
Abstract
Background. Prophylactic treatment with antiepileptic drugs (AEDs) has been recommended to prevent early seizure onset in patients with traumatic brain injury (TBI). However, the potential neuroprotective and/or detrimental effects of prophylactic AED treatment on behavioral and cognitive function after TBI are not well studied. Objective. To investigate the effects of a novel AED, levitiracetam (LEV), on behavioral and cognitive function after experimental TBI in rats. Methods. Adult male rats were administered LEV (intraperitoneal 50 mg/kg) or vehicle (saline; SL) daily for 20 days beginning 1 day after controlled cortical impact (CCI; 2.8 mm; 4 m/s) or sham surgery. Beam performance (days 1-6), Y-maze (day 7), and Morris water maze (days 14-19) postinjury testing was assessed. Results. Daily LEV treatment improved motor function, increased novel arm exploration in the Y-maze, elicited greater hippocampal cell sparing, and decreased contusion volumes compared with CCI/SL rats. Daily LEV administration also reversed a TBI-induced decrease in regional glutamate transporter expression and neuroplastic marker proteins present 20 days post-CCI. Also, daily LEV treatment decreased regional IL-1β expression after TBI. Conclusions. These results suggest that daily LEV treatment has beneficial effects on histological, molecular, and behavioral elements of neurological recovery after TBI, in part, via modulation of neuroinflammatory and excitatory pathways.
Keywords
Introduction
Traumatic brain injury (TBI) is a major cause of death and disability worldwide. Among those with severe TBI, ~12% develop posttraumatic seizures (PTS) or posttraumatic epilepsy. 1 Based on clinical treatment guidelines, patients with severe TBI should receive prophylactic treatment with phenytoin for 7 days after injury. 2 However, the efficacy of phenytoin administration beyond the acute postinjury period is not supported. 3 Multiple studies show that prolonged post-TBI treatment with phenytoin has adverse effects on cognition.4,5 Also, in our controlled cortical injury (CCI) model of experimental TBI, we have shown that daily administration of phenytoin for 3 weeks after injury has adverse effects on cognitive performance, hippocampal cell survival, and hippocampal markers of plasticity, 6 providing experimental evidence that phenytoin use should be limited to prophylaxis when possible.
Despite current treatment guidelines for limited phenytoin use, there is substantial variation among clinicians in the practice of PTS prophylaxis regarding indication for treatment, choice of drug, and duration of treatment. Recent studies suggest that levetiracetam (LEV) has neuroprotective effects in both humans7,8 and rodents 9 after TBI and other acquired brain injury. However, the relative costs of LEV are significantly higher than phenytoin,10-12 which raises concerns about the need for, and benefits of, LEV in seizure prophylaxis and/or chronic therapy after TBI.
Levetiracetam confers both antiseizure and antiepileptogenic properties via the inhibition of synaptic vesicle protein 2A, selective inhibition of calcium channels (leading to a blunted increase in [Ca++]I),and modulation of γ-aminobutyric acid and glycine receptors. 13 Also, LEV has neuroprotective properties, through its ability to upregulate the expression of glial Glutamate (Glu) transporters EAAT1/GLAST and EAAT2/GLT-1. 14 Each of these findings provide possible mechanisms by which more favorable neurological outcomes might be obtained. To determine how LEV therapy affects neurological recovery in experimental TBI, we used the CCI model to examine the effects of LEV on histological, molecular, and behavioral recovery measures and to examine possible mechanisms by which LEV can influence behavioral outcomes.
Methods
Animals
Adult male Sprague–Dawley rats (Hilltop, Scottsdale, PA) were housed as previously described. 6 All behavioral procedures were conducted by assessors blinded to injury and treatment status, and exploratory tasks were videotaped for assessment purposes. All experimental procedures were approved by our University Animal Care and Use Committee. Animals were randomly assigned to an injury vehicle group (saline, SL) (TBI + SL, n = 12), injury drug group (TBI + LEV, n = 12), sham surgery vehicle group (SH + SL, n = 10), and a sham surgery drug group (SH + LEV, n = 10). Rats received 50 mg/kg injections of either LEV or saline every 24 hours for the duration of the 20-day study. Injections were performed after daily testing, giving ~23 hours between the injection and the next day of testing.
Controlled Cortical Impact Surgery
The CCI injury device has been described previously 6 and consists of a small (1.975cm) bore, double acting stroke-constrained pneumatic cylinder with a 5.0 cm stroke. The impact was delivered to a depth of 2.8 mm at 4.0 m/s. Craniotomy (6 mm in diameter) was performed between bregma and lamda. The actual impact site is approximately −2.00 to −8.00 mm anteroposterior relative to bregma, with tissue deformation ~2.8 mm below the dural surface. Sham animals underwent the same procedures, including craniotomy, except the CCI injury.
Behavioral Performance
To assess motor deficits after surgery, both beam balance and beam walk tests were employed as previously described. 6 On days 1 to 6 postsurgery, rats were evaluated on both tasks by completing three trials/day and monitoring return to a previously established baseline. Animals were assigned a beam walk score based on how far across the beam they travelled. Each of 5 equidistant sections on the beam walk task was assigned a score, with 5 indicating entrance into the goal box and 0 indicating an inability to move forward on the beam. The animal was assigned the score associated with the section of the beam reached at the conclusion of each testing trial. If the animal fell off of the beam, it was given a score corresponding to the portion of the beam from which it fell. One Y-maze testing session was conducted for each rat on day 7 postsurgery, as previously described. 6 The total time spent in each of the arms was recorded. The amount of time spent in the novel arm was compared with the average time spent in the familiar arm (eg, [(time in familiar arm 1 + time in familiar arm 2)/2]). The Morris water maze (MWM), previously demonstrated to be sensitive to cognitive function/dysfunction, 6 was employed days 14 to 19 where the time to locate a hidden platform was assessed on days 14 to 18, and time to locate a visible platform (VP) was assessed on day 19. Averaged daily latencies for each rat were compared between treatment groups on each testing day.
