
Abstract
Background
There is high variability in post-stroke aphasia severity and predicting recovery remains imprecise. Standard prognostics do not include neurophysiological indicators or genetic biomarkers of neuroplasticity, which may be critical sources of variability.
Objective
To evaluate whether a common polymorphism (Val66Met) in the gene for brain-derived neurotrophic factor (BDNF) contributes to variability in post-stroke aphasia, and to assess whether BDNF polymorphism interacts with neurophysiological indicators of neuroplasticity (cortical excitability and stimulation-induced neuroplasticity) to improve estimates of aphasia severity.
Methods
Saliva samples and motor-evoked potentials (MEPs) were collected from participants with chronic aphasia subsequent to left-hemisphere stroke. MEPs were collected prior to continuous theta burst stimulation (cTBS; index for cortical excitability) and 10 minutes following cTBS (index for stimulation-induced neuroplasticity) to the right primary motor cortex. Analyses assessed the extent to which BDNF polymorphism interacted with cortical excitability and stimulation-induced neuroplasticity to predict aphasia severity beyond established predictors.
Results
Val66Val carriers showed less aphasia severity than Val66Met carriers, after controlling for lesion volume and time post-stroke. Furthermore, Val66Val carriers showed expected effects of age on aphasia severity, and positive associations between severity and both cortical excitability and stimulation-induced neuroplasticity. In contrast, Val66Met carriers showed weaker effects of age and negative associations between cortical excitability, stimulation-induced neuroplasticity and aphasia severity.
Conclusions
Neurophysiological indicators and genetic biomarkers of neuroplasticity improved aphasia severity predictions. Furthermore, BDNF polymorphism interacted with cortical excitability and stimulation-induced neuroplasticity to improve predictions. These findings provide novel insights into mechanisms of variability in stroke recovery and may improve aphasia prognostics.
Keywords
Background
Post-stroke aphasia severity and potential for language recovery are highly variable and difficult to predict. Common prognostic indicators of aphasia severity include inter-related variables such as neurological (eg, lesion size, location) and demographic (eg, age, time post-stroke) profiles. 1 However, standard prognostics do not include neurophysiological indicators or biomarkers of neuroplasticity. These are critical variables to examine because neuroplastic reorganization is necessary for biological and behavioral recovery after stroke or other forms of brain injury.2-8
This research examined the hypothesis that individual differences in biomarkers of neuroplasticity improve predictions of aphasia severity. In particular, we examined whether individual differences in neuroplasticity—including a genetic biomarker and neurophysiological indicators of cortical excitability and neuroplasticity—improved ability to predict aphasia severity in subjects with chronic post-stroke aphasia. Below we summarize evidence that indicates each of these factors may account for variance in post-stroke aphasia.
First, we examined brain-derived neurotrophic factor (BDNF), which is a protein encoded by the BDNF gene. BDNF is a neurotrophin critical for neural repair and plasticity. It exhibits activity-dependent release at synapses 9 and modulates long-term potentiation 10 and long-term depression processes. 11 The BDNF gene has a common single nucleotide polymorphism (SNP). There are genotype variations, where either one or two Met alleles are present. Met allele carriers show deficiencies in activity-dependent release of BDNF, which leads to a downregulation of long-term potentiation and diminished synaptic plasticity in animal models. 12 Human carriers of Met alleles exhibit reduced hippocampal volume and episodic memory storage13,14 as well as deficits in learning and memory, increased motor impairment post-stroke, 15 and worse subcortical stroke outcomes. 16 It is predicted that due to downregulated BDNF secretion, Met allele carriers have more severe post-stroke aphasia compared to homozygotes with Val66Val alleles.
There is sparse and inconsistent evidence regarding the effects of BDNF polymorphism on aphasia. de Boer and colleagues 17 tested 53 subjects approximately 1-month post-stroke to examine improvement on communication in daily life situations (Amsterdam-Nijmegen Everyday Language Test 18 ) and confrontation naming (Boston Naming Test 19 ). Participants were tested before and after speech-language therapy, which was individualized in an inpatient rehabilitation setting where treatment type and duration varied widely. Subjects showed significant improvement on both language measures. However, there were no group differences based on BDNF genotype and language performance varied at both timepoints regardless of polymorphism. Established prognostic factors like stroke severity and lesion size were not examined in this study, and could have accounted for additional variance in aphasia severity. 1 In contrast, a study by Fridriksson and colleagues 20 reported that Met allele carriers had more severe naming impairments and overall aphasia than Val66Val carriers. Notably, these studies’ participants differed in subacute vs chronic stages of aphasia. Thus, these discrepant findings could reflect potential differences in spontaneous recovery associated with BDNF polymorphism. One aim of the current study is to reconcile this inconsistent evidence regarding whether BDNF polymorphism is predictive of aphasia severity following stroke. Moreover, the current study examined whether BDNF accounts for additional variation above and beyond established predictors of aphasia severity, such as lesion size, age at stroke, and cortical excitability. We expected not only that BDNF polymorphism would predict aphasia severity, 20 but also that genotypes might interact with neurophysiological indicators of plasticity to provide a more accurate predictive model of aphasia.
