
Abstract
Gene manipulation technologies are part of the past, present, and future of the domestication of plants and animals. Scientific advances, however, have resulted in those changes being more rapid and precise than ever before. In this review, the authors discuss gene manipulation technologies currently in use, their mechanisms, and their potential biosafety issues, with a focus on mammalian laboratory animals.
Keywords
Recent years have seen remarkable technical advances in the ability to engineer the genome of organisms of all types. It is now within the realm of possibility to tailor any genome on demand so as to introduce any change imaginable, from changes in a single base pair (eg, to mimic a particular disease-causing mutation in humans in an animal model) to the insertion of large-scale changes (eg, replacing the genes encoding the components of the immune system in an animal model with the homologous human genes, thus producing a “humanized” animal). However, with this amazing opportunity comes ethical and biosafety concerns and responsibilities.
From the outset, as these techniques were first applied to the simple genomes of bacteria and viruses, it was clear that recombinant DNA technologies could pose biosafety concerns. 1 As these technologies were extended to the engineering of animal genomes, the potential for the accidental creation of biohazards did not lessen. Since most of these alterations first occurred in laboratory mice, the most commonly used model organism in science and the first mammalian model organism to have its genome fully sequenced, 2 ethicists and regulators were concerned about inadvertent release and human exposure or the entry of these animals into the wildlife food web. 3 Mice were created that expressed human, bacterial, and viral proteins. In some cases, the genomes of laboratory mice were modified to include the inserted genomes of entire organisms. For example, a viral genome such as that of hepatitis B is relatively small at 3.5 kilobases, and a transgenic animal expressing a replication-competent human virus is well within modern technical ability to create. 4
With this genetic revolution comes renewed concern over issues of safety: does the sudden ease and availability of these new genome modification technologies increase the potential to produce organisms that could pose a biosafety threat? In this work, we propose to review the history of the development of genome manipulation, describe the new technologies in detail, and discuss the potential dangers and regulatory response in the face of this new technological landscape.
Genome Manipulation in Animals
Humans have been manipulating the genome for thousands and thousands of years—ever since the recognition that one could select animals or plants with desirable traits and breed them together— resulting in many of these traits being passed on to offspring. This led to a wide variety of domesticated animal breeds and crop strains, exemplified for most people by the astounding number of modern dog varieties. 5 The domestic dog is a species derived from wild canines and has been selected for traits such as size (eg, Chihuahua vs Great Dane), behavior (eg, herding dog vs sled dog vs retriever), and head shape (eg, pug vs greyhound), among many others.
It was not until the discovery of the mutagenic properties of radiation and of certain chemicals that it was possible to deliberately induce changes in the genome, rather than waiting for spontaneous mutations or selecting desired traits and reinforcing them through breeding. Although this was a random process in the sense that one could not target a particular gene, artificially induced mutation could dramatically increase the genetic variability in the offspring of treated animals from which desirable traits could be selected. In the early 1980s, it became possible to produce specific, intentional mutations in the genome. Initially, these mutations consisted of the production of an artificial gene that was inserted randomly into the genome via a process called transgenesis. Shortly thereafter, it became possible to target a specific site within the genome and introduce a change there. More recently, the introduction of new technologies has resulted in a vast increase in the efficiency and decrease in cost for this kind of targeted mutagenesis.
Random Mutagenesis
The discovery by Muller in the 1920s 6 that irradiation of fruit flies with X-rays would result in the production of a variety of new phenotypes among the offspring and that these phenotypes were heritable was the first indication that mutagenesis could be induced by treatment of the organism with some sort of exogenous agent. Recall that this work was done after the rediscovery of Mendelian genetics but before the demonstration that DNA was important in heredity and well before the discovery of the structure of DNA. In short order, other forms of radiation 7 and chemical treatments 8 were shown to have the same effect. Creating such random mutations, in which offspring are screened for any phenotypical differences, has an advantage in that it is not predicated on any understanding of the form and function of a gene—instead, these agents randomly introduce a genetic change that causes a phenotypic alteration. Once the mutation itself is induced, the mutation is characterized to provide information about basic biological functions. Scientists soon moved on from insects to mammals, however, after discovering the sensitivity of mammalian germ cells to these mutagenic agents. 9 Discovering the exact nature of the changes induced by radiation or chemical mutagenesis required completion of the mouse genome sequence, so large-scale chemical mutagenesis projects did not come into their own until the end of the 1990s.
Random mutagenesis simply increases the frequency of mutation and does not produce mutations that are different from those found in nature. Mutations produced by ionizing radiation may involve large regions of DNA or be point mutations, while typical mutations produced by chemical agents such as N-ethyl N-nitrosourea are point mutations scattered throughout the genome. Any agent used to boost mutation rates is a priori dangerous because they are mutagens, teratogens, and carcinogens.
Transgenesis
In the early 1980s, research10 -12 revealed that DNA molecules injected into a pronucleus of a fertilized mouse embryo would randomly integrate into the genome. If this piece of transferred DNA contained a functional gene, that is, DNA encoding a functional protein or RNA along with the sequences that control its expression, this gene product would be expressed in the resulting animals. Animals carrying these exogenous transferred genes, or “transgenes,” became known as transgenic animals and the process itself as “transgenesis.” Production of transgenic animals is thus conceptually straightforward: one obtains 1-celled embryos at the pronuclear stage and injects a DNA construct. These embryos are then transferred into foster animals that carry the manipulated embryos to term, and the offspring are screened for the presence of the transgene (Figure 1). In mice, the efficiency is relatively high: 5% to 15% of pups will be transgenic depending on the strain of mouse being manipulated. 13 Rats have a lower efficiency of 1% to 5%. 14