Histological Assessments
Twenty days after CCI or sham surgery, brains were perfused and subsequently embedded and stained with cresyl violet as previously described. 6 One coronal section underlying the area of contusion (~3.5 mm posterior to bregma) from each rat in all groups was analyzed by a single technician blinded to treatment in order to determine injury and treatment effects on hippocampal (HC) CA1 and CA3 neurons. All data were reported as the percentage of total neurons in the ipsilateral CA1 and CA3 regions relative to the contralateral hippocampus. The area of the lesion (mm2) was calculated by outlining the missing cortical tissue for each section taken at 1 mm intervals (MCID, Imaging Research, Ontario, Canada), and the volume (mm3) of the lesion was determined by multiplying the sum of the lesioned area obtained from each section by the distance between sections (1 mm).
Western Blotting
Samples from freshly dissected tissues were prepared as previously described. 6 Aliquots of 50 µg of protein from each regional tissue sample (hippocampus and frontal cortex from ipsilateral hemisphere) were mixed with 4× sample buffer and boiled for 5 minutes. The proteins were resolved on a 10% SDS–polyacrylamide gel and transferred onto Hybond-PVDF membranes (Amersham, Arlington Heights, IL). Membranes were blocked using Tris-buffered saline with Tween-20 (150 mM NaCl, 10 mM Tris–HCl, pH 8.0, and 0.05% Tween-20) containing 5% nonfat milk for 1 hour, then probed with anti-EAAT1 antibody (1:1000 Santa Cruz Biotechnologies, Santa Cruz, CA) for 1 hour, anti-EAAT2 (1:1000 Santa Cruz), anti-EAAT3 (1:1000 Santa Cruz), anti-IL-β (1:5000, Abcam, Cambridge, MA), anti-synaptophysin (1:5000, Santa Cruz), or anti-GAP43 (1:2000 Santa Cruz) overnight at 4°C. After washing 3 times with Tris-buffered saline with 0.5% Tween-20, the membranes were probed with horseradish peroxidase conjugated antibody (1:10 000) to allow detection of the appropriate bands using enhanced chemiluminescence (Pierce Biotechnology, Rockford, IL). The ECL system was removed by incubating the blot in Restore Western Blot Stripping Buffer (Pierce Biotechnology, Rockford, IL) for 15 minutes. The membranes were then probed again with anti-β-actin antibodies (1:5000 Sigma, St Louis, MO) to assess protein loading. Band densities were normalized to the β-actin band. Total pixel intensities were analyzed with Image J (National Institutes of Health).
Immunohistochemistry
Interleukin1-β (IL-1β) and GLT-1/EAAT2 immunohistochemistry was conducted on paraffin-embedded tissue. Sections were preblocked with 10% normal donkey serum (NDS) and 0.1% Triton X-100 in 0.1 M phosphate-buffered saline (TX-PBS). Sections were incubated with primary antibody, Rabbit anti-IL-1β polyclonal antibody (1:1000; Abcam), goat anti-EAAT2 polyclonal antibody (1:1000; Santa Cruz), with 5% NDS and TX-PBS at 4°C for 16 to 24 hours. Goat anti-rabbit IgG and rabbit anti-goat IgG (1:50; Jackson ImmunoResearch Laboratories, West Grove, PA) were incubated as secondary antibodies with 5% NDS and TX-PBS at 4°C for 2 hours on a shaker. Rabbit and goat peroxidase–anti-peroxidase (PAP) soluble immune complexes 20 g/mL (Jackson ImmunoResearch Laboratories) were used to visualize immunoreactivity with 2% NDS and TX-PBS at 4°C for 3 hours. Tissue slides were rinsed between all steps with TX-PBS 3 times for at least 10 minutes each time. The peroxidase reaction was developed with DAB Substrate Kit (Vector Laboratories, Burlingame, CA) until a dark brown reaction product was evident. Sections were rinsed in water, dehydrated in alcohols, defatted in xylenes, and cover-slipped for light microscopic analysis. Control experiments were run in parallel to confirm specificity.
Statistical Analysis
Data are presented as mean ± SEM (standard error of the mean). For behavioral tasks that involved repeated test sessions (motor testing and MWM), repeated-measures analysis of variance (ANOVA) was used. Post hoc pairwise analysis, adjusting for multiple comparisons (Tukey correction), was used to assess behavioral performance between injury and sham groups and among injury treatment groups. Western blot data were evaluated using either single-factor ANOVA with a Tukey correction. Y-maze behavioral assessments were performed using multivariate ANOVA. Histological comparisons were assessed using Student’s t test. Statistical analyses were completed using IBM SPSS for Windows version 20.0. A P value, adjusted for multiple comparisons when appropriate, of P ≤ .05 was considered significant (*P < .05, **P < .01, ***P < .001).
Results
Motor Performance
For each of the beam tasks, sham rats exhibited a small deficit in performance on day 1 that resolved by day 2 postsurgery and has been observed previously with this model.15-17
There was an overall effect of injury, time, and an injury × time interaction (P < .001 all comparisons) on beam balance performance. Injured rats were transiently impaired on the beam balance task compared with sham controls. Post hoc contrasts showed daily LEV treatment improved motor function for TBI rats compared with TBI + SL rats (P = .035). No significant performance differences were noted between SH + LEV and SH + SL rats. TBI + LEV rats returned to baseline performance by day 4, whereas TBI + SL rats did not return to baseline until day 5 postsurgery (Figure 1A). For beam walking latency, there was an effect of injury, day, and injury × day interaction (P < .001 all comparisons). TBI rats were also transiently impaired on this task compared with sham controls. Post hoc comparisons showed that TBI + LEV rats performed better than TBI + SL rats (P = .003). TBI + LEV rats returned to baseline performance by day 5, while TBI + SL rats did not return to baseline until day 6 (Figure 1B). Similar results were noted for the beam walking score, where there was an effect of injury status, day, and an injury × day interaction (P < .001 all comparisons). TBI rats were transiently impaired on this task compared with shams. Post hoc contrasts showed that TBI + LEV rats performed better than TBI + SL rats (P = .002). Here, TBI + LEV rats returned to baseline performance by day 5 postsurgery, but TBI + SL rats did not return to baseline until day 6 (Figure 1C).