Second, we examined motor-evoked potentials (MEPs), as a neurophysiological indicator of cortical excitability and neuroplasticity. MEPs were measured before and after inhibitory continuous theta burst stimulation (cTBS)—a type of repetitive transcranial magnetic stimulation (rTMS) that requires less than a minute of application to induce comparable neuromodulatory effects 21 and is sensitive to intrinsic differences in cortical excitability and stimulation-induced neuroplasticity.22-24 In particular, we investigated MEP amplitudes at baseline to measure cortical excitability as well as cTBS-induced MEP suppression (ie, stimulation-induced neuroplasticity; difference between pre- vs post-cTBS MEP amplitudes) as a measure of neuroplasticity. Examining MEPs enables us to objectively assess propensity for response to non-invasive brain stimulation (NIBS),25-28 which is critical to understanding the mechanisms of efficacy and inter-individual variability in NIBS responsiveness for post-stroke language recovery and aphasia rehabilitation. We explored MEPs as an index of language recovery in aphasia due to evidence that MEPs index domain-general individual differences in cortical excitability and neuroplasticity. Neuroplasticity varies considerably in neurotypical populations, and MEPs are one way to elucidate those differences.29-31 Accordingly, we hypothesized that MEPs may predict recovery not only from damage to the motor cortex, but from any neural injury.
Last, we explored the degree to which BDNF polymorphism interacted with other metrics of plasticity to improve aphasia severity predictions compared to established predictors. In particular, the metrics of plasticity we examined included cortical excitability, stimulation-induced neuroplasticity, and age at stroke, which is an established predictor of post-stroke severity that is also associated with plasticity.32,33 Interactions were critical to examine based on evidence that BDNF polymorphism modulates stimulation-induced plasticity.20,34 Parchure and colleagues 34 examined whether BDNF polymorphism impacts cTBS-induced MEP suppression in chronic stroke patients. They found that Val66Val carriers exhibited immediate MEP suppression, which is the expected inhibitory response to cTBS.22,35,36 In contrast, Val66Met carriers exhibited a delayed cTBS response, such that MEPs decreased 30-minutes post-stimulation. This suggests that BDNF polymorphism influences MEP response to neuroplasticity-inducing stimulation protocols, such as cTBS, in chronic stroke patients. However, clinical applications of this finding have not been explored. The current work is, to our knowledge, the first to investigate the utility of genetic and neurophysiological biomarkers of neuroplasticity in predicting post-stroke aphasia severity. We hypothesized that individual variation in MEPs both before and after cTBS may predict severity, and that this effect may furthermore be modulated by BDNF polymorphism.
Indeed, BDNF polymorphism has predicted language outcomes after combined neurostimulation and behavioral aphasia treatment. In the study discussed above, Fridriksson and colleagues 20 found that BDNF polymorphism predicted response to anodal transcranial direct current stimulation (A-tDCS) during aphasia treatment. Sixty-six participants with chronic stroke aphasia were assigned to the A-tDCS or sham condition, followed by a 45-minute behavioral treatment, where participants indicated whether an audiovisual word matched a picture. Proportional changes in correct naming were compared at multiple time points. There was no significant effect of BDNF for the sham group. However, in the A-tDCS group, Val66Val carriers showed significantly more proportional change in correct naming compared to subjects with the Met polymorphism. This is consistent with predictions that NIBS enhances aphasia treatment effects by modulating long-term synaptic plasticity, which is a process that relies on BDNF secretion. We predicted the same mechanism may influence aphasia severity, and therefore that BDNF polymorphism may interact with cortical excitability, stimulation-induced neuroplasticity, and age at stroke to predict severity.
To summarize, our central hypothesis was that individual differences in neuroplasticity contribute to language recovery and should improve predictive models of post-stroke aphasia severity in the chronic stage. First, we assessed whether a genetic biomarker of neuroplasticity—BDNF polymorphism—improves prognostic estimates of aphasia severity from established predictors derived from a patient’s neurological (lesion size) and demographic (time post-stroke, age at stroke) profile. Second, we explored the degree to which BDNF polymorphism may interact with other metrics of plasticity (cortical excitability, stimulation-induced neuroplasticity, age at stroke) when established predictors like lesion volume and time post-stroke are controlled. Overall aphasia severity, measured by the Western Aphasia Battery Aphasia Quotient (WAB-AQ 37 ), served as a metric for the degree to which language functions recovered post-stroke.
Methods
Participants
Participant Demographics, Neurophysiological Characteristics, and Stimulation Parameters.
