
Abstract
Background. Peripheral axon regeneration is improved when the nerve lesion under consideration has recently been preceded by another nerve injury. This is known as the conditioning lesion effect (CLE). While the CLE is one of the most robust and well characterized means to enhance motor axon regeneration in experimental models, it is not considered a clinically feasible strategy. A pharmacological means to re-produce the CLE is highly desirable. Objective. To test whether chemodenervation with a clinical grade formulation of botulinum toxin A (BoTX) would be sufficient to reproduce the CLE. Methods. We examined the effects of a 1-week preconditioning administration of BoTX on motor axon regrowth in both a mouse tibial nerve injury and human embryonic stem cell (hESC)–based model. We assessed neuronal reinnervation in vivo (mice) with retrograde tracers and histological analysis of peripheral nerve tissue after injections into the triceps surae muscle group. We assessed motor neuron neurite outgrowth in vitro (hESC) after incubation in BoTX by immunohistochemistry and morphometric analysis. Results. We found that BoTX conditioning treatment significantly enhanced outgrowth of both murine motor axons in vivo and human MN neurites in vitro. Conclusions. BoTX preconditioning represents a pharmacological candidate approach to enhance motor axon regeneration in specific clinical scenarios such as nerve transfer surgery. Further studies are needed to elucidate the molecular mechanism.
Keywords
Introduction
Peripheral nerve injury (PNI) is a common cause of functional impairment.1,2 The causes of PNI are varied, ranging from penetrating trauma in combat to chronic compressive states such as carpal tunnel syndrome. Key to achieving optimal functional outcome after PNI is to minimize the duration of target denervation.3,4 Even when axons need only regenerate relatively short distances, such as in the case of a digital nerve injury, functional improvements are still relatively slow and often incomplete.
In the experimental setting, the conditioning lesion effect (CLE) has been one of the most well-studied and robust experimental strategies to enhance axon regeneration. 5 Briefly, the classic CLE paradigm results in accelerated axonal regeneration following an axotomy (test lesion) as a result of the axon having undergone a previous injury (conditioning lesion) 1 to 2 weeks earlier.6,7 It is widely believed that the CLE is largely attributable to early neuron intrinsic changes after denervation, 5 but it is generally viewed as infeasible for clinical translation based on its invasiveness and the temporal requirement for its application (ie, prior to nerve injury). To the second point, while a preconditioning treatment is generally not possible when a traumatic PNI is surgically managed solely with a primary repair (eg, suturing a lacerated nerve back onto itself), many peripheral nerve surgeries now involve a nerve transfer procedure given recent reports of superior clinical outcomes. 8 Nerve transfer surgery consists of the isolation and redirection of a functionally redundant, healthy donor nerve fascicle(s) located in close proximity to the denervated muscle targets of the damaged nerve with the goal to achieve the earliest muscle reinnervation possible. 9 Since the donor nerve has intact neuromuscular connections prior to transfer, it is amenable to a preconditioning treatment.
Based on several existing lines of evidence that highlight the similarities of effects between axotomy and botulinum toxin A (BoTX) on motor neurons (MNs),10-15 we hypothesized that preconditioning chemodenervation with BoTX prior to nerve repair would enhance motor axon regeneration. To increase the clinical relevance, we exclusively used a clinical grade formulation of BoTX. We tested this hypothesis using 2 separate models systems, a mouse model of in vivo nerve repair, as well as a human embryonic stem cell (hESC)–based model of motor neurite outgrowth in vitro.
Methods
Mice
Young adult (8-12 weeks old) male C57Bl/6 mice with initial weights between 18 and 25 g were obtained from Jackson Labs (Bar Harbor, ME). Mice were maintained in a 12/12 light/dark cycle with ad libitum access to nutrition in an AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care) accredited facility at Northwestern University with IACUC (Institutional Animal Care and Use Committee) approval of all procedures.
BoTX Injections
Mice were injected with OnabotulinumtoxinA (BoTX; Allergan, Irvine, CA) or 0.9% saline vehicle control unilaterally into the triceps surae muscle group. We did not use the contralateral limb for any control data. For the dose response experiments, mice (n = 4 per group) were administered 0.0 (ie, SHAM), 0.1, 0.25 or 0.5 units (U) of BoTX prepared in 50 µL. All injections were performed in transcutaneous fashion with a 31-gauge needle attached to a 0.3-mL insulin syringe (BD Medical, Franklin Lakes, NJ) into the triceps surae muscle group (see supplemental data for illustration). Based on our dose response experiments, a dose of 0.25 U was selected as the ideal treatment dose in our subsequent axon regeneration experiments.
Surgery
Mice were anesthetized with 1% to 2% isoflurane. An incision was made parallel to and below the femur to expose the tibial nerve just above the knee. For axon regeneration surgeries, the tibial nerve was crushed with #3 jeweler’s forceps (Figure 1A) holding down for 10 seconds with visual confirmation that all axons were severed as indicated by a resultant translucent appearance of the nerve segment. This was repeated once more for an additional 10 seconds to minimize the chance that any axons were spared. The crush site was marked with an 11-0 suture (Fine Science Tools, Vancouver, British Columbia, Canada) carefully tied through the epineurium so the site could be readily identified. For these studies, BoTX or SHAM injection into the triceps surae muscle group was performed 1 week prior to tibial nerve crush surgery. Since the tibial nerve innervates the triceps surae, the BoTX conditioning treatment was aimed at tibial neuromuscular connections.
