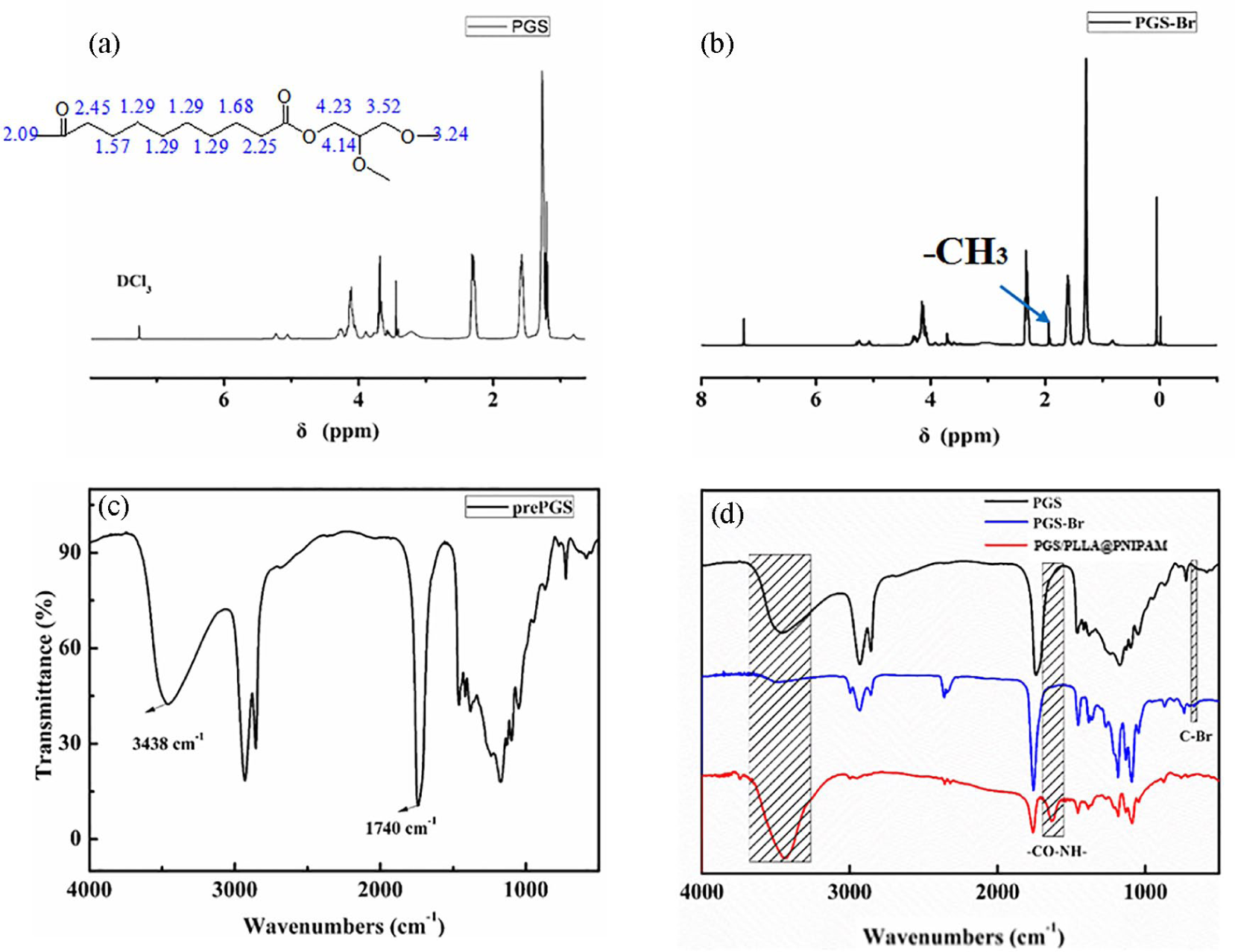
1H NMR spectrum of (a) PGS and (b) PGS-Br. The FTIR spectrum of (c) pre-PGS and (d) pre-PGS, PGS-Br and PGS/PLLA@PNIPAM.