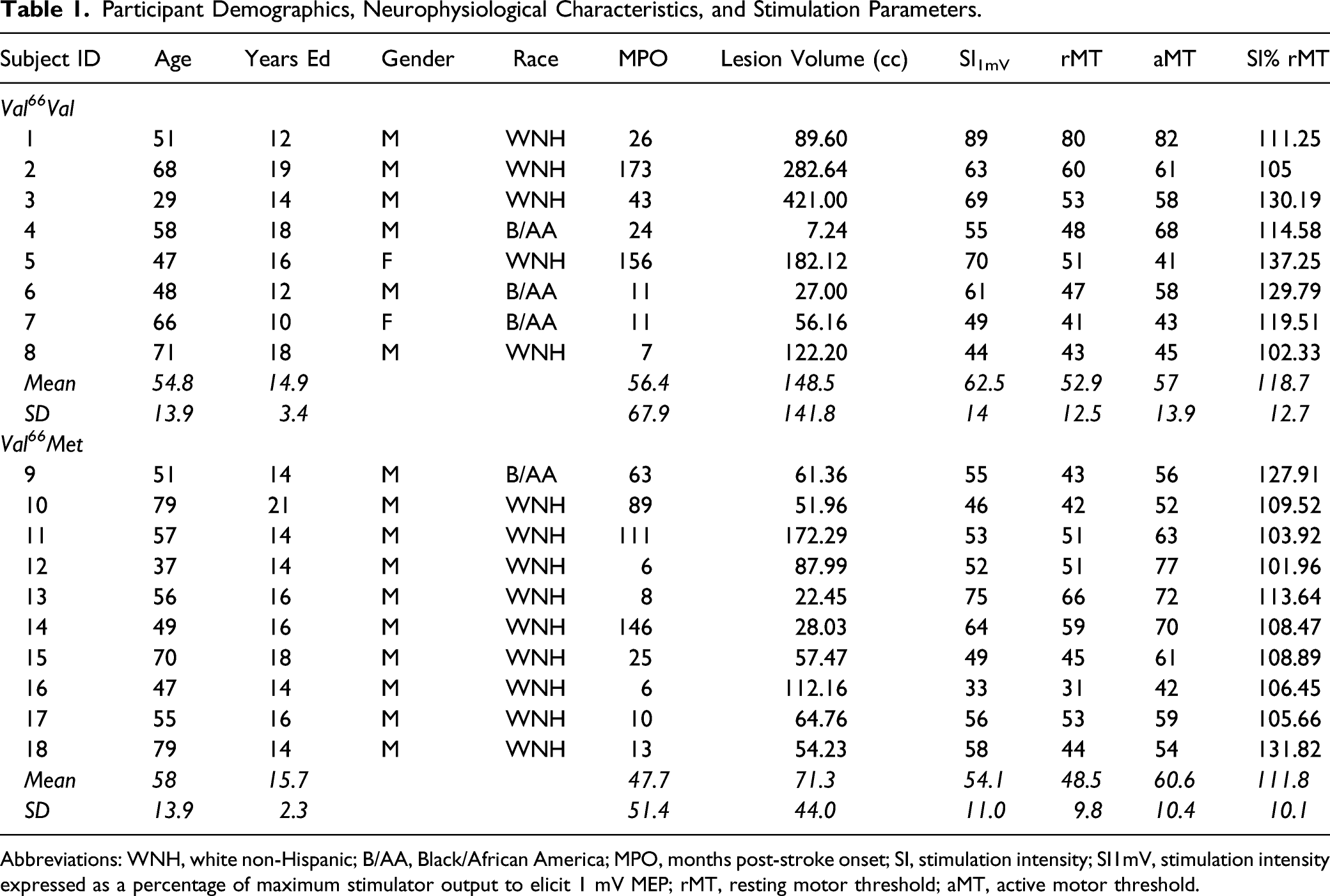
Abbreviations: WNH, white non-Hispanic; B/AA, Black/African America; MPO, months post-stroke onset; SI, stimulation intensity; SI1mV, stimulation intensity expressed as a percentage of maximum stimulator output to elicit 1 mV MEP; rMT, resting motor threshold; aMT, active motor threshold.
Overview of Experiment Design
WAB-AQ
37
was the outcome measure, measuring overall post-stroke aphasia severity. Each session began by determining the participant’s rMT and baseline MEPs. Next, we obtained the active motor threshold (aMT) to determine cTBS intensity. Thirty MEPs were obtained before cTBS (baseline) and 10-minutes post-stimulation. Refer to Figure 1. This analysis was embedded in a larger study of motor physiology, in which MEPs were gathered at multiple timepoints. The current analysis focused on the 10-minute post-cTBS MEPs because this timepoint showed peak stimulation effects.