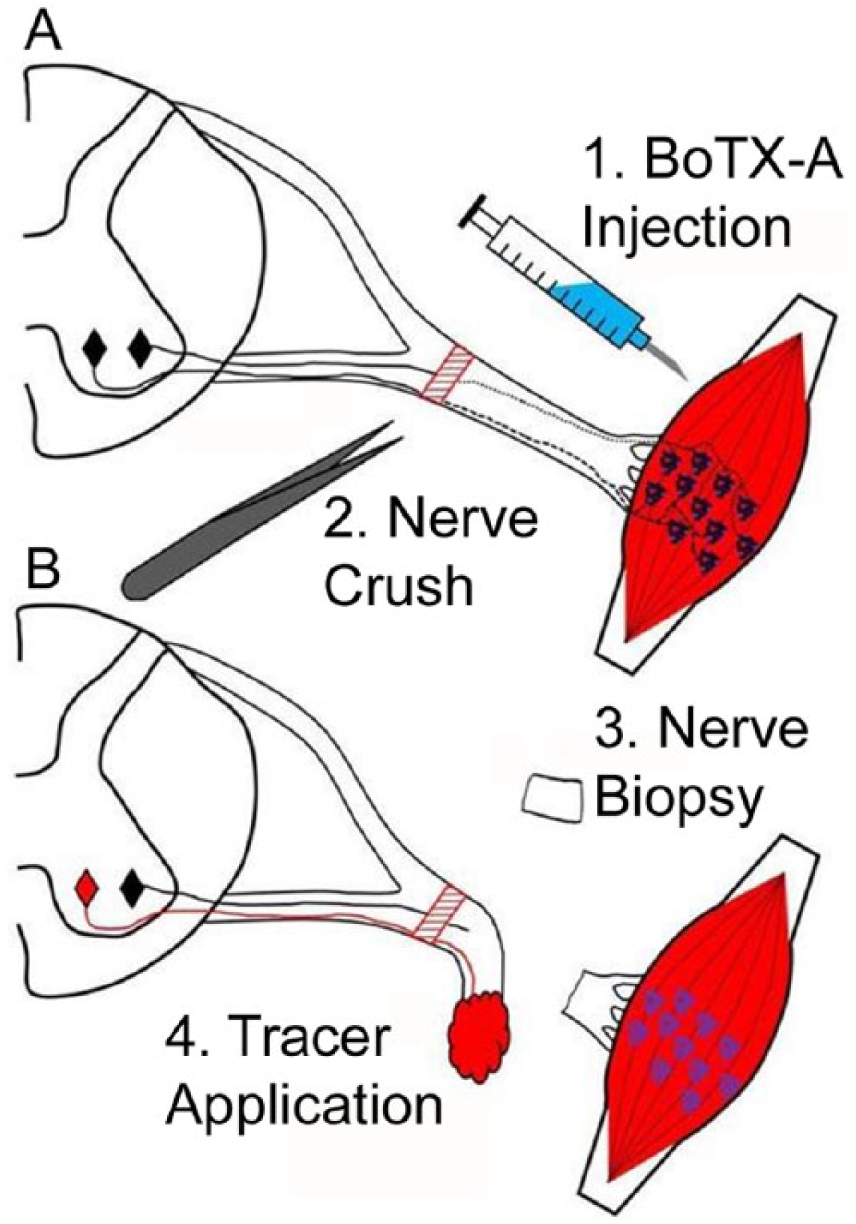
Experimental design for mouse tibial nerve injury model. (A) Intramuscular botulinum toxin A (BoTX) injection (1) was performed into triceps surae. One week later the tibial nerve was crushed (2). (B) One week after nerve crush surgery a nerve biopsy was taken distal to the injury site (3) and fluorescent tracer was applied (4).
In a separate cohort, the tibial nerve was completely transected without repair for the purpose of evaluating Wallerian degeneration (see Supplemental Data, available in the online version of the article).
Retrograde Labeling of Motor Neurons
After either 1 or 4 weeks, a second operation was performed to assess the number of MNs that had reinnervated the distal nerve. The tibial nerve was transected 10 mm distal to the crush site and 10% Fluoro-ruby dye (DS-1817, Thermo-Fisher, Waltham, MA) prepared in sterile saline was applied to the distal nerve with Gel Foam (Pfizer, New York, NY) as previously described 16 (Figure 1B). On day 7 after tracer application, mice were transcardially perfused with 4% paraformaldehyde (PFA) prepared in phosphate buffered 0.9% saline (PBS). The lumbosacral enlargement of the spinal cord was isolated and processed as described previously. 17 Spinal cords were sectioned on a cryostat in sagittal orientation at 60 μm. Retrogradely labeled MNs were counted in blinded fashion and quantified as described previously. 18
Myelinated Axon Counts
The 2 mm segment of tibial nerve immediately distal to the site of retrograde labeling (described above) was biopsied 1 week after nerve crush just prior to application of retrograde tracers (Figure 1B). Biopsy samples were fixed and processed for semithin sectioning as described previously. 19 One micrometer thick sections and were collected and stained with toluidine blue-O. Images were captured on a DM2500 LED microscope (Leica, Buffalo Grove, IL). Myelinated axons counts were made by a blinded observer using Image J (National Institutes of Health, Bethesda, MD).
Behavioral Analysis
For the BoTX dosing experiments we performed the Digit Abduction Score (DAS) assay, which is an observational evaluation of the BoTX-induced paresis as described by Aoki. 20 Briefly, a startle response was elicited by suspending mice hind limbs by grasping the tail, which produces a stereotypical reaction in which the mouse abducts its hind digits (Figure 2A). Varying degrees of digit abduction were scored on a 5-point scale (0 = normal to 4 = maximal reduction in digit abduction and leg extension) by an observer who was masked to treatment at baseline, then 3, 7, 14, and 21 days postinjection.