Preparation of PGS/PLLA@PNIPAM nanofiber membrane
The PGS/PGS-Br/PLLA-grafting-PNIPAM polymer was synthesized by AGET-ATRP reaction, the FTIR spectrum of PGS in Figure 1(d) shows the peaks for –CO–NH– at 1640 cm−1 and the peaks for –NH– at 3440 cm−1, which is completely consistent with the previous report. X-ray photoelectron spectroscopy (XPS) results show the co-presence of C, O, and H in the PGS/PLLA nanofiber membrane (Figure 2(a)). The high resolution C 1s shows that the peak more can be deconvoluted into four peaks (Figure 2(d)), O=C–O, C=O, C–O, and C–C, C–H, respectively. After grafting PNIPAM, it shows the co-presence of C, O, N, and H in the PGS/PLLA@PNIPAM nanofiber membrane (Figure 2(c)). The peak more can be deconvoluted into four peaks in the high resolution C 1s (Figure 2(f)), O=C–O, C=O, C–O, C–N, and C–C, C–H, respectively. 36 No significant difference can be obtained from the C 1s XPS spectra in the PGS/PGS-Br/PLLA nanofiber membrane and PGS/PLLA@PNIPAM nanofiber membrane. So N1s spectra of PGS/PLLA@PNIPAM nanofiber membrane was tested as shown in Figure 2(g). The high-resolution N 1s spectrum in Figure 2(g) shows the peak at 129.9 eV, revealing the presence of C–N bonds, which indicating the successful grafting of PNIPAM.
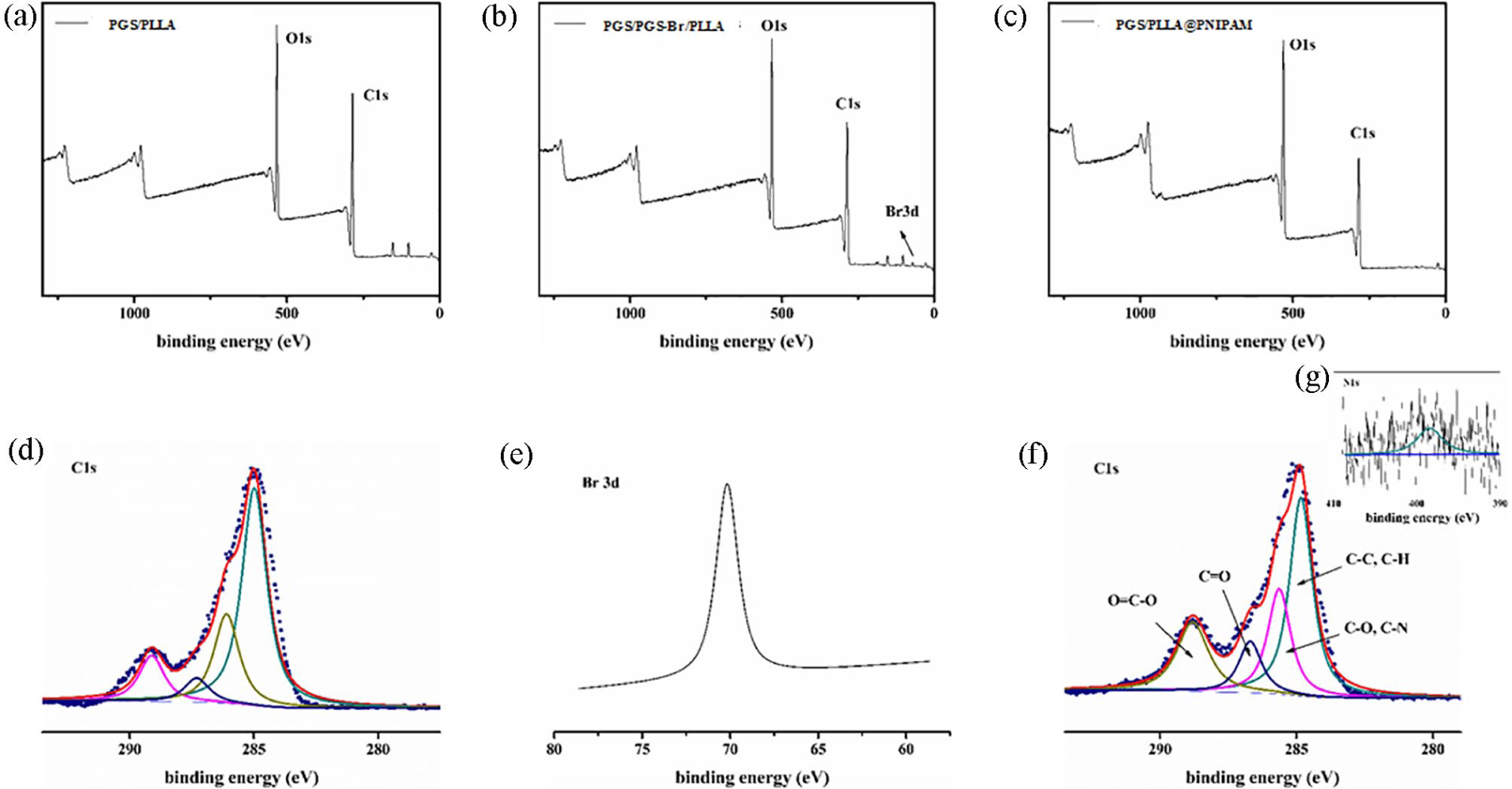
XPS survey spectra: (a) PGS/PLLA, (b) PGS/PGS-Br/PLLA, and (c) PGS/PLLA@PNIPAM nanofiber membrane; (d) high resolution C 1s spectrum of PGS/PGS-Br/PLLA nanofiber membrane, (e) high resolution Br 3d spectrum of PGS/PGS-Br/PLLA nanofiber membrane, (f) high resolution C1s, and (g) N1s spectrum of PGS/PLLA@PNIPAM nanofiber membrane.