34
Schematic of experimental design. Notes: Language testing occurred before cTBS. cTBS was administered for 40 seconds (600 pulses). 30 MEPs were recorded pre-cTBS and at 10 minutes after cTBS. Saliva samples were collected after all stimulation and multiple timepoints of MEP collection were complete.
Brain-Derived Neurotrophic Factor Genotyping
We collected genomic DNA from subject saliva samples, using Oragene® DNA collection kits. Genomic DNA was then isolated using the prepIT.L2Preagent (cat #PT-L2P-5, DNA Genotek Inc, Canada) and precipitated with ethanol according to the manufacturer protocol. The DNA samples were genotyped for BDNF (single nucleotide polymorphism rs6265) using the TaqMan SNP Genotyping Assay (C_11592758_10) designed by Thermo Fisher Scientific. Primers and probes were mixed with TaqMan® Universal PCR Master Mix (Thermo Fisher Scientific). 4.5 μL of genomic DNA (2.5 ng/μL) was transferred in triplicate to a 384-well plate, with each well containing 5.5 μL of the PCR mixture. PCR reaction was performed following a protocol provided by ABI. The allele was discriminated by post-PCR plate reading on the ViiA™ 7 System. Genotype data were processed using the ViiA™ 7 Software (Thermo Fisher Scientific).
Transcranial Magnetic Stimulation
We administered single-pulse TMS with a monophasic waveform to the primary motor cortex of the intact right hemisphere using a Magstim 2002 Stimulator with a 70 mm figure-eight coil (Magstim Co., Whitland, Dyfield, UK). We uploaded participants’ T1-weighted MRI scans to the Brainsight® Neuronavigation system (Rogue Research, Montreal) to identify the optimal scalp position within the right primary motor cortex for eliciting a MEP from the left first dorsal interosseous (FDI) muscle. In accordance with standard methods, we adjusted the intensity of stimulation such that resulting baseline MEPs for each subject had an average amplitude of ∼1 mV. 21 We acquired MEPs as subjects were seated in a chair with their arms resting on their lap or a pillow. Following standard procedures, we defined rMT as the minimum pulse intensity required to elicit MEPs with peak-to-peak amplitudes of at least 50 μV in five of ten consecutive trials with the FDI at rest.38,39 The starting stimulation intensity to acquire MEPs was 110% of rMT, after which the intensity was steadily increased by 1-2% until 10-12 consecutive MEPs were close to 1 mV in peak-to-peak amplitude. The coil position was maintained at the optimal scalp location and orientation during the acquisition of MEPs using the neuronavigation system. The single TMS pulses were delivered with an inter-stimulus interval of 6 seconds with a random jitter of 6%. See Table 1 for individual stimulation parameters.
Electromyography
We recorded EMG activity using surface electrodes panning the belly of the FDI muscle of each subject’s left hand, with the ground electrode placed along the left wrist. Signals were amplified and band-pass-filtered between 20-2000 Hz, digitized with a sample-rate of 5 kHz, and stored for off-line analysis using SIGNAL software (Cambridge Electronic Devices, Cambridge, UK).
Continuous Theta Burst Stimulation
We administered cTBS with a biphasic waveform using a Magstim SuperRapid2 Stimulator (Magstim Co., Whitland, Dyfield, UK). CTBS consisted of a continuous delivery of 50 Hz triplets of TMS pulses at 5 Hz for a total of 600 pulses and approximately 40 seconds. CTBS intensity was 80% of aMT, which is the minimum pulse intensity required to produce MEPs with peak-to-peak amplitudes of at least 200 μV in five of ten consecutive pulses while participants contracted the first dorsal interosseous (FDI) muscle at 20% of the maximum voluntary contraction. To ensure 20% contraction of the FDI, we recorded EMG of each subject’s contracting at maximal force and then practicing at a strength that filled 20% of the maximal EMG bounds. We used the same biphasic stimulator to determine aMT and administer cTBS. All subjects tolerated the cTBS with no adverse effects.