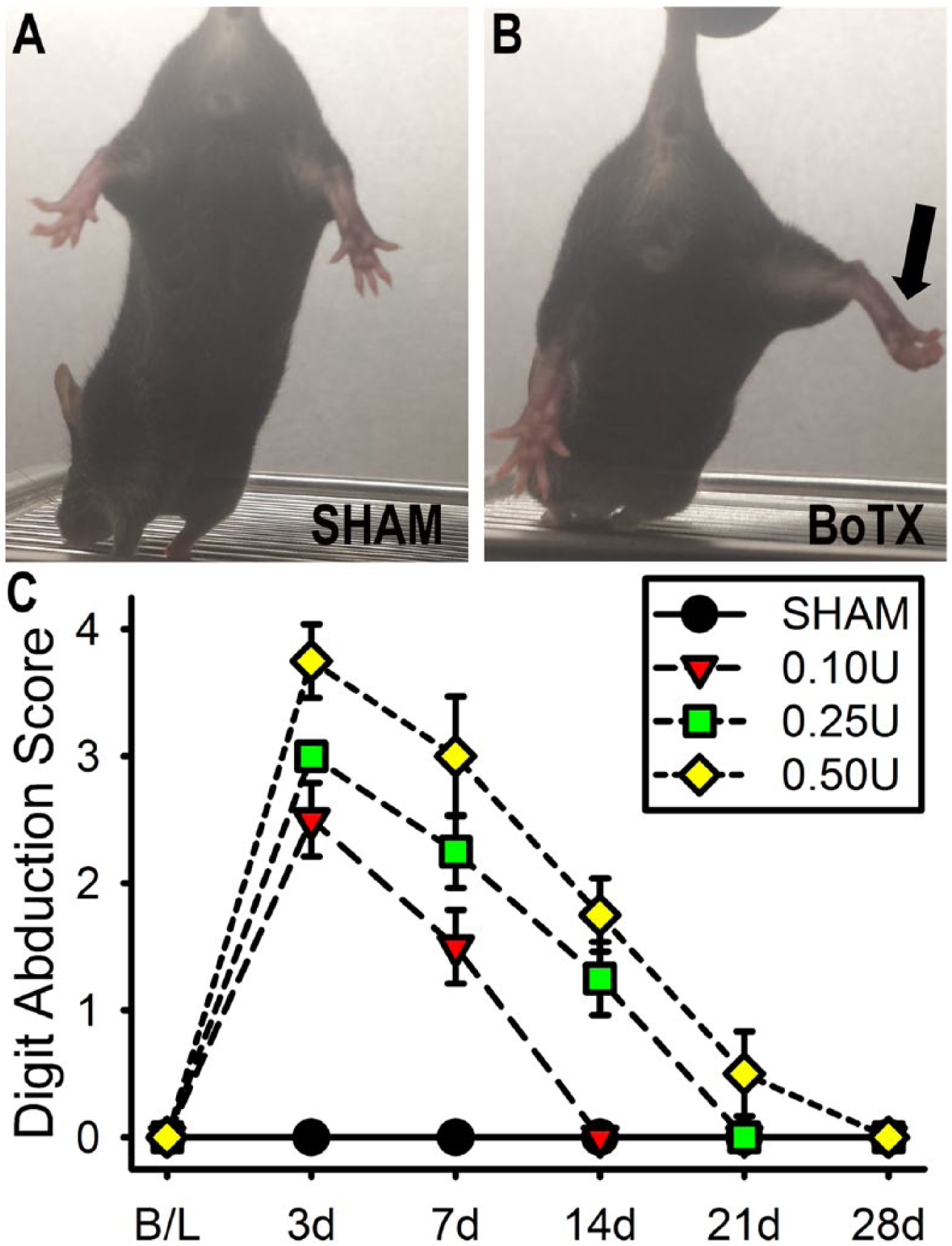
Dosing of botulinum toxin A (BoTX) injection. (A) The SHAM condition received a saline vehicle only injection and demonstrates a normal toe spreading response. (B) The 0.25 U of BoTX-A condition impairs toe spreading response 3 days after injection (black arrow). (C) Digit Abduction Score shows at the 0.25 and 0.50 U doses there was the most severe paresis that recovered to baseline over 3 to 4 weeks.
Neuromuscular Junction Staining
The triceps surae muscle group was isolated and dissected from the right leg of a PFA-perfused mouse, immersed in 20% sucrose-PBS mixed with OCT (1:2), pinned at its physiological resting length, flash frozen in dry-ice-cooled isopentane, and mid-point of the muscle was sectioned transversely with a cryostat on to slides at 20 µm thickness. The tissue was air dried overnight, blocked for 1 hour at room temperature in PBS containing 0.3% Triton X-100 and 2.5% bovine serum albumin (BSA), incubated overnight at 4°C in rabbit anti-neurofilament SMI-312 (1:1000; ab24574, Abcam, San Francisco, CA) diluted in blocking solution, washed several times with PBS, incubated overnight at 4°C in goat anti-rabbit IgG secondary antibody conjugated to Alexa Fluoro 488 (1:500; ThermoFisher) and rhodamine conjugated α-bungarotoxin (1:100; ThermoFisher) diluted in blocking solution, washed several times in PBS, and then mounted in ProLong Gold mounting media (ThermoFisher). Images were acquired with Leica DM2500 LED upright microscope as described above for MNs. The percentage of innervated neuromuscular junctions was determined based on the co-localization of the pre-synaptic (neurofilament) and postsynaptic (α-bungarotoxin) markers.
Culturing of Human Stem Cell–Derived Motor Neurons
MN differentiation was carried out as previously described 21 with modifications applied to a hESC line (HUES 64) that was obtained from Harvard Stem Cell Science Core facility. Refer to Supplemental Material (available in the online version of the article) for differentiation procedure.
Motor Neuron Neurite Assay and BoTX Treatment
Frozen MNs were thawed and plated as single cells onto 12-well cell culture plates (ThermoFisher) coated with poly-
Multielectrode Array Recordings of MN cultures
In some cases, 20K hESC-derived MNs were plated on 48-well multielectrode array (MEA) plates with 16 extracellular electrodes/well for recordings of spontaneous neural activity on the Maestro (Axion BioSystems) MEA recording amplifier with a head stage that maintained a temperature of 37°C. The MEA data were sampled at 15 kHz, digitized, and analyzed using Axion Integrated Studio software (Axion BioSystems) with 200 Hz high pass and 2500 kHz low pass filters and an adaptive spike detection threshold set at 5.5 times the standard deviation for each electrode with 1 second binning. These standard settings were maintained for all Axion MEA recording and analysis. Cell viability was assessed with the CytoTox 96 Non-Radioactive Cytotoxicity Assay according to manufacturer’s protocol (Promega, Madison, WI). This viability assay measures the release of lactate dehydrogenase (LDH), a stable cytosolic enzyme that is released on cell lysis. The percentage of cytotoxicity a tissue culture well can be determined by measuring LDH levels just before and after treatment with the manufacturer’s cell lysis reagent (ie, “100% kill”).
Statistics
Mean values (±standard error of the mean; SEM) are shown throughout. For the BoTX dose response on the DAS behavioral assay a Kruskal-Wallis test and if the null hypothesis was rejected a post hoc analysis was performed using the Dunn’s multiple comparison test with Bonferroni correction. Otherwise, the Student’s t test was used to make comparisons between time-matched BoTX and SHAM data.