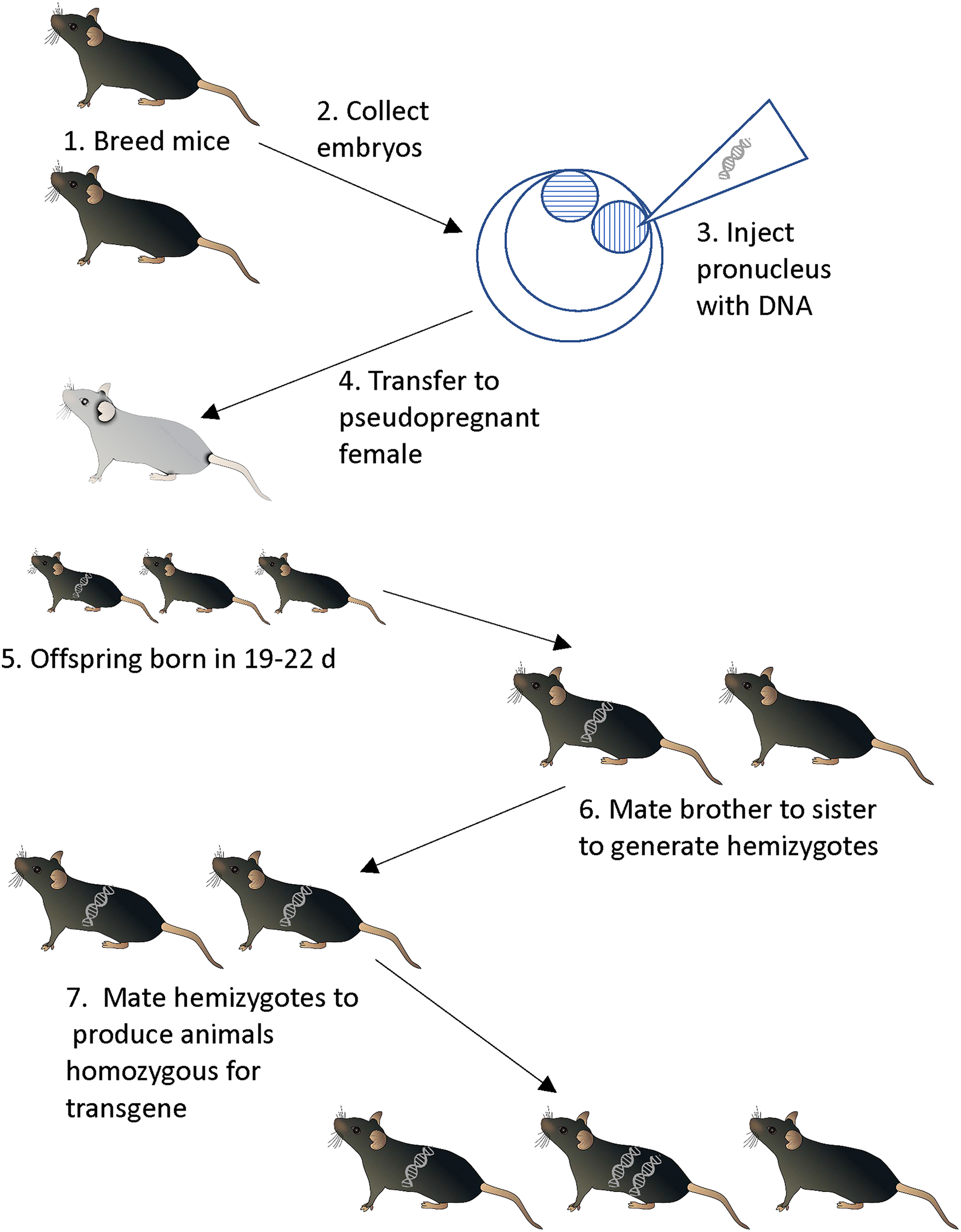
“Classical” transgenesis via pronuclear injection and implantation of manipulated embryos into host females.
Transgenes, integrated as they are into the genome, are passed to offspring in Mendelian fashion. It is important to note that the insertion of the transgene occurs in a random location in the genome. Furthermore, this insertion is itself a mutagenic event, interrupting as it does the endogenous genome, and transgenes are known to insert into and inactivate endogenous genes. 15 For example, in the authors’ experience, about 15% of all transgenic mouse lines cannot be bred to homozygosity, presumably because the insertion event has interrupted a gene necessary for embryonic development. Transgenesis introduces an exogenous gene, and although there are many details about when and where this gene is expressed in the animal, transgenesis results in the overexpression of the transgene product.
Other methods have been developed for transgene delivery. Notable among these is the use of lentiviral vectors. 16 Lentiviruses are a class of retroviruses with the ability to infect dividing and nondividing cells, including embryos, and whose genomes are not developmentally silenced. Lentiviral vectors have thus been harnessed as transgene delivery systems, and they are extremely efficient: it is possible to infect single-cell embryos and obtain offspring that are 100% transgenic. In fact, depending on the concentration of virus, many of these offspring will have the transgene inserted in multiple places, producing several lines as these founder animals are bred and the independent transgenes segregate. Despite this efficiency, lentiviral transgenesis has not been widely employed in the rodent transgenesis field for 2 reasons, although some animals have been created using this technology. 17 First, the size of the transgene that can be carried by these vectors is relatively small. 18 Second, there are several technical constraints, not the least of which is the human biosafety hazard presented by these vectors. 19 Nonetheless, this technique has found some utility in producing transgenic livestock. 20
Gene Targeting Using Embryonic Stem Cells
Rather than introducing an exogenous gene that overexpresses a product, gene targeting specifically changes the expression of endogenous genes. This was first accomplished using embryonic stem (ES) cells.21 -23 Using gene targeting, genes may have a loss of function (knockouts), expression of another function by gene replacement (knockins), or conditional expression, both by tissue and by the addition or removal of exogenous chemicals.
Embryonic stem cells are tissue culture cells derived from the blastocyst stage of mammalian embryogenesis, when the embryo consists of a hollow ball of cells surrounding the inner cell mass. The cells of the inner cell mass are the progenitors of all the cells in the adult animal, including the gonads and germ cells. For our purposes, they have 2 important properties. First, they can be propagated in vitro. Second, when combined with an intact mouse embryo either by being injected back into the lumen of the blastocyst or with an earlier, 8-cell embryo, these cells fuse with the host embryo and produce an animal that is a mix of cells derived from the cultured ES cells and the host embryo. Such an animal is known as a chimera. This is easily visualized if the ES cells are derived from a mouse strain with one coat color and the host embryo is of a different-colored strain (Figure 2). The important thing to note is that the chimerism extends to all the tissues of the animal, including the germline.

A chimeric dam and 2 knockout offspring. Note the differences in coat color; this is due to different levels of contribution to the female mouse from the injected embryonic stem (ES) cells and the host blastocyst. The offspring are a uniform color because the ES cells in the female mouse constituted the reproductive tract. Attribution: by staff at the National Institute of Mental Health’s Transgenic Core Facility: http://intramural.nimh.nih.gov/tgc/photogallery.html; public domain, https://commons.wikimedia.org/w/index.php?curid=7721249.
Most tissue culture cells, including ES cells, can be induced to take up exogenous DNA. Some will incorporate this DNA randomly into their genomes and thus become transgenic. However, if the DNA has sufficient homology with some part of that cell’s genome, at a low but finite frequency, that exogenous DNA will replace the endogenous DNA. It is possible to harness this mechanism to target a mutation to the genome. To do this, remove DNA from a region of the genome to be altered, then introduce the desired mutation somewhere in the middle of this region. This will result in long “homology arms” flanking the mutation, making it more likely the DNA will be incorporated. The naked DNA is then introduced to the cells in culture and then stimulating the cells through electroporation to take up the DNA and possibly incorporate it at cell division. The treated cells are screened for those in which the homologous recombination has taken place, introducing the specific mutation into the genome.
Homologous recombination is a very low-frequency occurrence, so the screening process is facilitated by a selectable marker, a gene whose expression renders the cell resistant to a drug. Usually, this gene is a version of the bacterial neomycin phosphotransferase gene, which confers resistance to members of the neomycin family of antibiotics, including G418, which kills mammalian cells. Thus, the DNA construct may be introduced with the selectable marker into several million cells, and then those cells are treated with G418 to kill all but the few hundred that have incorporated the DNA. These cells grow to form colonies of cells (clones) that still must be screened to distinguish those in which homologous recombination has occurred from the preponderance (approximately 99%) of cells that have randomly incorporated the DNA as a transgene. A second type of screening, a negative selection scheme, can be used to facilitate this selection of targeted cells. In this case, a gene that confers sensitivity to a drug is placed at one or both ends of the construct. This negative selection marker is commonly the herpes simplex virus thymidine kinase (hsv-tk) gene, which converts antiherpesvirus drugs such as ganciclovir into a toxin. Randomly incorporated DNA will retain this sensitivity gene, while homologous recombination will result in its loss, resulting in cell survival when exposed to acyclovir-type drugs (Figure 3).