Statistical Analysis
We conducted a series of linear regression models in R with aphasia severity (WAB-AQ) as the outcome measure. We used a hypothesis-driven, forward-fitting regression approach to determine which biomarkers of neuroplasticity improved predictions of aphasia severity. First, a base model included factors of interest that are common predictors of aphasia, including time post-stroke (log-transformed months post-stroke onset; LogMPO), age at time of stroke, and total lesion volume (LesVol). We assessed whether BDNF genotype (Val66Met vs Val66Val carriers) improved model fit over the established predictors. If so, this BDNF model would constitute a new base model that would serve as a baseline comparison for subsequent analyses assessing the prognostic value of other neuroplastic factors and their interaction with BDNF genotype. Second, in a series of three separate analyses, we examined interactions between BDNF and (1) age at time of stroke (AgeCVA), (2) baseline MEP (cortical excitability; MEPbase), and (3) cTBS-induced changes in MEP (stimulation-induced neuroplasticity, measured as the difference between pre- and 10-minutes post-cTBS; MEPd10). Within each analysis, models iteratively compared the relative contribution of each neuroplastic factor above and beyond the established base model, followed by the interaction between that factor and BDNF, while controlling for LogMPO and LesVol. Models exploring neurophysiological indicators of plasticity (MEPbase and MEPd10) additionally included AgeCVA as a covariate. BDNF interactions with LogMPO and LesVol were not examined because these variables showed unequal variance across BDNF genotype groups in our sample (Levene’s test, LogMPO: F = 930.7, P < .001; LesVol: F = 67.14, P < .001). Furthermore, LogMPO was not expected to be tied to neuroplasticity in this sample because all participants were in the chronic stage of stroke recovery. The final models were as follows: (1) base_model ← lm(WABAQ ∼ LogMPO + LesVol + AgeCVA + BDNF, data = df) (2) age*BDNF_model ← lm(WABAQ ∼ LogMPO + LesVol + AgeCVA*BDNF, data = df) (3) excitability*BDNF_model ← lm(WABAQ ∼ LogMPO + LesVol + AgeCVA + MEPbase*BDNF, data = df) (4) plasticity*BDNF_model ← lm(WABAQ ∼ LogMPO + LesVol + AgeCVA + MEPd10*BDNF, data = df)
Model comparisons assessed the degree to which each factor and interaction term significantly improved model fit. We computed variance inflation factor using the R car package, 40 and found no concerning collinearity between MEP measurements at baseline (VIF = 3.53) vs 10 minutes post-stimulation (VIF = 2.08).
Next, we used L1 regularization models to assess each model for overfitting using the glmnet method in the R caret package. 41 This is a machine learning approach also known as Least Absolute Shrinkage and Selection Operator (LASSO). For each of our three linear regressions, we specified a LASSO regression model using training data and a leave-one-out cross-validation framework. We centered and scaled all variables and used the optimal lambda (tuning parameter) for each model. Adjusted R2 was computed for each model by testing model predictions derived from the training set on the testing dataset. This approach adjusts for potential overfitting of our linear regressions, which was a concern given our limited sample size and multiple predictors of interest.