Results
Induction of Transient Muscle Paresis and Neuromuscular Sprouting With BoTX
The effect of BoTX injection unilaterally into the triceps surae muscle group was examined over a 4-week period using the Digit Abduction Score (DAS) motor behavior assay 20 (Figure 2A and B) that evaluates for the toe spreading reflex. We demonstrated a dose-dependent pattern in the DAS score with increasing concentrations of toxin leading to decreased digit abduction relative to vehicle treated control at days 3 to 14 postinjection (P < .01; Figure 2C). The peak DAS score for all BoTX doses (0.10, 0.25, and 0.50 U per mouse) studied was observed on day 3 postinjection and had already began to decline by day 7. By day 14, the DAS scores from the 0.10-U dose group had returned to baseline and both the 0.10 U and SHAM were significantly less the 0.50-U group (P < .05), but the 0.50 U and 0.25 U groups were not significantly different at any time point studied. Since 0.25- and 0.50-U doses were essentially equivalent, all subsequent in vivo experiments were based on a dose of 0.25 U/mouse to allow for a greater safety margin relative to the lethal dose (LD)-50.
To assess for terminal sprouting from chemodenervated neuromuscular junctions, we injected BoTX at a dose of 0.25 U into the triceps surae muscle group (Figure 3A and B). Two weeks following the injection we harvested the muscle and stained frozen sections for presynaptic (neurofilament) and postsynaptic (acetylcholine receptors). We found terminal sprouts (Figure 3B) present at 5.2% ± 1.3% (n = 5 mice, m = 250 endplates) in the BoTX-treated condition, whereas just 0.8% ± 0.5% (n = 5 mice, m = 250 endplates) in the SHAM-treated condition (Figure 3C; P < .01).
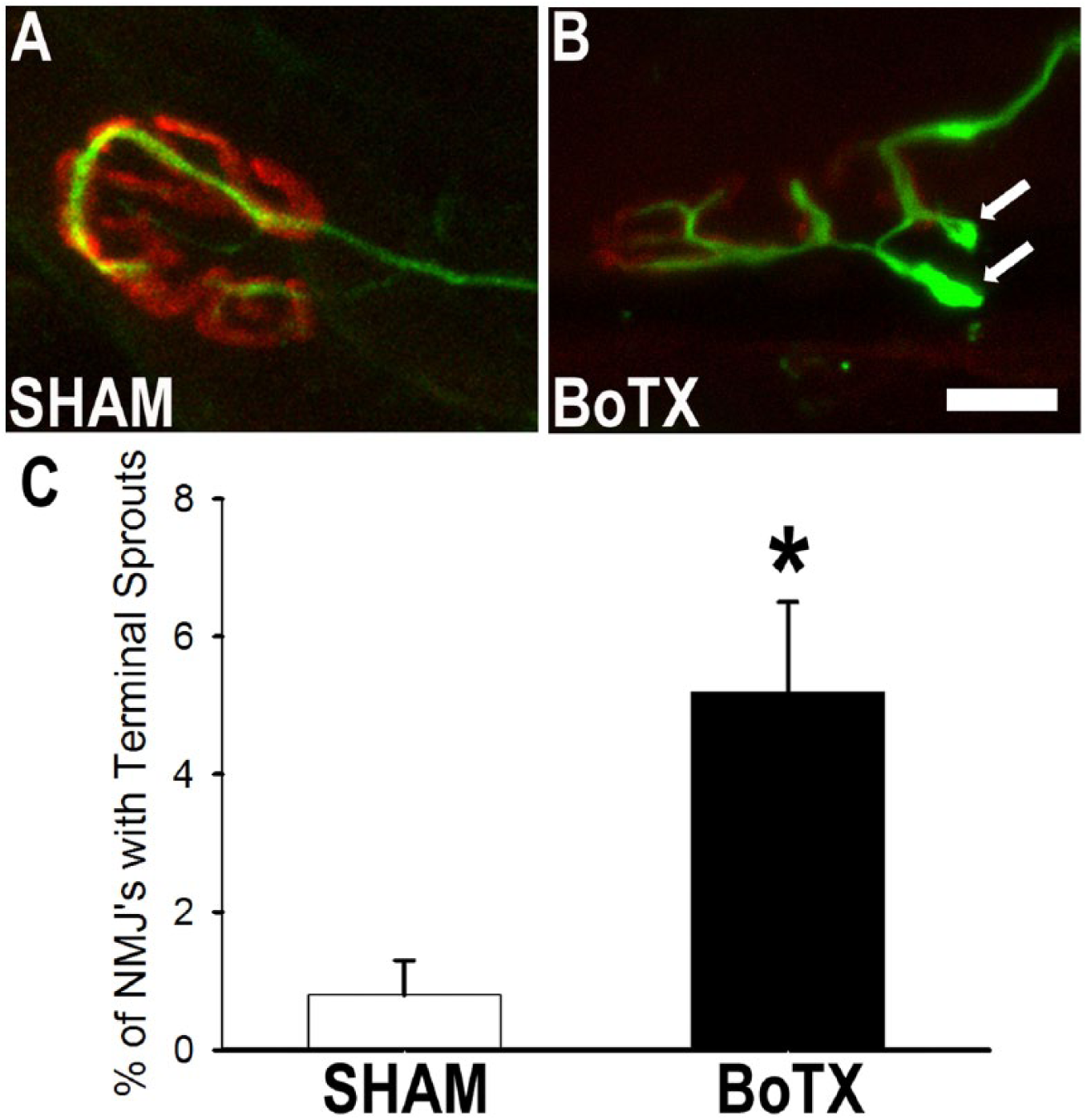
Botulinum toxin A (BoTX) injection induces neuromuscular sprouting. (A) Two weeks following 0.25 U BoTX injection, terminal axon sprouting is induced. (B) Controls neuromuscular junctions rarely had terminal sprouts (white arrows). (C) Significantly more terminal sprouts are identified after BoTX injection. Scale bar represents 10 μm. *P < .01.