Further, in order to prove the successful preparation of PGS/PLLA@PNIPAM, the prepared ground nanofiber membrane was characterized by SEM images and EDS elemental analysis, as shown in Figure 3. Figure 3(a) and (d) showed the SEM images of the nanofiber membranes before and after grafting PNIPAM polymer, and the distributions of N and Br elements were investigated on the nanofiber surface, respectively. From SEM images, as we can see the surface of nanofiber membrane changed from smooth to rough folded surface, which indirectly demonstrates the successful polymerization of PNIPAM polymer on the surface of the nanofiber. Figure 3(b), (c), (e), and (f) show the distribution of N and Br elements of PGS/PGS-Br/PLLA nanofiber membrane and PGS/PLLA@PNIPAM nanofiber membrane, respectively. It can be seen from the Figure 3(b) that there was no distribution of N elements before grafting PNIPAM on PGS/PGS-Br/PLLA nanofiber membrane, and the distribution of Br elements was relatively large. After grafting PNIPAM polymer, there had been appeared N elements. Accordingly, the distribution of Br elements was reduced. Therefore, PNIPAM polymer was successfully grafted onto the nanofiber membrane in the experiments.

The magnification SEM images (2000 and 15,000 times) and corresponding EDS element mappings of PGS/PGS-Br/PLLA and PGS/PLLA@PNIPAM nanofiber membrane: (a, d) SEM, (b, e) N element mapping, and (c, f) Br element mapping.
As shown in Figure 4, the wettability of nanofiber membranes were characterized by water contact angle tests. The PGS/PGS-Br/PLLA nanofiber membrane has a water contact angle of about 47°, while the nanofiber membranes with PNIPAM polymer modification were hydrophobic at room temperature with a water contact angle of about 129°. The difference are related to the increased surface roughness of electrospun nanofiber membranes at the nanoscale. After modification with PNIPAM polymer, water droplets were permeating into the membrane within 30 s at room temperature (20°C, <LCST) and showed a slow spreading. The main force for the absorption in this state is the hydrophilicity of PNIPAM polymer layer and the capillary effect in the microporous membranes. In addition, the pore size in nanofiber membranes increased upon passing the transition. Here, we draw the conclusion that regarding variations of permeability, the effect of surface roughness change caused by the increases of pore size is stronger than the effect of the permeability reduced by the hydrophobic force. 33

Wettability of different membranes at 20°C for 30 s.
Furthermore, the temperature sensitivity and application prospect of the drug release were studied by controlled release tests at different temperatures. As we can see from Figure 5, the maximum release is only 38.40% at 20°C (T < LCST), but 61.05% at 50°C (T > LCST). At the same time, comparing Figure 5(a) and (b), we found that the drug was released slowly at 20°C, while the drug was released suddenly at 50°C, which phenomenon is secretly related to thermosensitive PNIPAM polymer, it is hydrophilic swelling at low temperature, inter-molecular hydrogen bond strengthening at high temperature, and nanofiber surface fold exposing more nuclear layer surface, which enhances drug release rate. This results show that the core-shell PGS/PLLA@PNIPAM nanofibers can be used as drug carriers and realize temperature response control.

Drug release tests at different temperatures: (a) 20°C (T < LCST) and (b) 50°C (T > LCST).
Conclusion
A novel core-shell structure PGS/PLLA@PNIPAM nanofiber membrane was prepared by the combination of electrospinning and surface ATRP grafting technology. In the experiment, a macromolecule-initiator PGS-Br was prepared and characterized by FTIR and 1H NMR. The surface morphology, composition, element of the nanofiber was characterized by SEM, FTIR, and XPS. In addition, it is worth noting that the core-shell nanofiber membrane can respond to temperature in drug release tests. This work provides a new idea for the preparation of functional core-shell nanofibers.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the China Postdoctoral Science Foundation (Grant no. 2020M671359) and Natural Science Foundation of Jiangsu Province (Grant no. BK20200914).