Results
Brain-Derived Neurotrophic Factor Genotyping
Among the 17 subjects, 7 were BDNF Val66Val carriers, 10 were Val66Met allele carriers. Val66Met carriers were coded as 0 and Val66Val carriers were coded as 1 for our analysis. BDNF groups did not significantly differ in age (t = .75, P = .47), education (t = .85, P = .41), time post-stroke (t = −.42, P = .68), or lesion volume (t = −1.78, P = .12).
Linear Regression
Model Comparison Statistics.
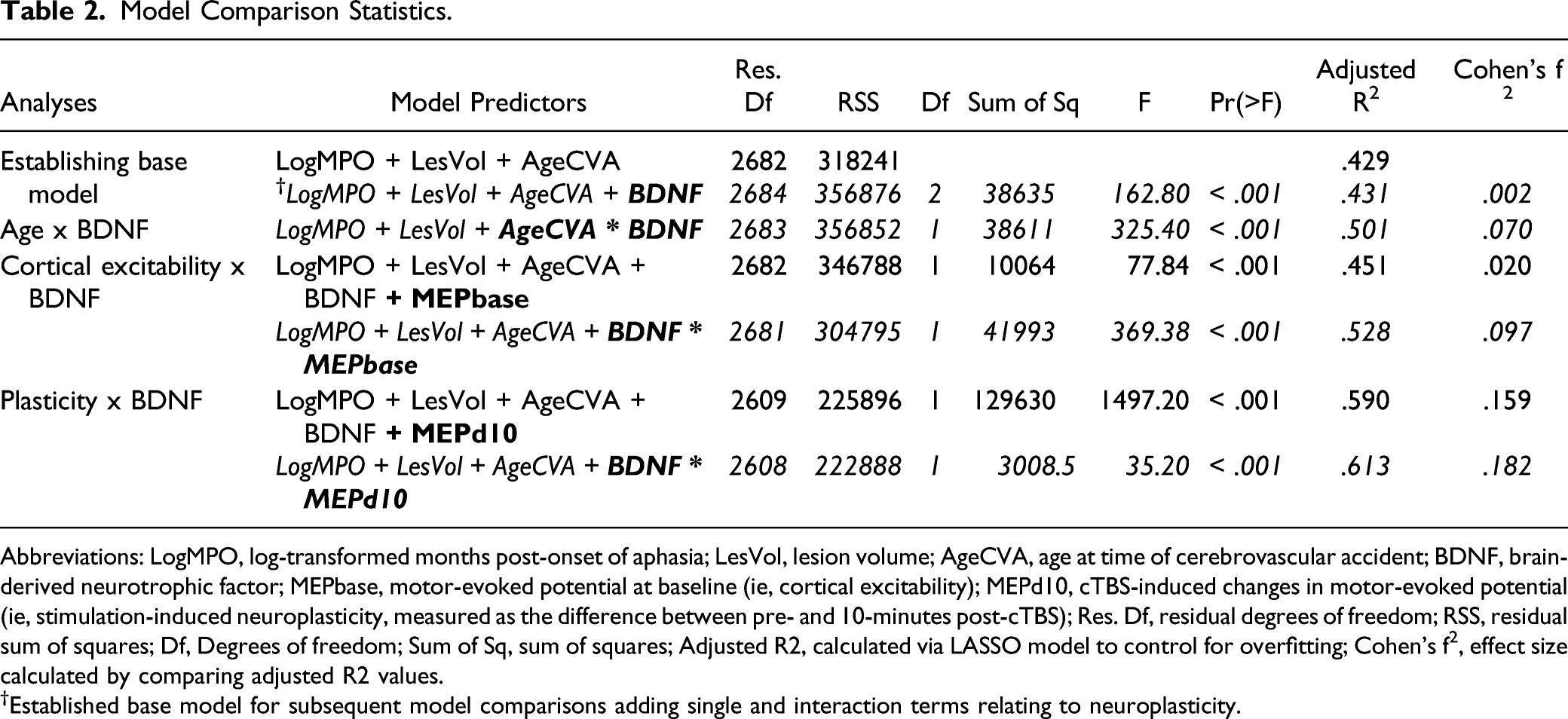
Abbreviations: LogMPO, log-transformed months post-onset of aphasia; LesVol, lesion volume; AgeCVA, age at time of cerebrovascular accident; BDNF, brain-derived neurotrophic factor; MEPbase, motor-evoked potential at baseline (ie, cortical excitability); MEPd10, cTBS-induced changes in motor-evoked potential (ie, stimulation-induced neuroplasticity, measured as the difference between pre- and 10-minutes post-cTBS); Res. Df, residual degrees of freedom; RSS, residual sum of squares; Df, Degrees of freedom; Sum of Sq, sum of squares; Adjusted R2, calculated via LASSO model to control for overfitting; Cohen’s f2, effect size calculated by comparing adjusted R2 values.
†Established base model for subsequent model comparisons adding single and interaction terms relating to neuroplasticity.
Age at CVA × Brain-Derived Neurotrophic Factor interaction
Adding an interaction between age at CVA and BDNF genotype significantly improved predictions from the established base model (Table 2). Increased age at stroke was associated with lower WAB-AQ for both groups, but had a stronger effect on aphasia severity for Val66Val carriers than Val66Met carriers (β = −.60, SE = .03, t = −18.04, P < .001). We conducted post-hoc analyses comparing BDNF genotype groups for the upper and lower terciles of age at CVA in the R package emmeans,
42
which revealed that this interaction effect was driven by a significant difference for individuals who were younger at time of CVA. Specifically, compared to Val66Met carriers, Val66Val carriers exhibited less severity at younger (β = −5.52, SE = .57, t = −9.71, P < .001) but not older age at CVA (β = .38, SE = .49, t = .79, P = .86; Figure 2A). BDNF interactions predict WAB-AQ. Notes: X axis is (A) age in years at time of cerebrovascular accident, (B) mean natural log-transformed MEP amplitudes at baseline as an index of cortical excitability, (C) difference between baseline and post-cTBS mean natural log-transformed MEP amplitudes. Y axis (A-C) is predicted WAB-AQ values. Val66Val carriers are represented in blue and Met allele carriers are in red.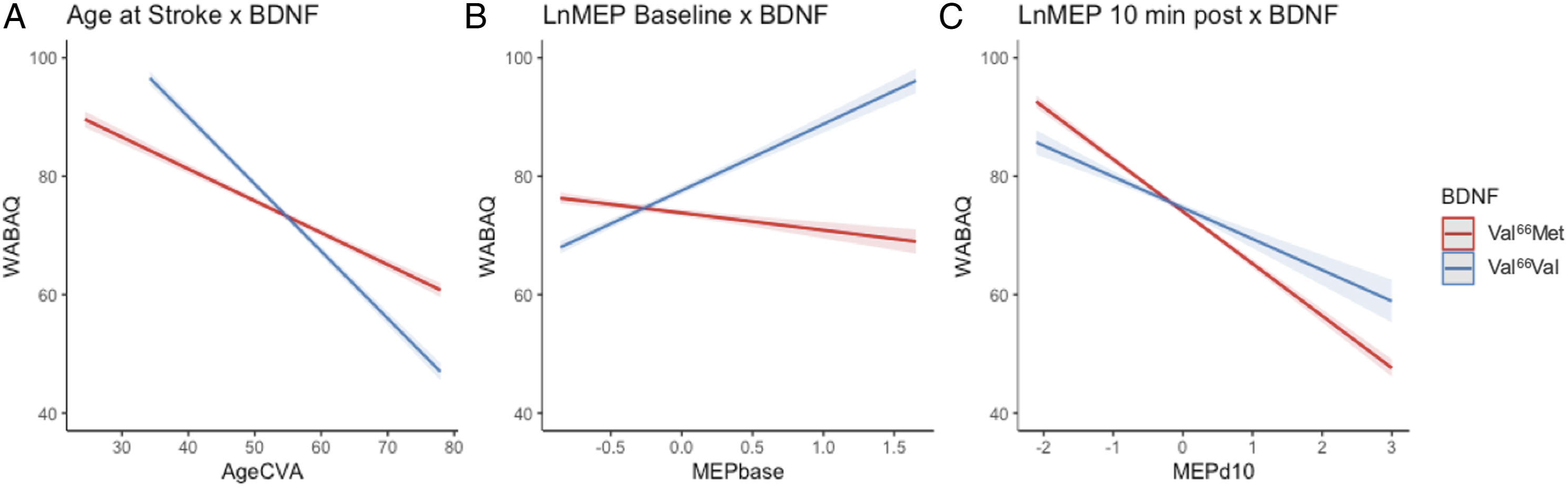
Cortical excitability × Brain-Derived Neurotrophic Factor interaction
Cortical excitability significantly improved model fit over the established base model, where model fit was further significantly improved by addition of the interaction between cortical excitability and BDNF (Table 2). Cortical excitability was positively associated with WAB-AQ for Val66Val carriers (β = 14.13, SE = .74, t = 19.22, P < .001), but negatively associated with WAB-AQ for Val66Met carriers (β = −2.86, SE = .49, t = −5.82, P < .001; Figure 2B).