BoTX Preconditioning Enhances Motor Nerve Reinnervation
We next studied the effect of BoTX pre-conditioning on motor axon regeneration. The preconditioning dose of 0.25 U was injected unilaterally into the triceps surae muscle group 1 week before we performed a tibial nerve crush injury (Figure 1A). Toluidine blue staining was performed on nerve biopsies to compare the number of myelinated axons that had reinnervated the distal tibial nerve 1 week following nerve crush. Qualitative assessment of the stained nerve biopsied revealed a consistent pattern where there appeared to be a greater density of myelinated axons in the nerves that had been pretreated with BoTX (Figure 4A and B). To accurately quantitate this observation, we performed blinded manual counts of the number of myelinated fibers that had reinnervated each nerve sample. The number of myelinated axon profiles counted 10 to 12 mm distal to the site of nerve crush in the BoTX (366 ± 24, n = 5) versus SHAM (193 ± 36, n = 5) was significantly enhanced (P = .004; Figure 4C and D). We could not attribute this difference in myelinated axonal counts to altered Wallerian degeneration, because in a separate set of experiments when we cut the tibial nerve without repair we identified no significant differences between BoTX (n = 3) and SHAM (n = 3) after 5 days in the loss of neurofilament or myelin staining (Supplemental Figure 1A and B, available in the online version of the article). However, both the transected BoTX and SHAM groups had significantly reduced neurofilament and myelin staining as compared with uncut controls (n = 4; P < .01).
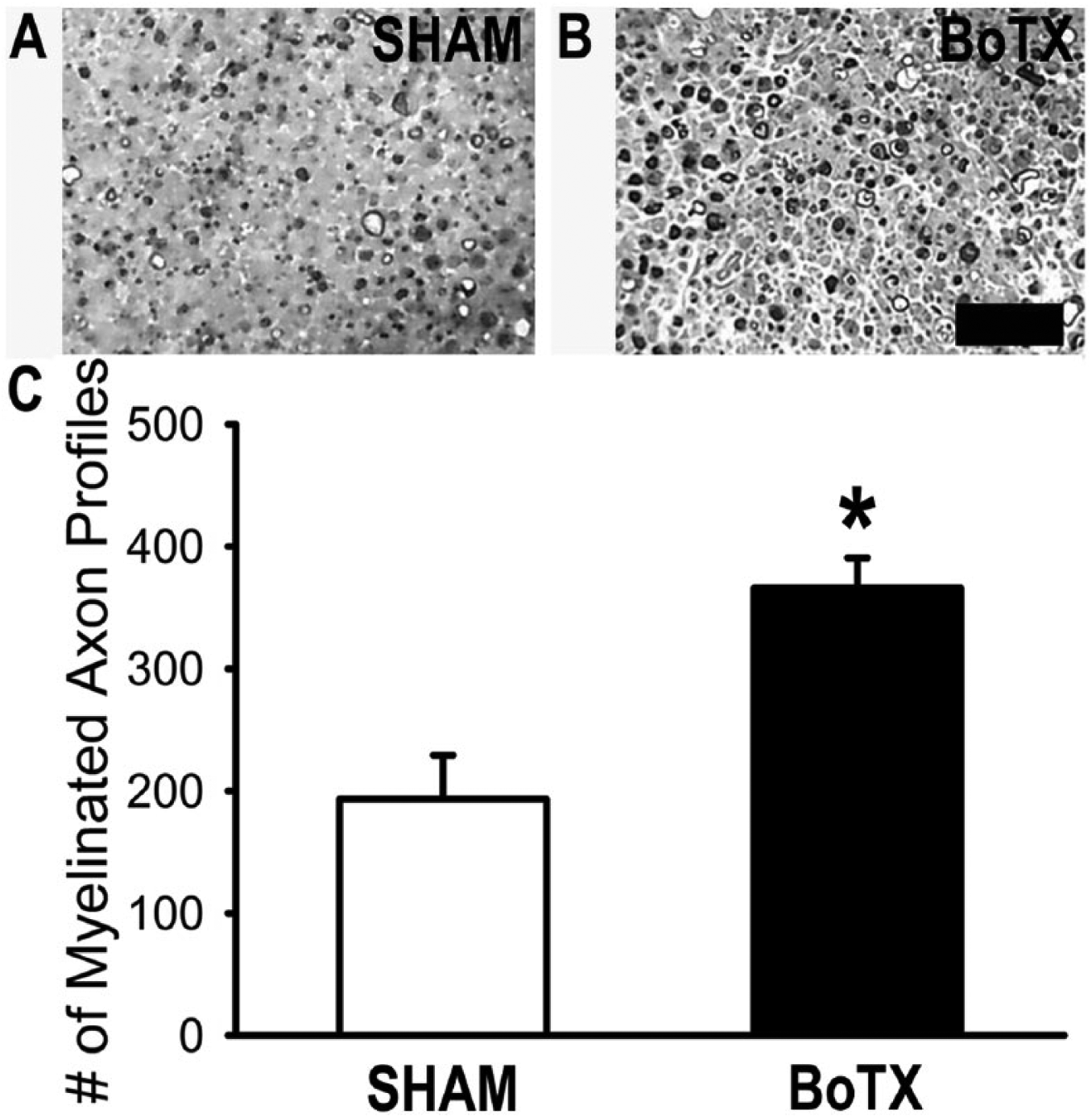
Botulinum toxin A (BoTX) conditioning increases axonal reinnervation. One week following BoTX injection more myelinated axons can be appreciated in the distal nerve stump then after control tibial nerve injury (A, B). This represents a significantly increased number of myelinated axon profiles in BoTX as compared with SHAM control (C). Scale bar represents 5 μm. *P = .004.
To examine the effects of BoTX pre-conditioning on motor reinnervation directly we counted the number of retrogradely labeled MNs that picked up fluoro-ruby dye applied 10 mm distal to the repair (Figure 1B). We have employed this technique previously 16 and have demonstrated that it effectively labels MNs in the ventral spinal cord that are quantifiable (Figure 5A). We performed blinded manual counts of the number of MNs labeled in each treatment group after 1 or 4 weeks. The number of labeled MNs in the BoTX (236 ± 18, n = 11) versus the SHAM (172 ± 19, n = 10) group was significantly higher at 1 week (P = .02; Figure 5B). By 4 weeks of recovery, when the number of regenerating MNs was expected to plateau, there was no difference in the number of labeled MNs between the BoTX (342 ± 12, n = 3) versus the SHAM (347 ± 18, n = 3) group (P = .83; Figure 5B).