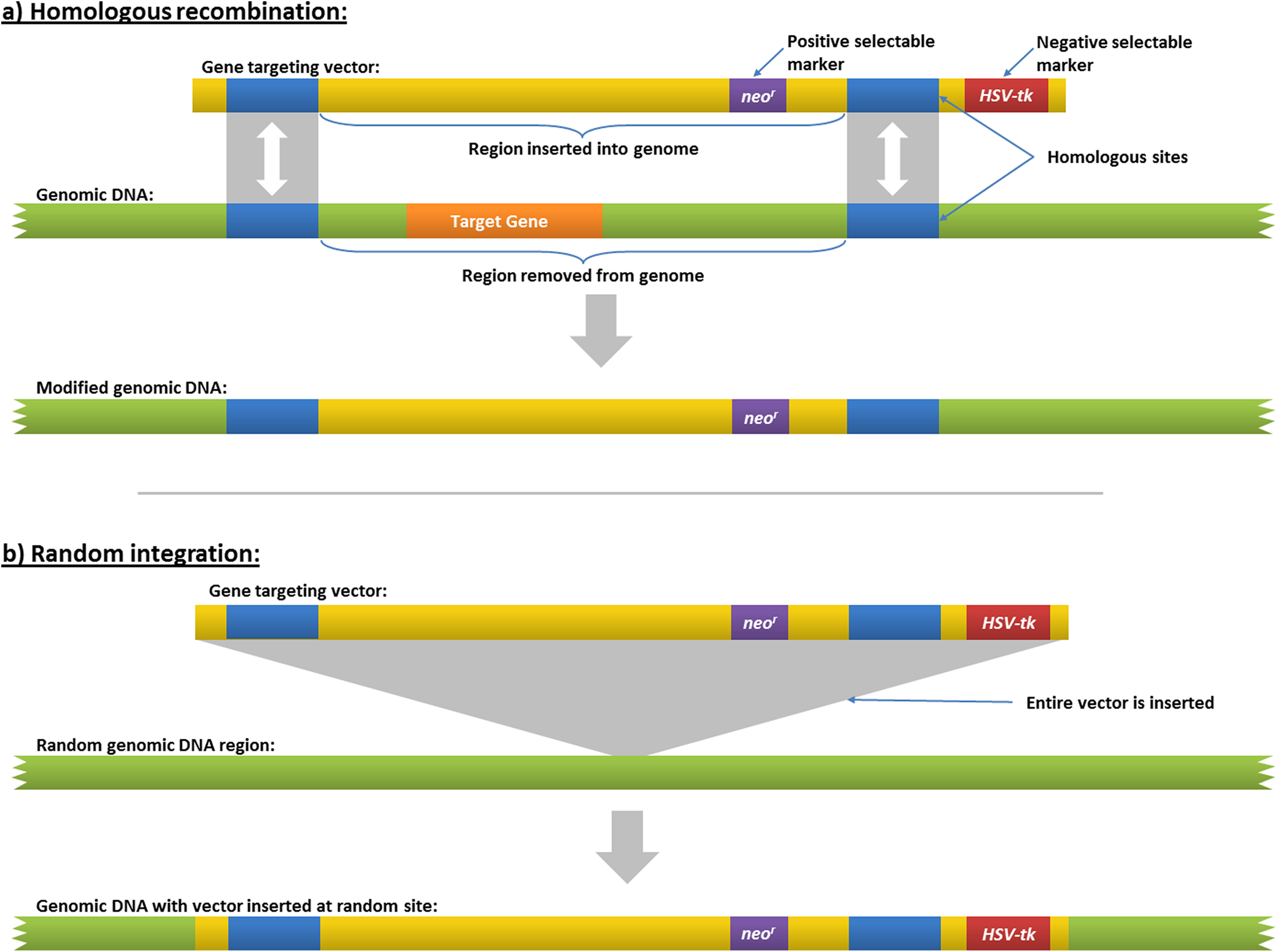
Homologous recombination in embryonic stem (ES) cells. This figure also illustrates how positive and negative selection markers are used to screen transformed ES cells. By BioStu, CC BY-SA 3.0 (https://creativecommons.org/licenses/by-sa/3.0), from Wikimedia Commons.
Once clones of cells have been obtained that carry the targeted mutation, these are expanded and then combined with appropriate host embryos using micromanipulation technology to place the ES cells in the inner cell mass of a 3.5-day blastocyst. The manipulated embryos are transplanted into foster mothers. The resulting chimeras will be a mixture of cells derived from the host embryos and the mutant ES cells, and at sexual maturity, these are then mated with wild-type females (for the ES cells are almost always genetically male). 24 First-generation offspring (F1 pups) resulting from ES cell-derived sperm are then identified (usually by coat color), and these are screened for the presence of the mutation.
The process has been quite effective, and methodologies for high-throughput targeted mutagenesis were eventually developed and harnessed into large-scale collaborative efforts, such as the Knockout Mouse Project, or KOMP, to inactivate all the known genes in the mouse genome and create a bank of ES cells from which mice carrying any of these mutations could be produced.25,26 Simply “knocking out” all the genes in the genome was not enough, however. Other types of targeted mutations are desirable, for example, ones in which specific base changes are made or in which specific domains of a gene are removed or replaced, or the gene is modified in such a way that it is only inactivated under certain conditions, for example, in particular cell lineages or organs or at particular times in development or when the animal is treated with a particular drug or chemical. For this, both transgenesis and targeted mutation are necessary as well as natural matings to produce the desired mutation.
As successful as this approach has been, however, it depends critically on the existence of ES cells.
Early in the adoption of this technology, germline-competent ES cells were only available for mice and only for mice from a very few inbred mouse strains. Culture methods were eventually developed that extended the ability to produce ES cells to most mouse strains 27 and even to rats.28,29 In other mammalian species, the development of somatic cell nuclear transfer (SCNT), in which the nucleus of a somatic cell is microinjected into an oocyte that is then activated to form an embryo, allowed manipulation of these species via that targeting and selection done in the donor cells before SCNT, but this methodology comes with its own set of technical difficulties.30,31
Additional technical details also are important when targeting genes in ES cells. Not all ES cell clones can create chimeric animals or contribute to the germline of those chimeras, so it is important to produce and use multiple ES cell clones for any given project. More important, the process is time- and labor-intensive. The first step is to produce the targeting construct, which can often take many weeks if the construct is large and complex and not subject to easy synthesis. Once the construct is produced, it can take another 4 weeks to produce, screen, and isolate targeted clones, although as mentioned above, in some cases, an appropriately targeted clone may be available from the KOMP consortium. Elapsed time then becomes a matter of biology. After combining the ES cells with the embryos and transplanting these to foster mothers, pups are born after 3 weeks and reach sexual maturity at around 8 weeks of age. Thus, it is about 12 weeks before chimeras may be bred with wild-type animals. Three weeks beyond that, the first pups are born, and the resulting mutant animals (assuming that any are found) again need 8 weeks to reach sexual maturity. Thus, assuming starting from the very beginning, it will be on the order of 6 to 8 months before animals can be used as the foundation of a colony for experimental animal production.