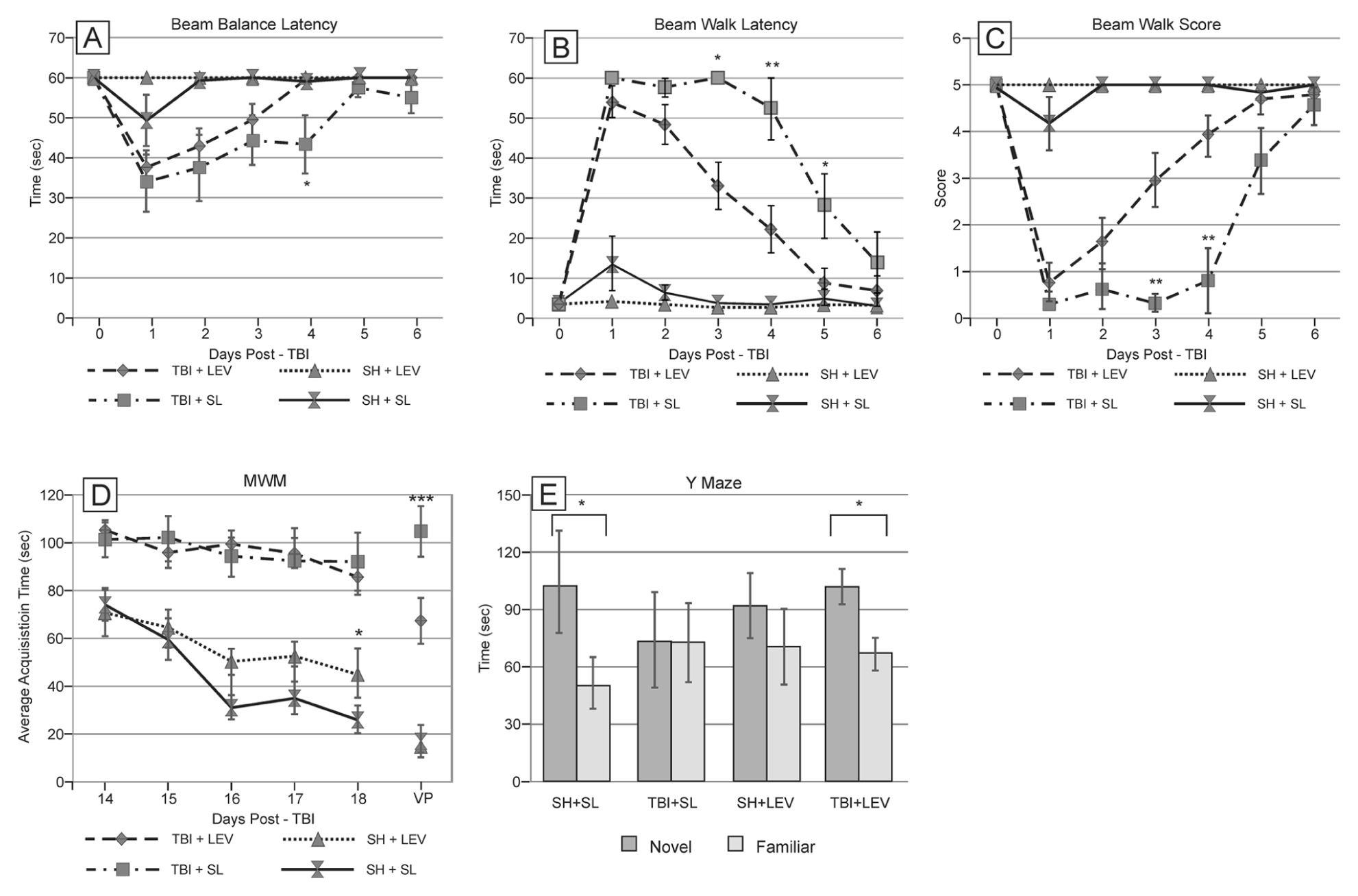
Effects of levetiracetam (LEV) treatment on the behavioral tasks. (A) Beam balance latency ***P = .001: TBI + LEV versus TBI + SL at day 4. (B) Beam walking balance latency **P = .002: TBI + LEV versus TBI + SL day3; ***P = .001: TBI + LEV versus TBI + SL day4; **P = .01: TBI + LEV versus TBI + SL day 5. (C) Beam walking score ***P < .001: TBI + LEV versus TBI + SL at days 3 and 4; (D) Morris water maze hidden platform latencies days 14 to 18 postsurgery. Day 19 visible platform (VP) *P = .044: TBI + LEV versus TBI + SL VP trial. (E) Y-maze *P = .041: SH + SL and P = .024: TBI + LEV time in novel versus familiar arm exploration. SH + SL, sham injured rats and daily saline administration; TBI + SL, traumatic brain injury rats and daily saline administration; SH + LEV, sham injured rats receiving daily levetiracetam; TBI + LEV, traumatic brain injury rats receiving daily levetiracetam administration.