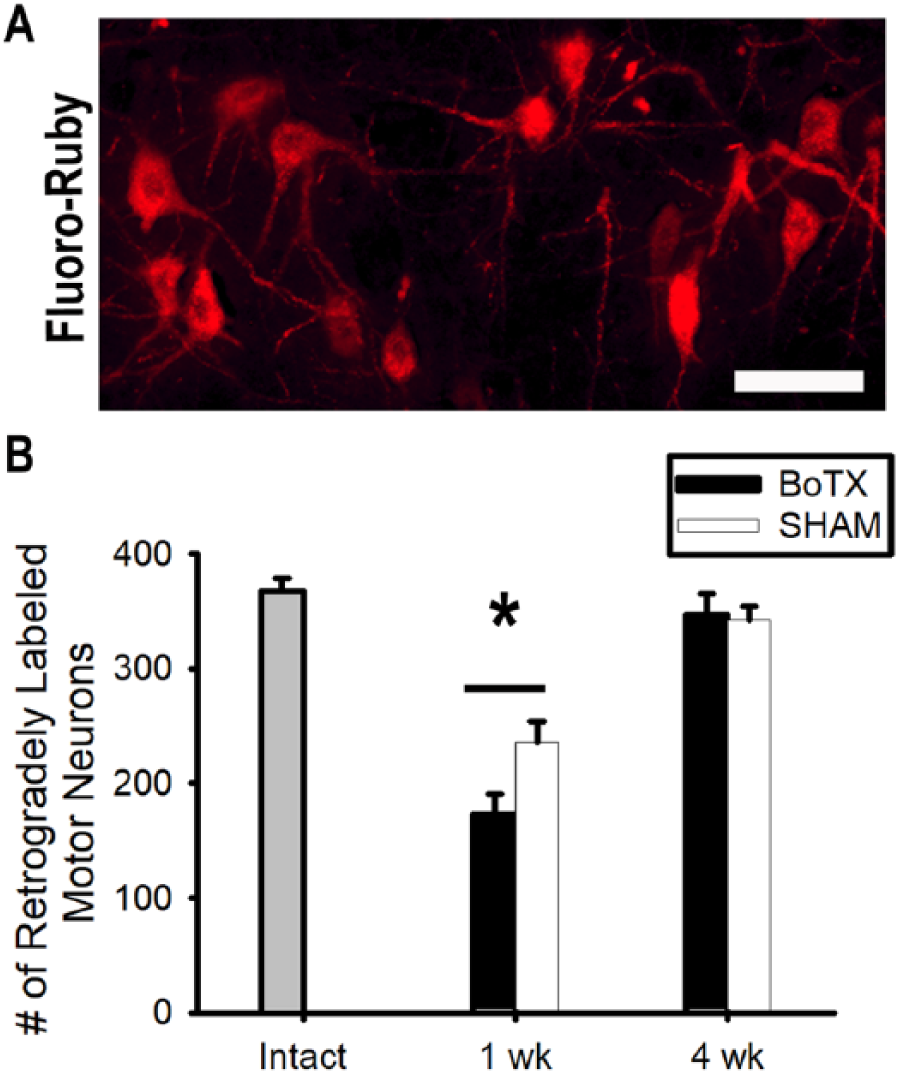
Botulinum toxin A (BoTX) conditioning increases motor reinnervation. (A) One week following fluorescent tracer application motor neurons that had reinnervated the distal nerve at different time points were easily quantifiable. (B) The number of retrogradely labeled motor neurons 1 or 4 weeks after tibial nerve crush. Scale bar represents 100 μm. *P = .02.
BoTX Preconditioning Enhances Human Motor Neuron Neurite Outgrowth
Given the substantial effects we found from BoTX preconditioning on motor axon regeneration in mice, we asked whether a similar effect would be seen in a human preclinical model of MN-neurite outgrowth. To block neurotransmission in this culture system we applied BoTX 2 weeks after plating MNs (Figure 6A). Treatment with BoTX at 2 weeks blocked neural activity within 48 hours (Figure 6B). It did so without causing any detectable change in neuron viability as assessed by raw colorimetric measurement of LDH assay (BoTX 0.014 ± 0.001 vs SHAM 0.013 ± 0.001, n = 9 per group) or percentage cytotoxicity calculated by normalization against maximum LDH release after treatment of same wells with manufacturer’s lysis buffer (Figure 6C; BoTX 25.9% ± 2.3% vs SHAM 26.3% ± 2.4%).