Double-Stranded DNA Nucleases and the Gene Targeting Revolution
Double-stranded DNA breaks are inimical to the survival of the cell, and all cells thus have an ability to repair such breaks. When a chromosome is broken, there are 2 predominant mechanisms for its repair (Figure 4). In the first, known as nonhomologous end joining (NHEJ), the 2 ends are simply rejoined. This is an error-prone process: insertions and/or deletions (known as indels) of DNA are found at the repair site. If these occur within the coding sequence of a gene, these changes will usually inactivate the gene. The second mechanism is known as homology-directed repair (HDR) and is also referred to in some circles as homology-mediated repair or simply homologous repair. HDR uses the intact gene on the homologous chromosome or sister chromatid as a template for error-free repair.
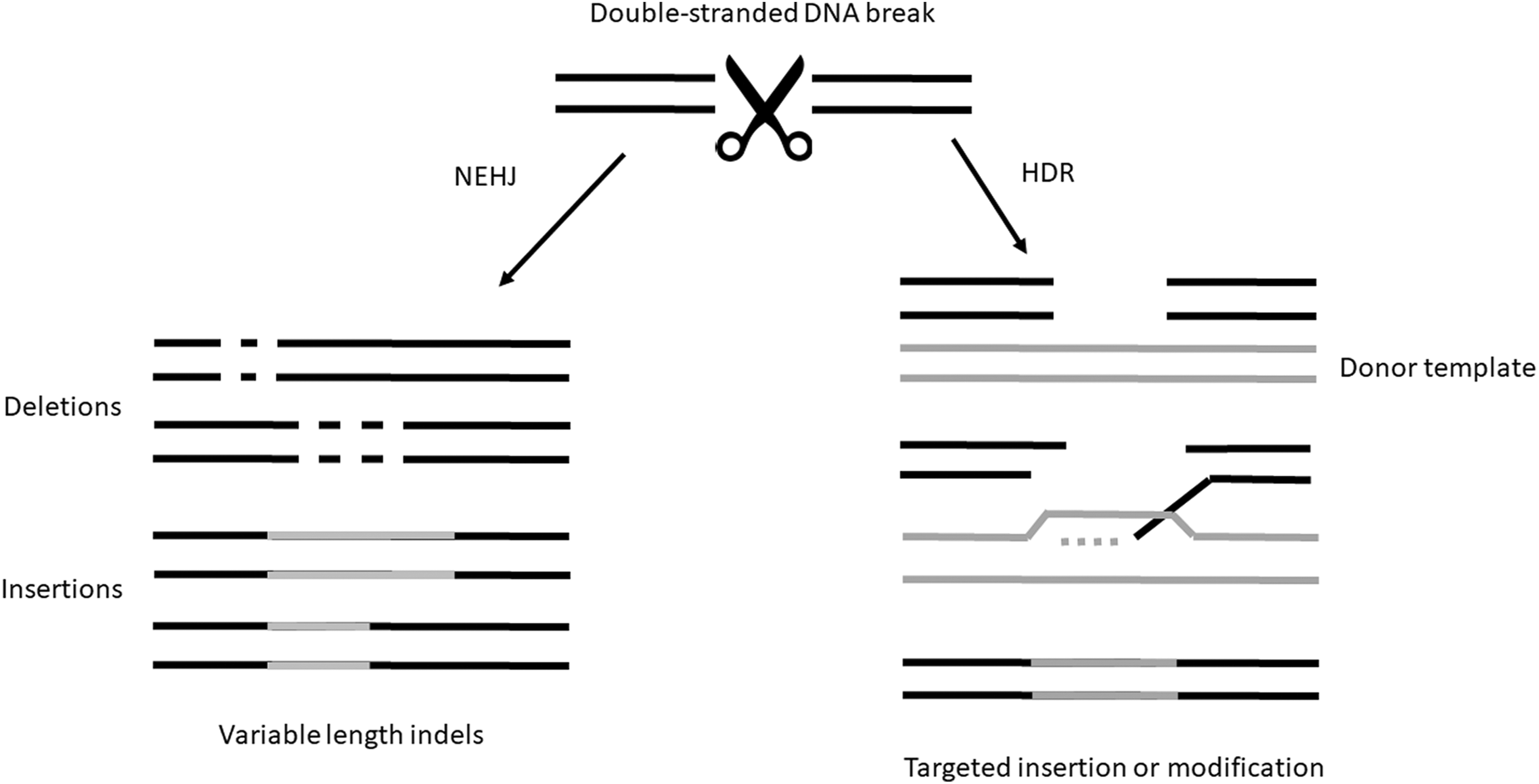
When double-stranded DNA breaks occur, there are 2 common repair methods used by the cell. In nonhomologous end joining (NHEJ), the 2 ends are simply rejoined. This is an error-prone process: insertions and/or deletions (known as indels) of DNA are found at the repair site. The second mechanism is known as homology-directed repair (HDR). HDR uses the intact gene on the homologous chromosome or sister chromatid as a template for error-free repair of double-stranded breaks.
A mechanism by which a double-stranded break could be targeted to a specific site in the genome would make exquisitely targeted mutagenesis possible. To inactivate a particular gene would only require breaking the chromosome at that gene and letting NHEJ introduce errors. If the introduction of a specific mutation was desired, a site-specific chromosome break in the presence of an exogenous repair template (ie, a piece of DNA with the requisite amount of homology but also carrying the desired mutation) would make this possible. In the early 2000s, the ability to produce such site-specific double-stranded DNA nucleases was developed, launching the current genome modification revolution.
The first of these engineered nucleases developed for practical use were the zinc finger nucleases (ZFNs). These artificial enzymes comprise a sequence-specific DNA binding domain attached to the nuclease domain from the restriction endonuclease Fok1. 32 The sequence-specificity derives from the use of zinc fingers, well-known protein domains that recognize a specific 3 base-pair DNA sequence. A selected set of these domains is combined to form a unit that specifically binds to a 9 to 18 base-pair target. 33 A ZFN pair is designed to a pair of these binding sites that flank a spacer sequence; when the ZFN is bound to DNA, the Fok1 nuclease domains dimerize and the DNA in the spacer is cleaved (Figure 5a). Although each zinc finger binds a trinucleotide, binding is context dependent (ie, binding also depends on surrounding sequence). As a result, not all sequences can be targeted, but in practice, targets can be identified at densities of 1 target every 100 to 1000 nucleotides, depending on the system used. Furthermore, ZFNs are prone to cutting elsewhere in the genome at sites with some homology to the intended target. 34 ZFNs have been used to genetically modify zebrafish, 35 rats, 36 and farm animals, 37 to name but a few.