Morris Water Maze
There was an effect of group and days (P < .001 all comparisons) on hidden platform latencies. TBI rats performed worse than shams, regardless of treatment. TBI + LEV rats did not perform differently than the TBI + SL group. When assessing daily performance, SH + LEV rats tended to perform worse than sham + SL beginning on day 16. VP testing on day 19 showed injury-related differences in platform latencies compared with shams. Latencies were similar between SH + LEV and SH + SL groups. However, TBI + LEV rats performed better than TBI + SL controls (P = .044; Figure 1D).
Y-Maze
For Y-maze performance, there was an arm effect (P < .001) and injury × arm effect (P < .001) observed. No treatment × injury interactions were identified. Post hoc contrasts showed that there was increased time spent in the novel arm, compared with familiar arm, in the SH + SL group (P = .041). Similarly, TBI + LEV rats demonstrated a significant increase in time spent in the novel arm compared with familiar arm exploration (P = .024). Neither the TBI + SL nor the SH + LEV group showed any preference for the novel arm (Figure 1E).
Cortical Contusion Volume and Hippocampal Neuron Quantification
The mean cortical contusion volume of the TBI + SL was 41.65 ± 3.18 mm3, and the mean cortical contusion volume of the TBI + LEV group was smaller at 28.56 ± 2.83 mm3 (P = .007; Figure 2A). All TBI rats had a significant loss of morphologically intact HC neurons in the ipsilateral hemisphere. There was increased CA1 neuron survival with LEV versus vehicle-treated TBI rats (P = .003), whereas there was no difference in CA3 neuron survival (Figure 2B).
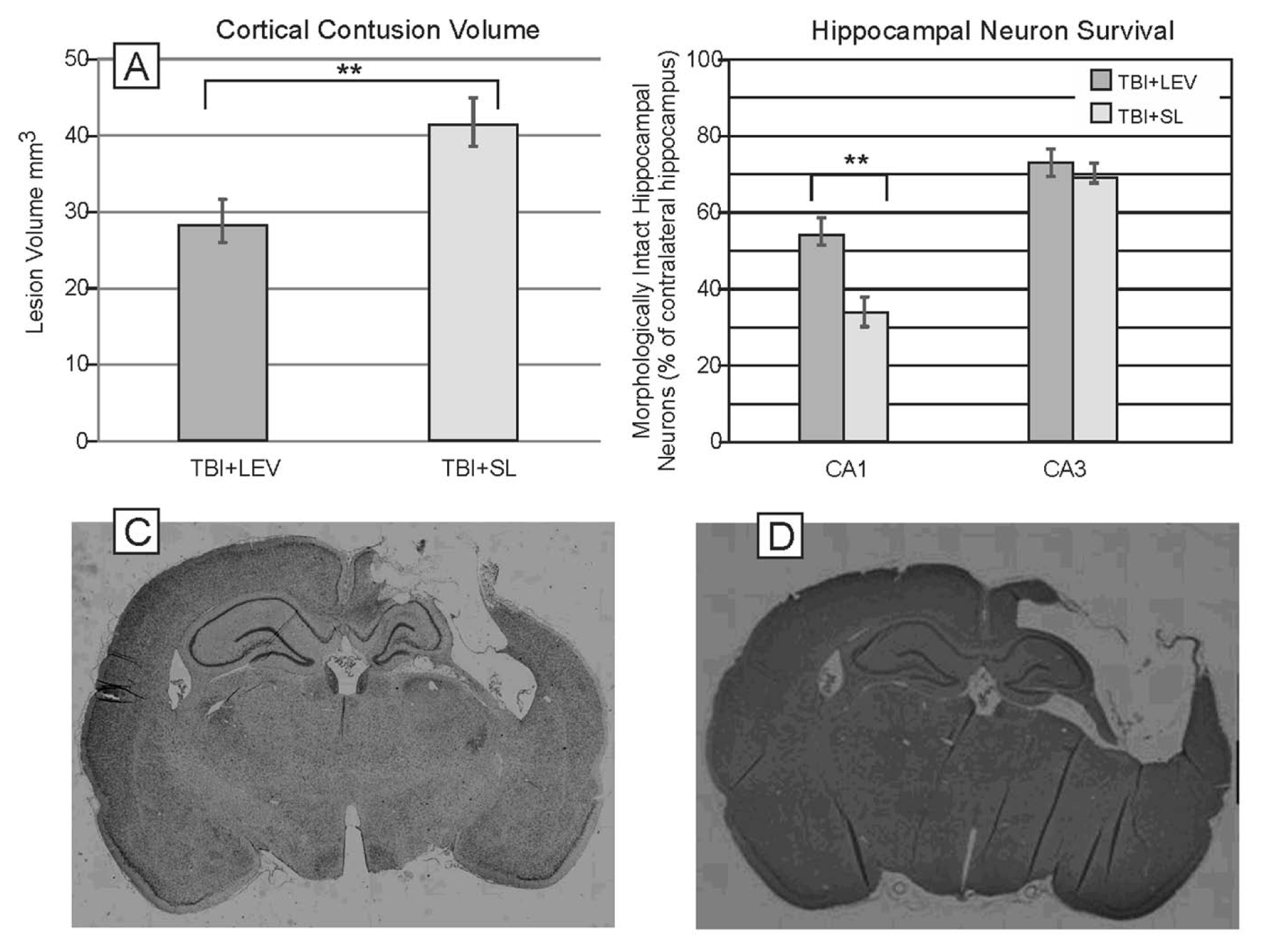
Neuroprotective effects of levetiracetam (LEV). (A) Cortical contusion volume: **P = .007 TBI + SL versus TBI + LEV. (B) Hippocampal neuron survival (**P = .003) percentage survival (ipsilateral/contralateral) CA1 neurons TBI + SL versus TBI + LEV. SH + SL, sham injured rats and daily saline administration; TBI + SL, traumatic brain injury rats and daily saline administration; SH + LEV, sham injured rats receiving daily levetiracetam; TBI + LEV, traumatic brain injury rats receiving daily levetiracetam administration. (C) Representative photomicrographs showing cresyl violet staining of tissue sections taken from the TBI + SL group. (D) Representative photomicrographs showing cresyl violet staining of tissue sections taken from the TBI + LEV group.