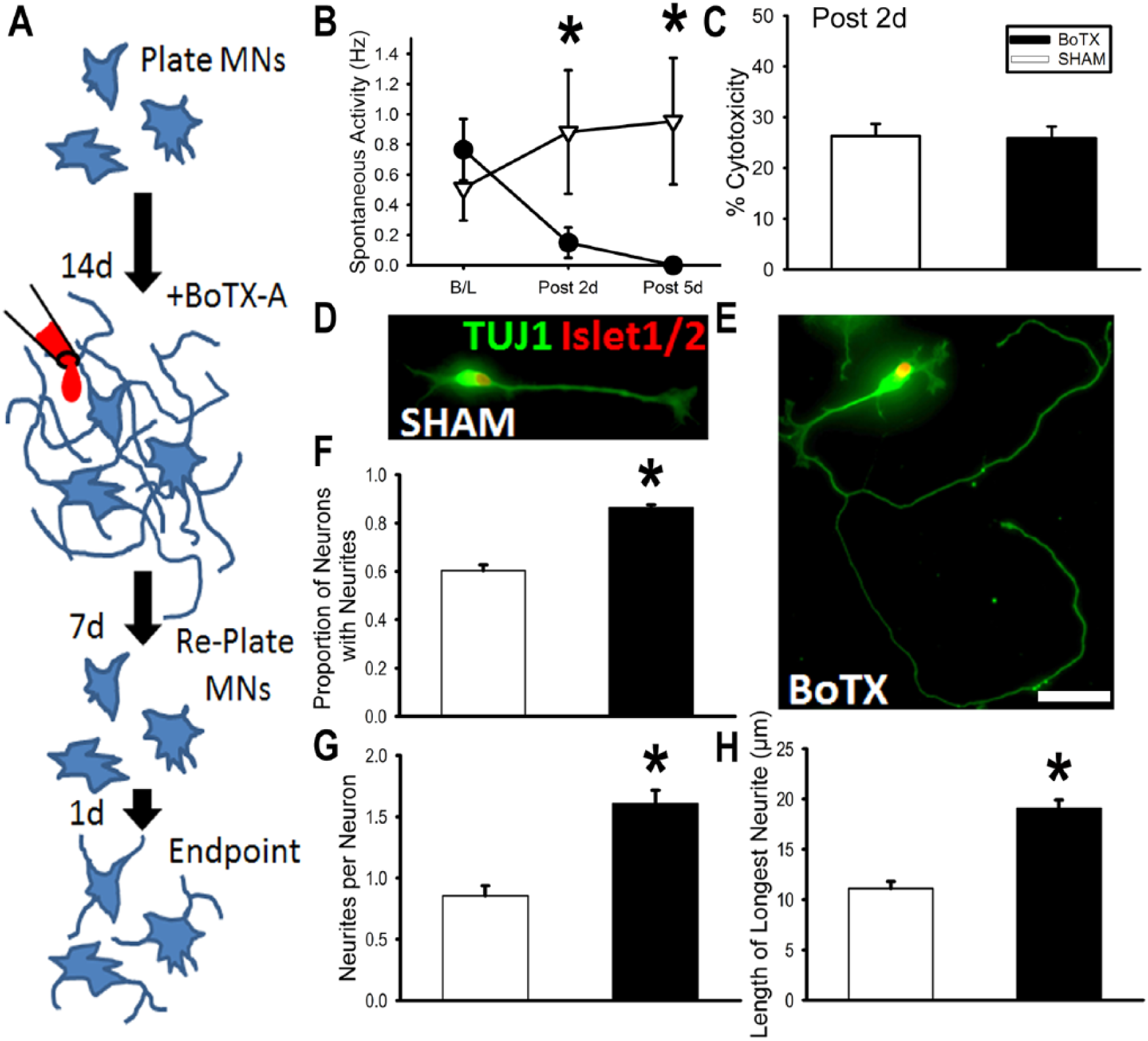
Botulinum toxin A (BoTX) conditioning increases human embryonic stem cell (hESC)–derived motor neuron (MN) neurite growth. (A) Treatments and timing of human MN neurite regrowth assay. (B) Spontaneous electrical of MN cultures at 2 weeks (baseline) and 2 to 5 days following BoTX or SHAM treatments. (C) Lactate dehydrogenase (LDH) cytotoxicity assay was performed 2 days after treatment and did not reveal a difference between BoTX versus SHAM controls. (D, E) Representative photomicrographs of SHAM and BoTX-treated MNs. (F) Proportion of neurons with neurites at endpoint. (G) Mean number of neurites per neuron at endpoint. (H) Mean length of the longest neurite formed per neuron at endpoint. Scale bar represents 10 μm. *P < .001.
BoTX or saline-vehicle SHAM preconditioning was applied to hESC-derived MN cultures on day 14. Analogous to our in vivo paradigm, we then waited for one additional week (ie, day 21) before detachment and then replating the hESC-derived MNs on a PDL-laminin coated glass coverslip. We allowed the replated hESC-derived MNs to grow for 24 hours (Figure 6D and E), since this early time point was associated with limited outgrowth for untreated hESC-derived MNs. MNs were analyzed in the BoTX (166 cells; 5 replicates) and SHAM (186 cells; 4 replicates) groups for their proportion with initial process formation (neurite initiation) (Figure 6F), the number of neurites per neuron with a neurite process equal to or longer than cell diameter in length (Figure 6G), and the longest neurite (Figure 6H). 19 Only MNs with neurites present were included in analysis for longest neurite length (n = 114 cells for BoTX, n = 107 cells for SHAM). The proportion of MNs with initial process formation in the BoTX group was 0.86 ± 0.02, which was significantly greater (P < .001) than the SHAM group (0.60 ± 0.04; Figure 6F). The mean number of neurites per MNs was significantly higher (P < .001) in the BoTX group, 1.61 ± 0.11, as compared with the SHAM group, 0.86 ± 0.08 (Figure 6G). Finally, the mean length of the longest neurite after BoTX was 19.1 ± 0.8 µm and SHAM was 11.1 ± 0.7 µm, which was significantly different (P < .001; Figure 6H). These findings identified a direct impact on MN-neurite outgrowth that mirrors the enhanced outgrowth observed we found with our in vivo murine model of axon regeneration.