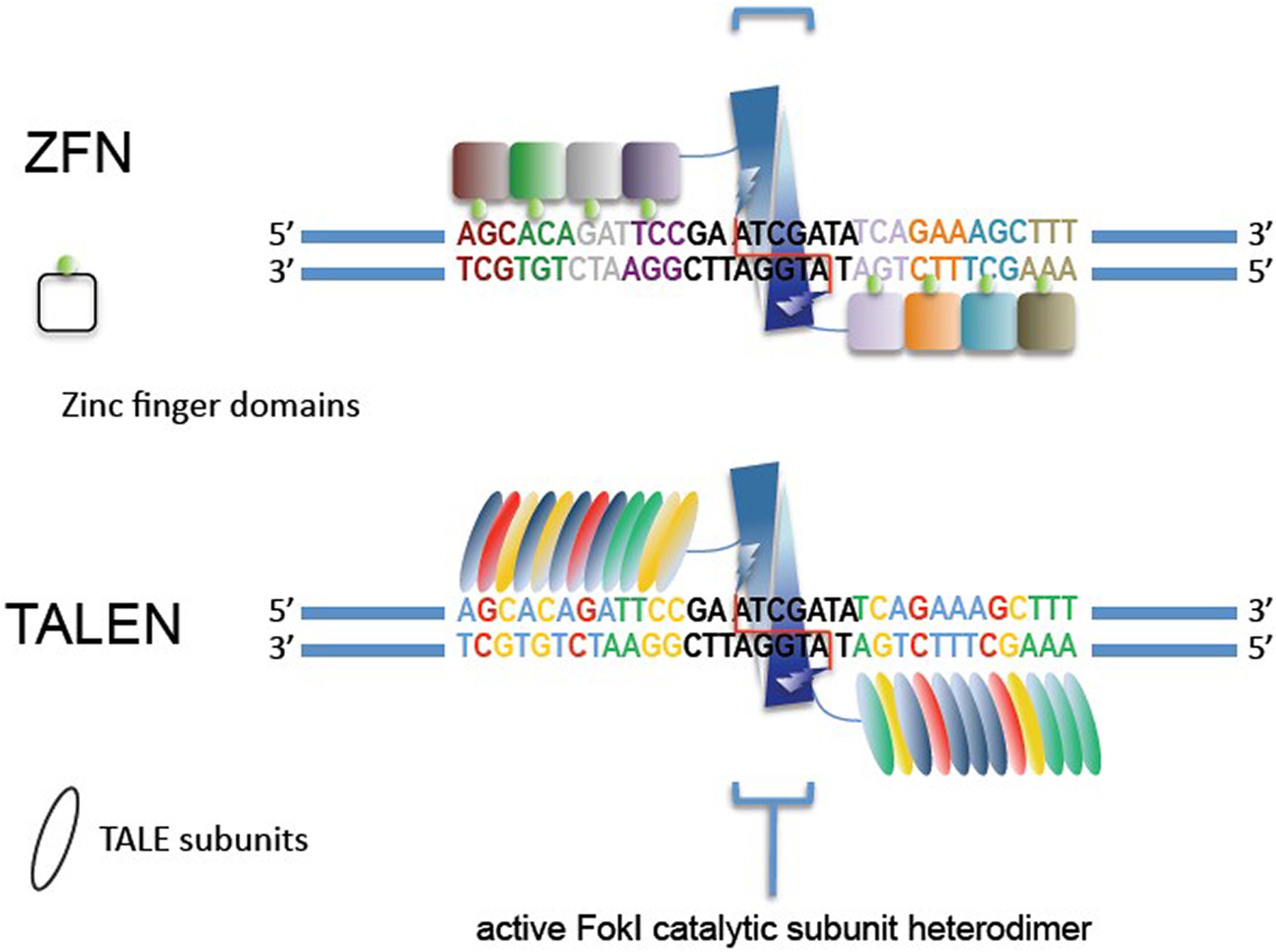
Zinc-finger nucleases (ZFN) and transcription activator-like effector nucleases (TALENs). Both programmable nucleases act in a similar manner, with the ZFN or TALEN domains binding to DNA and the attached Fok1 endonuclease cleaving the target DNA. Illustration by Farzad Jamshidi, https://en.wikipedia.org/wiki/File:Engineered_Nucleases.jpg.
Transcription activator-like effector nucleases (TALENs) were developed next. These are like the ZFNs, again with a sequence-specific DNA binding domain coupled to a Fok1 nuclease domain, but they differ from ZFNs in their DNA binding domains. TALEN DNA binding domains are derived from DNA binding proteins of Xanthomonas spp., a plant pathogen, known as transcription activator-like effectors (TALEs). 38 These proteins bind to DNA with a set of 33 to 35 amino acid repeat sequences known as effector binding elements (EBEs), each of which recognizes a single base pair 39 in the folded protein groove. 40 Again, like ZFNs, a set of EBEs is assembled to bind to a pair of specific sequences of 15 to 18 bases flanking a spacer sequence, so that the Fok1 nuclease cleaves the DNA in the spacer (Figure 5b). Because the binding of each EBE is not dependent on surrounding DNA, TALENs can be designed to target any sequence. As initially described, TALENs could not bind and cut methylated DNA, 41 although TALENs can now accomplish this feat. 42 Because TALENs cut with a higher target density and are simpler to generate, and because ZFN intellectual property rights were onerous, TALENs quickly supplanted ZFNs for most genome engineering uses.43,44
The most recently developed of these programmable nucleases are the RNA guided nucleases, commonly referred to as clustered regularly interspaced short palindromic repeats (CRISPRs) 45 (Figure 6). These were derived from a bacterial system that combines short pieces of RNA with homology to invading phage or plasmid DNA with a protein to form a nuclease that then digests the invading foreign DNA. The acronym CRISPR describes the regions of the bacterial genome that encode these targeting RNAs. The protein initially used was derived from the bacterial Cas9 protein, and thus the system is sometimes called CRISPR-Cas9. 46 Other proteins from different bacteria have also been adapted to the system, so CRISPR-CPF1 or CRISPR-Cas13 may also be seen; in general, these fall under the larger category of CRISPRs, and they all function in a similar manner. TALENs have been used to target genes in chickens, 47 zebrafish, 48 and mice, as well as in many other animal and plant species. 49
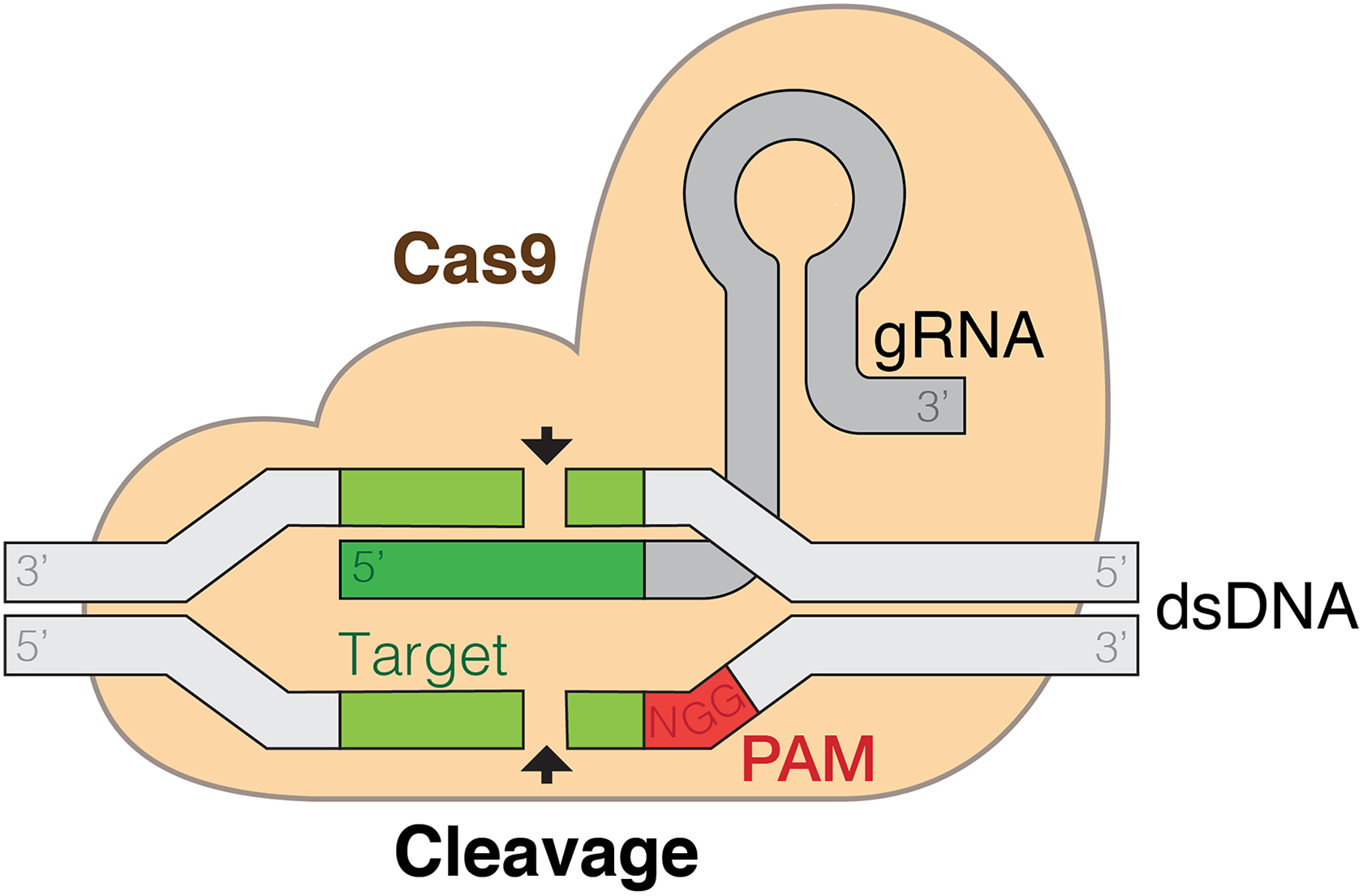
The clustered regularly interspaced short palindromic repeats (CRISPR)–Cas9 system. CRISPRs are a ribonucleoprotein complex consisting of a protein, Cas9, and a guide RNA (gRNA). This gRNA has 2 functions: it binds to Cas9 and guides it so that the complex binds at a stretch of complementary target sequence in the genome. This complementary sequence is 10 to 17 bases long and terminates in a protospacer adjacent motif (PAM). At the complementary DNA sequence, the double-stranded DNA is cleaved, allowing for nonhomologous end joining or homology-directed repair and thus genetic modification. By Marius Walter, CC BY-SA 4.0, https://commons.wikimedia.org/w/index.php?curid=62766587.
Unlike ZFNs and TALENs which are proteins, CRISPRs instead are a ribonucleoprotein complex consisting of a protein, Cas9, and a guide RNA (gRNA). This gRNA has 2 functions: it binds to Cas9, and guides it so that the complex binds at a stretch of complementary target sequence in the genome. This complementary sequence is generally 10 to 17 bases long and terminates in a protospacer adjacent motif (PAM), usually the nucleotides NGG (N is not a nucleotide but rather a representation that the nucleotide could be A, T, G, or C). Once bound to the target, the complex cleaves the DNA at the NGG. The target density is lower than that of the TALENs but much higher than that of the ZFNs. 44