Western Blotting
In the ipsilateral frontal cortex, we examined injury and LEV-related changes in glutamate transporters (Figure 3A-C). Using ANOVA, we identified a group effect for GLAST, GLT-1, and EAAC-1. CCI was associated with decreased GLAST, GLT-1, EAAC-1 expression (P < .001 all comparisons), whereas LEV treatment increased glutamate transporter protein expression among TBI groups (P < .001 all comparisons) such that there were no differences between SH + SL treated groups and TBI + LEV treated groups for GLAST and EAAC-1. However, GLT-1 levels for TBI + LEV were higher than for SH + SL treated rats (P = .004). Interestingly, daily LEV treatment had opposite effects on shams compared with TBI, where GLAST (P = .007) and EAAC-1 (P = .005) levels were lower for SH + LEV rats compared with the SH + SL group.
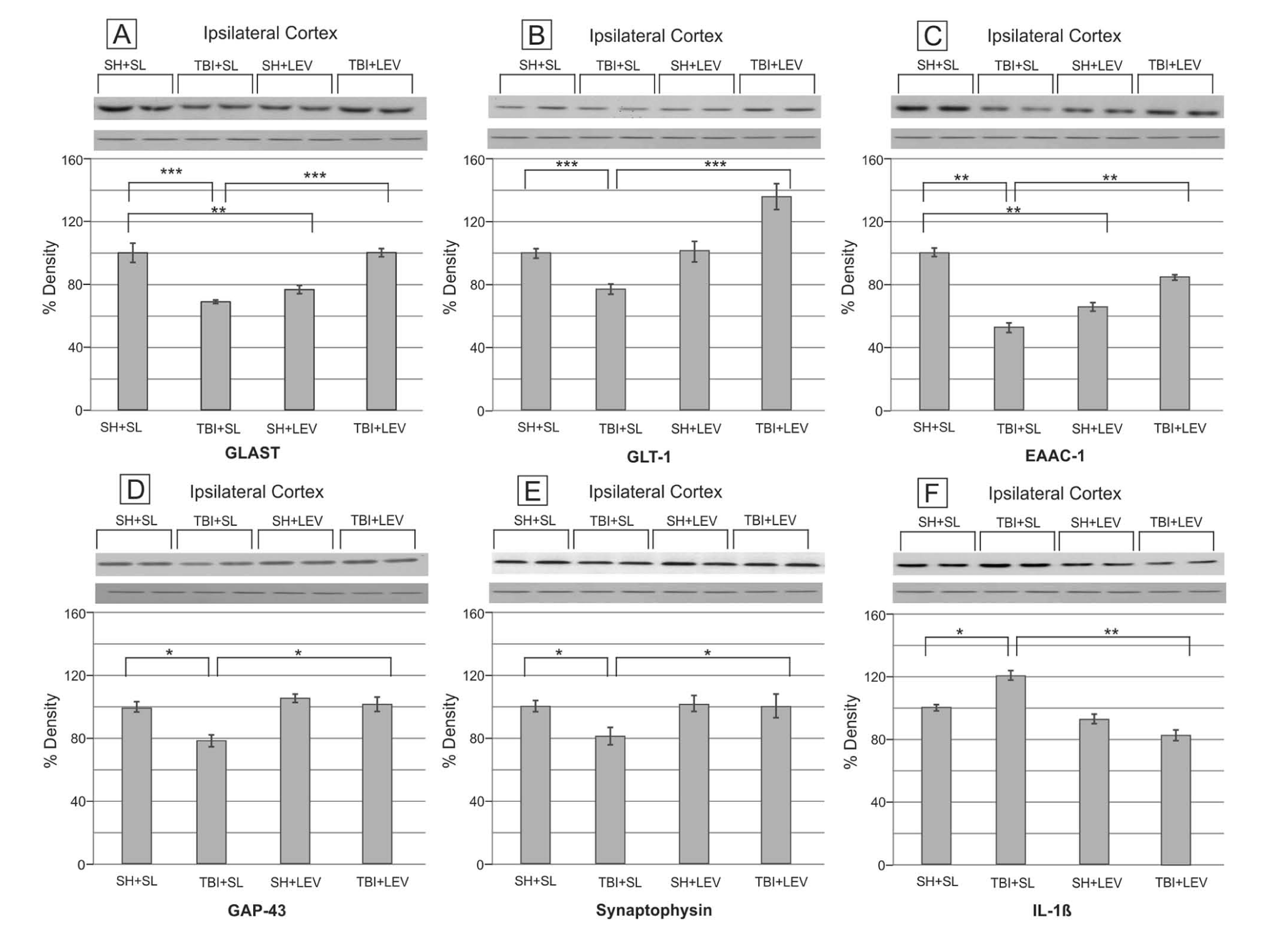
Effects of levetiracetam (LEV) on the EAATs, neuroplastic markers, and IL-1β at the ipsilateral frontal cortex. (A) Ipsilateral frontal cortex GLAST expression. (***P < .001) SH + SL versus TBI + SL, (***P < .001) TBI + SL versus TBI + LEV, and (**P = .007) SH + SL versus SH + LEV. (B) Ipsilateral frontal cortex GLT-1 expression. (***P < .001) SH + SL versus TBI + SL and (***P < .001) TBI + SL versus TBI + LEV. (C) Ipsilateral frontal cortex EAAC-1 expression. (***P < .001) SH + SL versus TBI + SL, (***P < .001) TBI + SL versus TBI + LEV, and (**P = .005). (D) Ipsilateral frontal cortex GAP-43 expression. (*P = .022) SH + SL versus TBI + SL and (**P = .017) TBI + SL versus TBI+LEV. (E) Ipsilateral frontal cortex synaptophysin expression. (*P = .024) SH + SL versus TBI + SL and (*P = .018) TBI + SL versus CCI + LEV. (F) Ipsilateral frontal cortex IL-1β expression. (*P = .035) SH + SL versus TBI + SL and (**P = .008) TBI + SL versus TBI + LEV. SH + SL indicates sham injured rats and daily saline administration; TBI + SL, traumatic brain injury rats and daily saline administration; SH + LEV, sham injured rats receiving daily levetiracetam; TBI + LEV, traumatic brain injury rats receiving daily levetiracetam administration.