Stimulation-induced neuroplasticity × Brain-Derived Neurotrophic Factor interaction
Stimulation-induced neuroplasticity significantly improved model fit over the established base model, and the interaction between stimulation-induced neuroplasticity and BDNF genotype significantly improved predictions from the model including stimulation-induced plasticity as a single term (Table 2). Less severe aphasia (higher WAB-AQ) was associated with greater MEP suppression (ie, the expected cTBS aftereffects) in both BDNF genotypes. This effect was stronger for Val66Met than Val66Val carriers (β = 3.56, SE = .60, t = 5.93, P < .001; Figure 2C).
Discussion
This study provides evidence that individual differences in neuroplasticity contribute to aphasia severity. In a sample of participants with chronic post-stroke aphasia, we replicated main effects of established aphasia predictors, including lesion volume, age post-stroke, and time post-stroke. Moreover, we found that BDNF polymorphism, a genetic biomarker of neuroplasticity, accounted for a substantial amount of variance in aphasia severity and improved estimates of aphasia severity from established predictors alone. In addition, BDNF polymorphism interacted with other metrics of plasticity, including age at stroke,32,33 cortical excitability,29,30 and stimulation-induced neuroplasticity27,28 to predict aphasia severity. These findings and their implications are discussed below.
The current results indicate that BDNF polymorphism is a critical predictor of variance in aphasia, beyond established predictors. We found that when controlling for the effects of total lesion volume, age post-stroke, and time post-stroke, Val66Val carriers showed less severe aphasia than Val66Met carriers. This is in line with Fridriksson and colleagues’ 20 findings that Val66Met carriers presented with more severe chronic aphasia than Val66Val carriers. Although there are also reports that BDNF polymorphism did not influence post-stroke aphasia recovery, these studies differed in critical ways including recovery stage and outcome measures. As discussed in the introduction, De Boer and colleagues 17 examined patients with acute stroke and observed no differences in everyday language or confrontation naming abilities based on BDNF polymorphism. In addition, they did not control for or examine potential effects of critical factors like aphasia severity and lesion size. Notably, our findings are consistent with our mechanistic hypothesis: because Val66Val carriers show stronger propensity for neuroplastic change than Val66Met carriers,12,35 they will be more likely to recover and present with mild aphasia. Our results are also consistent with a meta-analysis and multiple independent reports that BDNF polymorphism influenced general stroke recovery, 43 as well as post-stroke motor 44 and functional recovery, 45 such that Val66Met carriers demonstrate worse rehabilitation outcomes than Val66Val carriers.