Because target specificity derives only from this short sequence of RNA, CRISPRs are extremely simple to produce; a short guide RNA that can be combined with the Cas9 (or similar) protein is all that is needed. The synthesis of a short piece of DNA that is plugged into a backbone encoding the rest of the gRNA, followed by the production of the RNA itself, is a process that takes only a few days. Since the same protein is used regardless of target, it is possible to use multiplex targeting. Multiple gRNAs may be combined with the protein to target several sites at once. CRISPRs may have significantly more off-target cutting50,51 (although this is controversial)52,53 than ZFNs and TALENs, but for reasons outlined above, they are the current programmable nuclease of choice for most genome engineering applications.
Programmable nucleases have been used in a variety of systems to modify the genomes of a number of species, including nematodes, insects, plants, frogs, fish, and mammals such as rats, mice, rabbits, pigs, cattle, and nonhuman primates; they have also been used to modify many types of cells in culture, including human stem cells.34,44,54 -57 Recent work outlining preexisting adaptive immunity to Cas9 proteins in humans, however, may illustrate the limits of utility of CRISPR/Cas9 systems in humans according to a preprint at bioRxiv. 58 Germline modification only requires the delivery of the nuclease, which is typically done by microinjecting or otherwise introducing either DNA encoding the components or, in the case of CRISPRs, the reagents themselves, along with a targeting construct to affect the modification, into 1-cell embryos.
Each type of programmable nuclease has its advantages and disadvantages, such as varying success rates (24%-99%; much higher than any other genetic engineering method used previously), target densities, mutation rates (10%-20%), and off-target effects. 34 Nonetheless, the CRISPR system, because of the ease of design and manufacture of the reagents and its efficiency, is most commonly used. But more important, they are highly efficient in mice. In fact, in the hands of some, targeted mutation of multiple genes resulted in biallelic knockout of multiple genes reported at efficiencies approaching 100%, which is to say that almost all the resulting pups are homozygous mutants at several loci. 59
This astounding efficiency serves to dramatically decrease the time for producing targeted mutations. In contrast to the minimum of 6 to 8 months to produce heterozygous animals for use as foundation stock using ES cells, one can produce CRISPR reagents, apply them to embryos, and have mutant heterozygotes and even homozygotes in 12 weeks. 59 Because of this efficiency, this methodology has largely supplanted ES cells as the route to targeted mutagenesis in mice and rats. After microinjection of the nucleases as either plasmids or as messenger RNA (mRNA) into the pronucleus, it is possible to obtain knockout animals in the offspring at rates as high as 80% to 90%, depending on the locus. Inclusion of targeting vectors in this microinjection to obtain knockins is also possible, and CRISPRs are efficient enough that multiple alleles can be targeted simultaneously for modification. 60 This is clearly faster than modifying ES cells in vitro, then making chimeric animals that must then be bred another generation to produce founder animals. In addition, the limitation of working in only those strains for which ES cells exist is removed, as embryos of any inbred strain or line can be targeted in this manner.