In the frontal cortex, injury status and LEV treatment influenced GAP-43 and synaptophysin expression (P < .001 all comparisons). Pairwise comparisons (Tukey test) show reductions in GAP-43 (P = .022) and synaptophysin (P = .028) as a result of TBI (Figure 3D and E). However, daily LEV treatment reversed the injury induced changes and resulted in an increase in cortical GAP-43 (P = .017) and synaptophysin (P = .018) expression postinjury compared with TBI + SL rats.
In the ipsilateral hippocampus, there was also a group effect for GLT-1 and EAAC-1 (P < .001 all comparisons). Pairwise comparisons showed decreased glutamate transporter expression for GLT-1 (P < .001), but not EAAC-1 and GLAST expression, as a result of injury (Figure 4A-C). LEV treatment, however, increased both GLT-1 (P < .001) and EAAC-1 expression (P = .023) after TBI compared with TBI + SL rats GLT-1 (P = .043). EAAC-1 levels were lower for SH + LEV rats compared with the SH + SL group (P = .037).
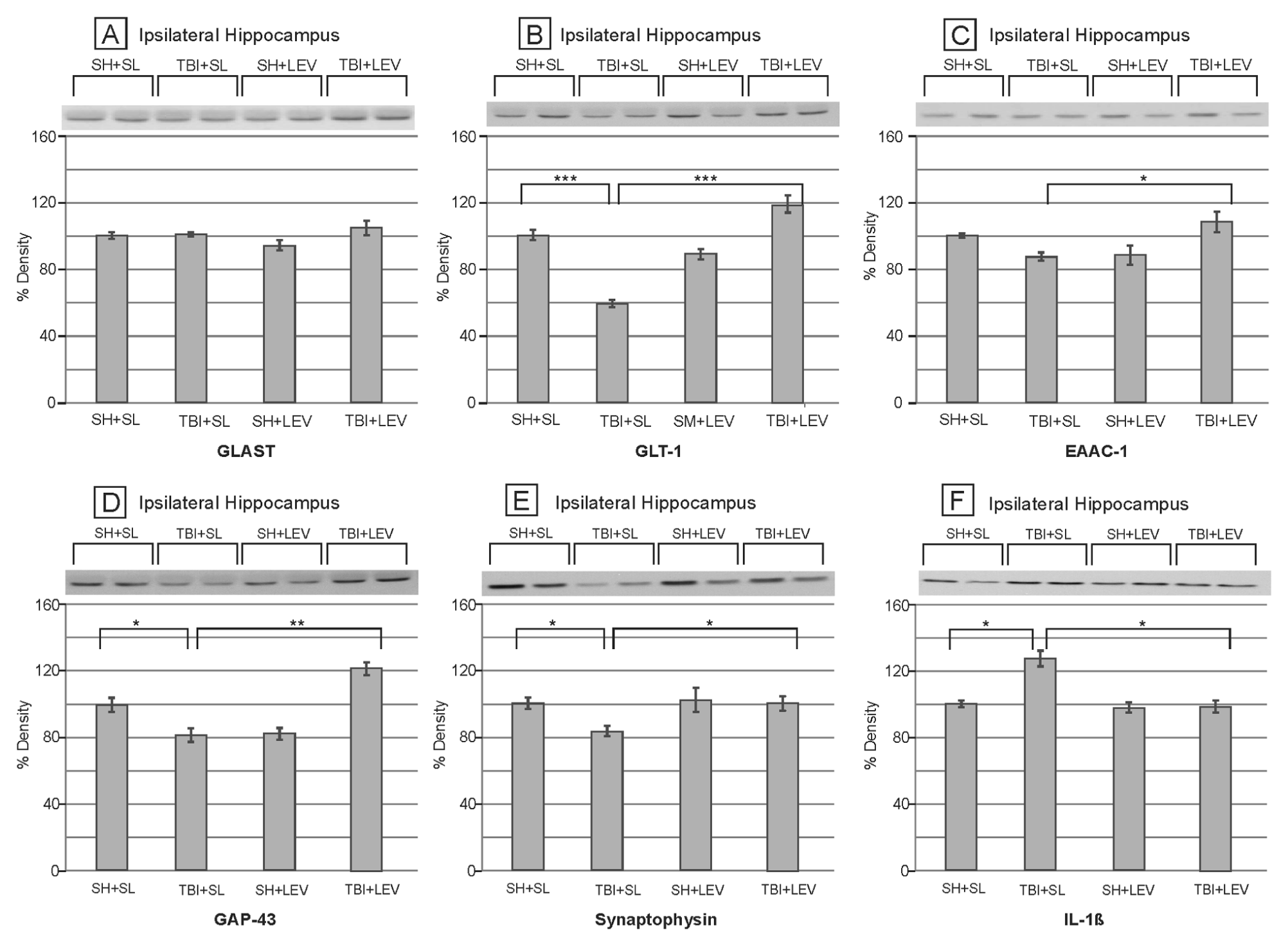
Effects of levetiracetam (LEV) on the EAATs, neuroplastic markers, and IL-1β at the ipsilateral hippocampus. (A) Ipsilateral hippocampus GLAST expression. (B) Ipsilateral hippocampus GLT-1 expression. (***P < .001) SH + SL versus TBI + SL and (***P < .001) TBI + SL versus TBI + LEV. (C) Ipsilateral hippocampus EAAC-1 expression. (*P = .023) TBI + SL versus TBI + LEV. (D) Ipsilateral hippocampus GAP-43 expression. (*P = .038) SH + SL versus TBI + SL and (**P = .006) TBI + SL versus TBI + LEV. (E) Ipsilateral hippocampus synaptophysin expression. (*P = .029) SH + SL versus TBI + SL and (*P = .037) TBI + SL versus TBI + LEV. SH + SL, sham injured rats and daily saline administration; TBI + SL, traumatic brain injury rats and daily saline administration; (F) Ipsilateral hippocampus IL-1β expression. (*P = 0.027) SH + SL versus TBI + SL and (*P = .016) TBI + SL versus TBI + LEV. SH + SL indicates sham injured rats and daily saline administration; TBI + SL, traumatic brain injury rats and daily saline administration; SH + LEV, sham injured rats receiving daily levetiracetam; TBI + LEV, traumatic brain injury rats receiving daily levetiracetam administration.