Discussion
We report a novel strategy to augment motor axon outgrowth on the basis of data corroborated by 2 distinct models: (a) an in vivo mouse tibial nerve injury and (b) an in vitro hESC-derived MN neurite regrowth assay. Specifically, we found that preconditioning with a clinical grade formulation of BoTX 1 week prior, augmented motor axon growth in both the reinnervation of the distal tibial nerve stump (mouse) and the neurite outgrowth and branching of cultured MNs (human). We propose that BoTX preconditioning is a clinically feasible candidate treatment to enhance motor axon regeneration in scenarios such as nerve transfer surgery, in which healthy donor nerves are surgically redirected to restore function after nerve damage for conditions ranging from peripheral nerve injuries, plexopathies, and spinal nerve root avulsions. 22
The present findings that treatment with BoTX caused transient muscle paresis (Figure 2) and induced motor terminal axon sprouting (Figure 3) were of no surprise,20,23,24 but these experiments were essential to establish appropriate dosing for our subsequent axon regeneration studies since the axotomy-like effects induced by BoTX have been shown to be dose dependent. 14 The extent of the sprouting we observed was in line, albeit on the lower end of the range, with past studies that used similar formulations and delivery of BoTX.24-26 We then proceeded to test our main hypothesis that motor axon regeneration would be enhanced in a manner similar to the CLE one week after BoTX chemodenervation. Our hypothesis was predicated on the basis of multiple lines of evidence to suggest that BoTX chemodenervation mimics key aspects of axotomy including the interruption of neurotransmission,14,15 induction of motor axon sprouting, 27 synaptic stripping of MN inputs, 28 as well as upregulation of regeneration associated proteins.10-12 Overall, we took this as evidence that BoTX pretreatment switches MN phenotype from a “signaling” mode to a “growth” mode, 29 which in general terms is understood to underlie the CLE itself. 5
Consistent with our hypothesis, we found that BoTX preconditioning resulted an 89% increase in the number of myelinated axons (Figure 4) and a 39% increase in the number of MNs (Figure 5) that initially reinnervated the distal nerve stump 1 week after the tibial nerve test lesion. As anticipated based on previous reports on recovery from a mouse tibial nerve crush,30,31 by 4 weeks both groups had full axonal reinnervation of the distal nerve stump (Figure 5). Interestingly, only a limited number of prior studies have looked at the effect of intramuscular injection of BoTX prior to a nerve crush in vivo, but to our knowledge there has not been a previous report of enhanced motor axon reinnervation. The reason for this discrepancy on the acceleration of motor axon regeneration appears to be attributable to key differences in experimental design. In one case, Brown and Hopkins 32 found no change in mouse soleus muscle reinnervation after BoTX injection using a very distal nerve test lesion site, just 1 mm proximal to the soleus nerve–muscle entry point, which would make the comparison of axon regeneration rate difficult given the very short distances. Also, they relied on physiological outcome measurements of motor unit number and muscle tension, 32 both of which would have been dependent on the rate of synaptogenesis in addition to the rate of axon regeneration. Finally, they had a very small sample size (n = 6 per group) split between different endpoints (1-5 days postinjury) therefore increasing the risk of type II statistical error. In 2 other studies that found no effect on axon regeneration after BoTX pretreatment, the intervals between injection and nerve injury were ill-timed to elicit the CLE with one being too brief (ie, between 1 and 4 days), 33 and the other being too prolonged (ie, 36 days) 34 since it has been established that the optimal interval between conditioning and test lesions for the CLE paradigm is 1 to 2 weeks.35,36
We found that BoNT-A had a neurite stimulatory effect of hESC-derived MNs grown in culture based on an increased number and length of the neurites formed when the MNs were dissociated and then replated 1 week after BoTX conditioning (Figure 6). These stimulatory effects of BoNT-A were associated with blockade of the spontaneous electrical activity in our MN cultures (Figure 6) and were in general agreement with past works on cultured MNs isolated from embryonic mouse spinal cord 37 and co-cultured chick ciliary ganglion neurons with skeletal muscle. 13 However, these results are at variance with reports that BoNT-A may actually inhibit neuritogenesis in different neuron subtype, including L1 cell adhesion molecule-positive embryonic cortical mouse neurons, 38 PC12 cells and embryonic mouse dorsal root ganglion neurons. 39 The reasons for these discrepant results are not immediately clear, but may be a dose-related phenomenon 37 and/or represent neuron subtype specific differences in the effect of BoTX on neuron intrinsic growth state. Reassuringly, in both of the MN types studied to date, that is, primary mouse embryonic spinal MNs 37 and hESC-derived MNs (Figure 6), BoTX conditioning had a stimulatory effect on neurites.
Despite many decades of nerve regeneration research that has led to a much deeper understanding of this complex process, 40 there remains no proven drug therapies to improve peripheral axon regeneration. It seems evident that we should take steps to reassess preclinical modeling of nerve injury. This will likely include greater utilization of human pluripotent stem cell-based models of degeneration and regeneration, 41 combined with avoiding the overreliance on any single model of axon regeneration since they each have their own set of limitations. 42 With this in mind, we sought to increase the clinical relevance of the motor axon growth enhancement seen in our mouse model by corroborating with a human preclinical model. To our knowledge this is the first time a peripheral axon regeneration therapy has been tested concurrently in a mouse in vivo and human in vitro models of axon regrowth. This is particularly exciting since we performed all of these experiments with an Food and Drug Administration–approved formulation of BoTX, and there are currently no medications known to improve axon regeneration in humans.
Future studies aimed at characterizing the molecular mechanisms that underlie the BoTX mediated enhancement of motor axon growth will be of great interest. These investigations should probably be made vis-à-vis the classic CLE paradigm, which has been linked to an accelerated rate of the axonal transport slow component B 43 as well as to an elevated level of cyclic AMP that appears pivotal to overcoming inhibitors of axon outgrowth.44,45 However, the need for more mechanistic studies does not preclude the possibility of simultaneously making efforts for clinical translation of BoTX for axon regeneration. There are multiple clinically approved BoTX formulations available in the United States and they have established safety profiles.46-48 We anticipate dosing requirements for application to nerve transfer surgery will be on the low end of what is already deemed safe to use in the clinical management of muscle spasticity, 49 since these surgeries often only require a nerve fascicle from a single muscle. 9 Given that chronic axotomy-related changes can be devastating to muscle, neuron, distal nerve pathway, and ultimately the functional outcome after PNI,2,7,33,34 identifying a pharmacological approach to enhance recovery speed would address an unmet clinical need and likely provide a meaningful difference in the rehabilitation of many patients.
Supplemental Material
Supplemental_Materials – Supplemental material for Botulinum Toxin Conditioning Enhances Motor Axon Regeneration in Mouse and Human Preclinical Models
Supplemental material, Supplemental_Materials for Botulinum Toxin Conditioning Enhances Motor Axon Regeneration in Mouse and Human Preclinical Models by Colin K. Franz, Alyssa Puritz, Lewis A. Jordan, Jeffrey Chow, J. Alberto Ortega, Evangelos Kiskinis and Charles J. Heckman in Neurorehabilitation and Neural Repair
Footnotes
Acknowledgements
We are grateful for the technical assistance provided Dr Jack Miller (Department of Physiology, Northwestern University).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by a grant to C.K.F. from the Craig H. Nielsen Foundation and intramural support from the Shirley Ryan AbilityLab (formerly the Rehabilitation Institute of Chicago). L.A.J. and J.C. were supported by the Shirley Ryan AbilityLab medical student externship program. Semithin nerve sectioning and toluidine blue staining was performed by Mr. Lennell Reynolds Jr. at the Northwestern University Cell Imaging Facility, which is an institutional core facility generously supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center. The lab of E.K. is supported by Les Turner ALS Foundation at Northwestern University.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.