Gene Drives
If a CRISPR targets a particular genomic site and has targeting constructs that consist of DNA exactly homologous to that site flanking a payload, the CRISPR will cut the genome and the cellular repair machinery will insert the payload into the target site. That insertion will also obliterate the target by changing its sequence. Suppose that the payload is DNA that encodes the CRISPR reagent itself, that is, it encodes Cas9 plus the targeted guide RNA. If this payload is inserted into the genome, it will then begin to produce more CRISPR reagent, and even if only 1 allele is initially targeted, the other allele will soon be cut, and the modified allele will serve as the template for HDR. If this modification takes place in the 1-cell embryo, then the animals will be homozygous for this allele. Consider what happens, though, when this animal is bred. Since it is essentially homozygous, all of its offspring will carry the transgene and at conception will be heterozygous for the transgene. Although this result is no different from any other mating between a homozygous and wild-type individual, the transgene will convert the other parental allele to the mutant allele, and all of the offspring will be made homozygous via the action of the transgene. If any of these offspring mate with wild-type animals, all those offspring will also be homozygous. Thus, the transgene will rapidly spread through an interbreeding population. This use of CRISPR technology to favorably bias the inheritance of a gene or set of genes is called a gene drive (Figure 7).
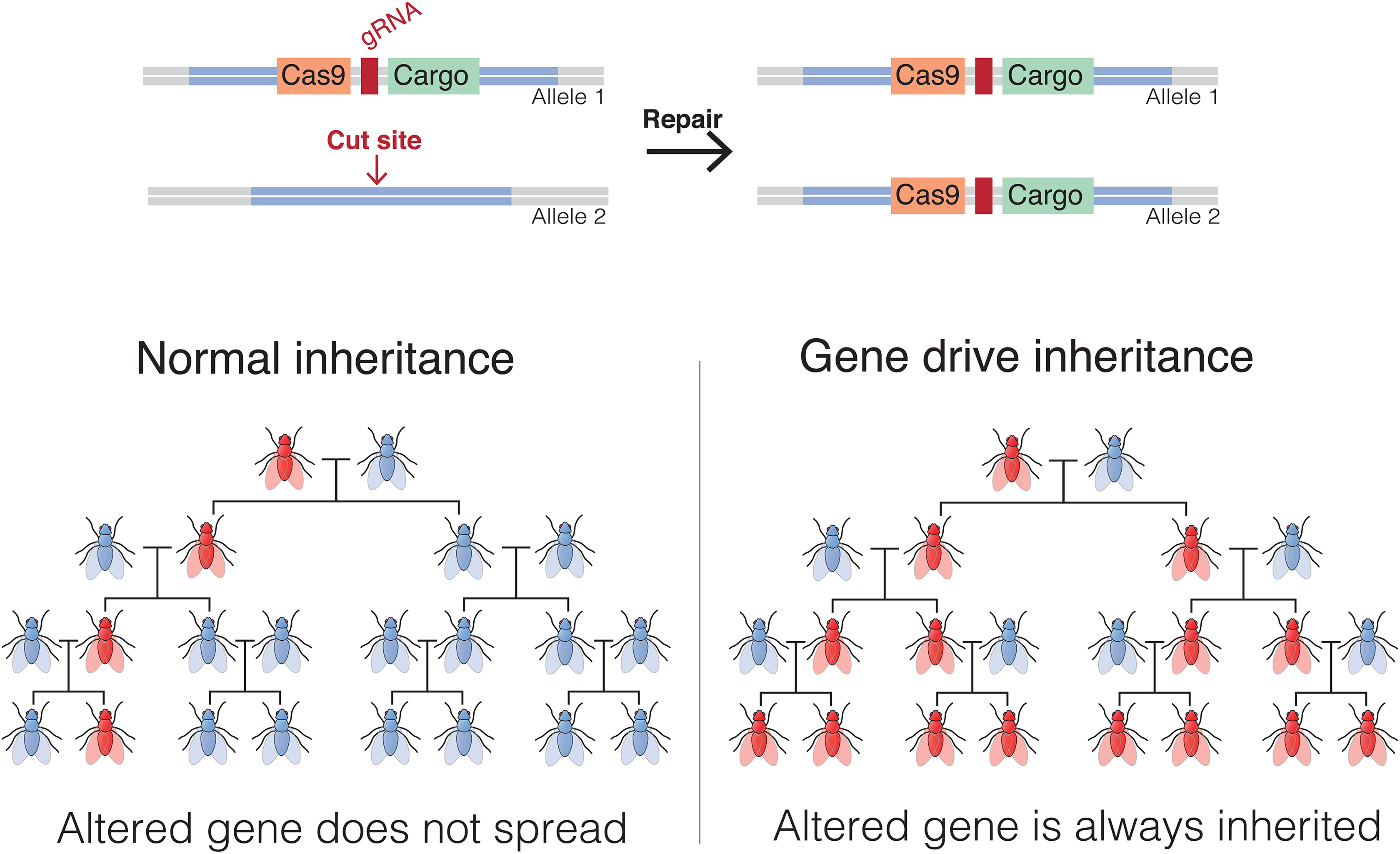
Gene drives are used to bias the inheritance of genetic material in sexually reproducing species. An endonuclease-driven gene drive cuts a chromosome at a specific site that does not include the drive. This induces the cell to repair the damage by copying the drive and its payload onto the damaged chromosome. The payload could be an allele that increases or, more typically for theoretical applications in nuisance species, decreases fitness or the ability of a pathogen to reproduce in the animal or plant. Gene drive illustration after one by Marius Walter, CC BY-SA 4.0, https://commons.wikimedia.org/w/index.php?curid=62766590.
An example of how this might be applied has been proposed for the control of disease-carrying mosquitos. This control would be via designing a gene drive that will interrupt a gene necessary for female fertility 61 or necessary for the mosquito to serve as an intermediate host to a pathogenic organism such as those that cause malaria, 62 dengue fever, or Zika, thus either eliminating the mosquito or eliminating the mosquito as a microbial vector. Gene drives, then, are potentially useful, but biocontainment of animals containing a gene drive transgene becomes imperative, especially if it also contains a potentially biohazardous transgene in addition to the drive.
Dangers and Biosafety Responsibilities
The invention of recombinant DNA technology—that is, the ability to combine random fragments of a genome with a vector that would replicate this piece of DNA when introduced into bacteria where they would replicate and expand—was a revolutionary step in modern biology and genetics, but as almost as soon as these techniques were developed, the potential dangers were recognized. Recall that our knowledge of genes and gene expression was still in its infancy at the time. Nonetheless, we knew of the existence of antibiotic resistance, cancer-causing, and toxin-producing genes, so it was immediately obvious that the cloning of one of these genes might inadvertently produce a new and extremely dangerous pathogen. As a result, leaders in the field of molecular biology called for a voluntary moratorium on these types of experiments until the dangers could be assessed; requested that the director of the National Institutes of Health (NIH) establish an advisory committee to oversee, review, and regulate recombinant DNA work with special attention to its biohazard potential; and called for an international conference of experts to meet and evaluate the field. 63 This moratorium was honored with very few exceptions while experts convened, and the dangers were further evaluated.