Similar to that observed in frontal cortex, GAP-43 and synaptophysin expression in the hippocampus was influenced by group (P < .001 all comparisons; Figure 4D and E). Reductions in both GAP-43 (P = .038) and synaptophysin (P = .029) were observed with TBI compared with sham, whereas daily LEV treatment reversed these decreases in GAP-43 (P = .006) and synaptophysin (P = .037) expression for TBI rats compared with saline-treated groups. Interestingly, GAP-43 was also reduced in SH + LEV rats (P = .041).
Injury and LEV treatment also were associated with persistent changes in IL-1β expression in both the frontal cortex and the hippocampus (P < .001 for both comparisons). Pairwise contrasts show that TBI caused a persistent increase in IL-1β expression in the cortex (P = .035) and hippocampus (P = .027) when compared with regional SH + SL groups. This increase in IL-1β expression was reversed after daily LEV treatment in the cortex (P = .008; Figure 3F) and hippocampus (P = .016; Figure 4F).
Immunohistochemistry
We performed immunohistochemistry analysis characterizing changes in inflammation and the primary glutamate transporter GLT-1. An increase in IL-1β immunoreactivity was observed in the ipsilateral pericontusional area for TBI rats (Figure 5A). However, LEV treatment attenuated IL-1β expression compared with TBI + SL controls (Figure 5B). Decreased GLT-1 immunoreactivity was observed in the ipsilateral pericontusional area for the TBI + SL (Figure 5C), whereas LEV-treated rats showed a relative increase in GLT-1 expression compared with TBI + SL controls (Figure 5D).

Photomicrographs of IL-1β immunohistochemical staining in the pericontusional region of (A1-A3) a saline-treated TBI rat, (B1-B3) levetiracetam-treated TBI rat, (C1-C3) GLT-1 immunohistochemical staining in the pericontusional region of a saline-treated TBI rat, and (D1-D3) levetiracetam-treated TBI rat. A1-D1, 4× magnification; A2-D2, 4× magnification. A3-D3, 10× magnification. Scale bar = 100 µm (panels 1 and 2) and 50 µm (panel 3).
Discussion
Despite accepted guidelines for prevention and treatment of PTS, current knowledge is relatively lacking regarding if/how antiepileptic drug (AED) prophylaxis may influence behavioral and cognitive function. One issue with current AED prophylaxis is that the duration of treatment varies in practice and is often prolonged past the point of known benefit. Likewise, multiple AEDs are used for prophylaxis despite the fact that their effectiveness in preventing PTS has not been well studied. Thus, the evaluation and implementation of a targeted, optimized AED prophylaxis regimen without adverse effects on neurological recovery is needed. Clinical trials with phenytoin therapy were central to the guideline of 1 week PTS prophylaxis, with treatment beginning immediately after severe TBI.2,18
In contrast, LEV is a relatively new and popular antiepileptic drug that is attractive for use in PTS management because of its favorable pharmacokinetic characteristics, including excellent bioavailability, linear kinetics, minimal plasma protein binding, and rapid achievement of steady state concentrations.
19
A recent small clinical study suggests that LEV has neuroprotective qualities that may translate to improved outcomes for patients with severe TBI and subarachnoid hemorrhage, compared with treatment with phenytoin.
8
The mechanism of antiepileptic action of LEV is not fully elucidated, but it does target the synaptic vesicle protein 2A.
19
Anticonvulsant and antiepileptic mechanisms for LEV specific to posttraumatic epilepsy are not well studied. However, given LEV’s ability to upregulate glutamate transporters in seizure and nonseizure models,
14
and as shown in this study after CCI, it may be that glutamate transporter modulation is a mechanism by which LEV confers neuroprotective and antiepileptic effects, particularly early postinjury when excitotoxicity is prominent. Extracellular glutamate levels increase after both TBI
20
and ischemic brain injury,
21
which causes overactivation of N-methyl-
Under physiological conditions, extracellular glutamate concentration is mainly regulated through reuptake of glutamate from the synaptic cleft via glutamate transporters. 22 Five different glutamate transporters have been identified to date: GLAST, GLT-1, EAAC1, EAAT4, and EAAT5. 23 GLT-1 constitutes the major glutamate transporter subtype in cortex, its major function is to remove glutamate from synapses. 24
Previous work in ischemia models shows that upregulation of glutamate transporters provides neuroprotection, whereas in vitro knockdown studies, particularly those reducing the glutamate transporter GLT-1, exacerbates brain damage.25-27 Previous studies demonstrate reduced GLAST/EAAT1 and GLT-1/EAAT2 expression at early time points after experimental brain injury, 28 a phenomenon that also occurs in neurodegenerative disorders such as amyotrophic lateral sclerosis, Alzheimer’s disease, and Huntington’s disease.29,30 Transient middle cerebral artery occlusion also causes decreased GLT-1/EAAT2 at both 24 and 72 hours of reperfusion. 26 Furthermore, hippocampal cell survival when using in vitro models of hypoxic–ischemic insult 31 is associated with glutamate transporter expression. Taken together with this literature, our data support the large impact for glutamate transporters on cellular damage in several neurological diseases, including TBI. This literature also suggests that early after TBI, reduced glutamate transporter expression may occur in the setting of cell death. Also, reduced glutamate transporter expression may also exacerbate excitotoxic injury, further perpetuating cell death.