Furthermore, BDNF polymorphism interacted with other indicators of neuroplasticity to improve predictions of aphasia severity. First, BDNF polymorphism interacted with age at the time of stroke. Older participants had more severe aphasia for both BDNF genotypes, but Val66Met carriers below the age of 50 exhibited worse outcomes when compared to their homozygous Val66Val counterparts. This finding again supports predictions that individuals with stronger, more intact neuroplasticity (Val66Val carriers and younger individuals) will be more likely to recover and present with mild aphasia in chronic stroke. Furthermore, these results are consistent with findings that Val66Met carriers show greater adverse effects of age on performance than Val66Val carriers in non-linguistic cognitive domains, as measured by subjective memory complaints, associative and episodic memory performance, and response to increased executive demands.46,47 This indicates that genetic effects may have a stronger influence on cognition when neural resources are reduced, as in old age. 47
Second, BDNF polymorphism interacted with two MEP indicators of neuroplasticity: cortical excitability and stimulation-induced neuroplasticity. Both indicators of neuroplasticity were particularly informative of aphasia severity for Val66Met carriers. We observed distinct patterns for cortical excitability: Greater baseline excitability was associated with less severity for Val66Val carriers, while greater cortical excitability was associated with greater aphasia severity for Val66Met carriers. Notably, evidence from neurologically healthy adults also shows strong interactions between BDNF polymorphism and baseline MEPs even when stimulation intensity is individually adjusted to minimize differences in baseline MEP amplitudes (eg, 1 mV). 24 In addition, greater stimulation-induced neuroplasticity (measured by cTBS after-effects of MEP suppression) was associated with greater aphasia severity for Val66Met carriers, and the degree to which individuals showed typical MEP suppression correlated with their potential for recovery. In contrast, stimulation-induced neuroplasticity was not associated with aphasia severity for Val66Val carriers. To coalesce these findings, we ran post-hoc correlations, which revealed that higher cortical excitability was associated with greater stimulation-induced neuroplasticity for Val66Val carriers, whereas the opposite was found for Val66Met carriers. For Val66Val carriers, higher baseline excitability may provide more potential for MEP suppression, similar to neurologically healthy response, 21 while more excitable Val66Met carriers may instead be less likely to show the expected response to neurostimulation. 34 Further work may explore the mechanisms driving this effect, such as potential associations between cortical excitability and delayed MEP suppression following neurostimulation in Val66Met carriers.
These findings provide evidence that BDNF polymorphism contributes to individual differences in cortical excitability and stimulation-induced neuroplasticity, indicating a prognostic utility of previous findings that BDNF polymorphism modulates individual responsiveness to stimulation.20,34 Furthermore, this study showcases how TMS can be used to measure neuroplasticity by leveraging neurophysiological indices of plasticity that inform functional prognostics. MEPs are informative neurophysiological indicators of plasticity that may reflect domain-general individual differences in cortical excitability and plasticity. Future work may explore the utility of these indicators in relation to domains of non-motor functional recovery following neural injury.
A potential limitation of this study is the modest sample size used to examine the behavioral effects of a genetic polymorphism. However, our sample size is comparable to other studies investigating BDNF and/or MEPs in aphasia.34,48,49 Despite our modest sample, our results show small but significant effects of multiple neuroplastic factors that account for separate variance in aphasia severity. In addition, it remains unclear whether BDNF polymorphism is only a significant predictor of chronic but not acute post-stroke aphasia. Additional work is required to characterize potential differences in spontaneous recovery associated with BDNF polymorphism. Such investigations could elucidate differences between studies finding no effects of BDNF polymorphism in acute aphasia 17 vs robust effects of BDNF in chronic recovery stages. 20 Likewise, neurophysiological responses such as MEP-suppression following NIBS may differ in acute to chronic stages of stroke recovery. Longitudinal investigations may help determine the optimal prognostic time at which biomarkers of neuroplasticity have the strongest predictive power of long-term functional outcomes. Additional predictors, such as stroke location, may further interact with neuroplastic indicators to contribute to the prognostic strength of the current approach. 50 Future work should explore all predictors investigated here in a single model—an analytic approach not suitable for the current study due to limited sample size. This will create a more parsimonious account of neuroplastic factors that affect aphasia recovery. Finally, future work should also seek to replicate the current findings in larger samples of stroke patients with and without aphasia.
Conclusion
BDNF polymorphism is a substantial predictor of chronic post-stroke aphasia severity, above and beyond established predictors. Furthermore, BDNF polymorphism interacted with other indices of neuroplasticity to predict aphasia severity. In particular, BDNF polymorphism interacted with age at stroke, cortical excitability, and stimulation-induced neuroplasticity to improve ability to predict aphasia severity from established descriptive factors. This study is among the first to demonstrate that including biomarkers and neurophysiological indicators of neuroplasticity (ie, cortical excitability and stimulation-induced neuroplasticity) significantly improves ability to predict aphasia severity. These findings provide novel insights into potential sources of variability in stroke recovery that may improve aphasia prognostics.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from the National Institute on Deafness and Other Communication Disorders (R01-DC012780) and National Institute of Child Health and Human Development (T32-HD071844).