The NIH responded with what became the Recombinant DNA Advisory Committee and the NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules (NIH Guidelines). The NIH Guidelines “specify the practices for constructing and handling: (i) recombinant nucleic acid molecules, (ii) synthetic nucleic acid molecules, including those that are chemically or otherwise modified but can base pair with naturally occurring nucleic acid molecules, and (iii) cells, organisms, and viruses containing such molecules.” 64 The NIH Guidelines formally apply to NIH-funded research and institutions that receive NIH funding but are often applied to other research conducted in the United States. By the same token, these or similar guidelines have been adopted internationally and updated regularly. Besides describing the scope and applicability of the document, the NIH Guidelines also cover safety considerations, define the types of experiments covered, and explain the roles and responsibilities of NIH and the institution. The institution itself is charged with establishing an Institutional Biosafety Committee (IBC), which reviews all research proposals with biohazard potential and establishes and oversees any required containment.
In considering safety issues, the NIH Guidelines are concerned with risk assessment and the setting of appropriate containment for experiments. In undertaking risk assessment, they separate agents in this case, the source of the transgene DNA, into risk groups (RGs) of escalating concern from RG-1 to RG-4 (Table 1). After risk is determined, the appropriate level of containment is chosen among a set of defined biosafety levels (BSLs) (1-4) that consist of combinations of laboratory practices and techniques, safety equipment such as biosafety cabinets, waste management (in this case, animal carcasses and waste from animal housing rooms), and laboratory facilities appropriate for the operations performed.
Explanation of Risk Groups for work with biohazardous agents, including as sources of DNA.

Abbreviation: RG, risk group.
By the time transgenic technologies were developed for mammals, the initial fears had abated somewhat, but the concerns engendered by transferring foreign genes to bacteria also applied to transfer into rodents and other animals, and the NIH Guidelines were amended to include sections concerning the production of transgenic animals. These concerns are contained in Section III-D-4, Experiments Involving Whole Animals, and Section III-E-3, Experiments Involving Transgenic Rodents. Importantly, Appendix Q provides guidance on containment (BSL-1 to BSL-4) and disposal of animals covered by the NIH Guidelines.
Most transgenic animals are produced and maintained under BSL-1, which corresponds to standard biological laboratory safety practices. No special containment is required, and such animals can be freely exchanged between laboratories. Transgenic animals may have higher classifications, however, based on the transgene inserted and the method of insertion used. As noted earlier, it is possible to insert a transgene consisting of an entire viral genome, resulting in a transgenic animal that can produce the intact virus. If that transgene was inserted using a viral vector, it may have a different classification than an animal with the same transgene inserted via direct pronuclear injection. In this manner, one might investigate the disease pathway in a model organism (eg, the mouse) that is not normally infected by the virus. If the virus is a pathogen, then the resulting transgenic animals can represent a biosafety hazard whose severity is determined by the virus RG. In this case, containment level rises to match the RG, and the containment pertains to not only housing and handling the animals but also their waste and their disposal. The best reference for containment and NIH Guidelines classification may be found at https://osp.od.nih.gov/wp-content/uploads/Animal_Activities_Table.pdf (last modified in 2017).
An additional consideration is the interbreeding of 2 strains of transgenic animal, each of which may only require BSL-1 containment and may result in the creation of a strain that now requires significantly higher levels of containment. For example, many strains of mice carry a transgene that will be expressed only in certain conditions, for example, at certain locations within the animal or at certain times of development or under certain environmental conditions. Such “conditional” expression is usually controlled via a second transgene supplied by breeding. Thus, it is possible to envision a scenario in which an inactive and therefore harmless transgene is combined with an activator transgene, and the activated transgene now represents a biohazard. Alternatively, one might have 2 strains of mice, each of which contains half of a viral genome, and combining these strains now results in a complete viral genome and generation of intact viral particles in mice.
In addition to the oversight of recombinant DNA research per se, animal research is also regulated. In the United States, national laws (specifically, the Animal Welfare Act [AWA] of 1966 and its amendments and the Health Research Extension Act of 1985) mandate the formation of an Institutional Animal Care and Use Committee (IACUC) and have also required the development by the NIH of a Guide for the Care and Use of Laboratory Animals (the Guide). 65 The IACUC is charged with reviewing and approving all animal research, ensuring that the care and use of animals conforms to the AWA and the Guide and that animal use is appropriate with regard to minimizing pain and suffering, the number of animals used, and that the research is not unnecessarily duplicative. As with recombinant DNA regulatory oversight, similar structures exist in most countries.
Genome modification in animals thus falls under the purview of both the IBC and the IACUC, and the approval of both is required for an animal genome modification project to move forward, regardless of the lab producing or using the animals. In many cases, a central core produces genetically modified animals (IBC/IACUC) using DNA provided by labs (IBC), and the animals produced are then transferred back to the laboratory (IBC for the genetically modified animal/IACUC for the actual use of the animal) for experimentation. Thus, in a typical laboratory setting, although it is possible that a genetically modified animal could be a biohazard risk, any risk should have been mitigated by the implementation of appropriate containment protocols based on multiple reviews of the work by independent bodies. Thus, the likelihood of the accidental creation of a biohazard via legitimate research is small. This assumes, of course, that a project is not undertaken by rogue actors. After all, the new genome modification technologies such as CRISPR-Cas9 are simply tools, and like any tool, they can be harnessed for good or for ill.
As always, worker safety remains a paramount consideration. Typical dangers in working with genetically modified rodents include needle sticks, bites, and scratches. Exposure to similar hazards, including the possibility of greater trauma through bites or kicks, is possible if working with larger genetically modified animals. If needles contain infectious, toxic, or radioactive agents, this is a standard laboratory hazard and is typically dealt with via usual channels, such as an occupational health program at the facility. Animal bites and scratches are also typically managed in the same fashion. If genetically modified animals are humanized, 66 screening of cells or cell lines implanted in these animals should include screening for human blood-borne pathogens. Animals permissive for any xenotransplantation should be monitored for agents transmissible from or to either donor or recipient species as well. Transmission to workers of cells from cell lines used in animal-based research is another possibility, and care should be taken with animals harboring human tumor lines. 67
Gene drives deserve special mention when considering the dangers of the new genome engineering technologies. Because of the potential that a gene drive has to rapidly propagate throughout a breeding population, there are significant environmental concerns in addition to ones of biosafety. Much in the same way that the scientific community came together to confront the risks of recombinant DNA technology as the methodology first emerged, there have been calls for careful evaluation of risks 68 and possible regulation, 69 and meetings have been convened to address the issues and produce recommendations. 70 Meanwhile, continued research suggests that factors already exist that can impede the propagation of an artificial gene drive through a population. 71
Conclusion
The manipulation of animal genomes via new technologies generally only simplifies or speeds up the process of genome modification. Thus, most modifications are little different from those provided by more traditional methods (ie, natural spontaneous mutations) and do not present any particular safety hazards. Even the special case of gene drives is not a biohazard concern sensu stricto, although it does raise legitimate environmental and biosecurity questions. The current regulatory framework seems to address potential hazards through a multipronged regulatory approach, but nimbleness of regulations will always lag compared with scientific innovation.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.