Interestingly, our data showed that TBI-induced reductions in glutamate transporters persist over weeks, which may be relevant to long-term TBI rehabilitation and recovery. When considering that glutamate transporter reductions are linked to neurodegenerative disease processes, and if chronically elevated glutamate levels are a result of injury induced reductions in regional glutamate transporter expression, LEV treatment may (in addition to early PTS prophylaxis) reduce glutamatergic induced evolution of epileptogenesis and slow the progression of neurodegenerative changes known to occur after TBI. 14
However, recent work suggests in a CCI model that pericontusional glutamate levels are reduced at 14 days post-CCI. 32 Thus, an alternative hypothesis to enhanced early neuroprotection with LEV-induced normalization of glutamate transporters expression is that injury-induced reductions in regional glutamate transporter expression by 3 weeks postinjury is a compensatory response to a low glutamate environment. Consistent with this interpretation, early neuroprotection with LEV may result in less injury, and thus, more homeostatic glutamate physiology at later time points postinjury that do not require compensatory changes in transporter expression.
Interestingly, LEV treatment also reduced some glutamate transporter expression in sham rats compared with saline-treated shams. This reduction may be one contributing factor to deficits in SH + LEV animals during MWM testing and for reduced novel arm exploration with Y-maze analysis. The consequences of glutamate transporter reduction with LEV treatment in sham animals are not clear; however, increased glutamatergic states have been linked to cognitive dysfunction.33,34
Although there were no significant improvements in hidden platform latencies during MWM acquisition trials, VP performance was improved in TBI + LEV rats compared with TBI + SL controls. As the VP trial is similar to other proximal cue paradigms, where nigral–striatal system function is critical to performance,35,36 significant improvements in the TBI + LEV may also implicate LEV in striatal neuroprotection after TBI. While regional hippocampal counts are linked to MWM performance in other rodent models of neurodegenerative disease, 37 and some CA-1 sparing was conferred with LEV after TBI, these histological improvements did not translate to improved place learning. However, TBI affects multiple extra-hippocampal regions associated with nonspatial elements of navigation in the MWM (eg, thalamus),38,39, and future work should focus on additional characterization of LEV effects on these regions and on the spatial and nonspatial contributions to MWM after CCI. 40 Faster resolution of motor deficits occurred in LEV-treated TBI rats compared with vehicle-treated controls. Although speculative, we hypothesize that these reductions in transient motor deficits after CCI are linked to LEV associations with smaller contusion volumes. Reductions in pericontusional edema in surrounding primary motor cortex post-injury with LEV may be another mechanism for motor improvements to explore in future work.
GAP-43 and synaptophysin are neuroplastic markers linked to axonal reorganization and synaptic plasticity, and hippocampal reductions have been previously described in CCI. 41 In this study, we found regional decreases in GAP-43 and synaptophysin expression in the ipsilateral frontal cortex and HC with TBI + SL rats that were not observed in TBI + LEV treatment groups. These results suggest that LEV reverses injury-related decreases in neuroplasticity markers in the cortex and HC, and this phenomenon is consistent with HC cell sparing and cortical contusion volume reductions in TBI + LEV rats compared with TBI + SL controls. However, the specific role of LEV in reducing injury effects, versus promoting neuroplasticity post-TBI, is not specifically delineated with this report.
In addition to glutamate transporters, glutamate homeostasis is also regulated by inflammatory molecules, including interleukins. 42 IL-1β can inhibit astrocyte glutamate transport in the spinal cord during viral encephalomyelitis, 43 and extracellular cytokines, such as IL-1β, are also known to inhibit EAAT-2/GLT-1 transcription. 44 These studies provide a link between inflammation and glutamate-mediated excitotoxicity. Although neuroinflammation is observed acutely after TBI, there might still be prolonged residual neuroinflammation days after TBI. We have previously shown that pericontusional elevations in IL-1β can occur up to 12 days after CCI. 45 Here, we show that prolonged (20 days post-TBI) regional increases in IL-1β occur after CCI and accompany decreased glutamate transporter expression. Furthermore, daily LEV administration reversed the chronic increase in IL-1β observed after CCI, a finding which might contribute to the normalization of EAAT expression, and may further contribute to the neuroprotective properties of LEV.
Although daily LEV therapy appears to be promising for neuroprotection and recovery, more work is required to understand the effectiveness of this drug in mitigating PTS risk compared with other (less costly) agents. Future work should also address whether the same benefits can be obtained with a temporally limited LEV treatment regime using a treatment window that is reflective of common clinical PTS practice. Dose response and pharmacokinetic studies to optimize treatment effects should be considered. Studies exploring relationships between excitotoxicity, inflammation, plasticity and epileptogenesis after TBI may help identify unique pathology involved in how TBI leads to epilepsy, and how LEV might target epileptogenesis and improve long term recovery. Although the costs for LEV are higher than phenytoin, the potential benefits of daily LEV on over all recovery, even in the context of a time limited PTS prophylaxis period, merits further evaluation.
Footnotes
Acknowledgements
We would like to thank Mary Synnott for the help with the figure production.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the University of Pittsburgh Department of Physical Medicine and Rehabilitation.