
Abstract
Background:
The utilization of live biological agents as vectors for gene delivery systems is becoming more prevalent in clinical research. Often, these agents are handled in patient care environments, such as hospitals and ambulatory sites. While expanding the use of these agents from nonclinical to clinical research, Northwell Health first established a robust process to standardize biological agent research and protect patients, employees, and visitors.
Methods:
A collaborative, multidisciplinary approach was employed to develop an organizational-wide approach to using live biological agents in clinical research. The needs of both investigators and patient subjects were addressed while outlining a process to safely conduct clinical research, which was rapidly established with project management support and methodologies.
Results:
An organizational-wide Institutional Biosafety Committee (IBC) was established within 6 months. In addition, a comprehensive Biosafety Program standardized Northwell Health’s approach to biological agent research, including agent handling and containment, training and education of providers and subjects, and an audit process for compliance.
Conclusion:
During clinical research involving biological agents, potential risks must be assessed throughout the agent’s chain of custody. Northwell Health successfully implemented a robust process in a large organization that other health systems can replicate to safely conduct research in this rapidly expanding area.
Keywords
Biological agents have evolved as an important therapeutic tool for cancer and other diseases. Recently, this field has expanded with research into the utilization of live biological agents as the vector for gene delivery systems. Although health care organizations have extensive experience in clinical research, very few have done clinical research using live vectors, such as viruses that may have the potential for replication in those who receive or are inadvertently exposed to the agents. In addition, the lentiviral vectors derived from the human immunodeficiency virus (HIV) integrate into the infected cells genome, thus carrying the potential for genotoxicity and ongogenesis. 1
Northwell Health (Northwell) is a 22-hospital integrated health care delivery system across the Greater New York metropolitan area that includes more than 62 000 employees, over 550 ambulatory sites, the Feinstein Institute for Medical Research (FIMR) that received $29 million in new research awards in 2016, the Hofstra-Northwell School of Medicine, and an oncology network that provides care to about 16 000 new patients annually. A central component of Northwell’s mission and vision is research; by advancing disease-oriented, basic scientific, and clinical research, Northwell strives to improve the health of surrounding communities and promote the delivery of innovative therapies.
Research involving biological agents is traditionally done in a laboratory setting at FIMR. However, with significant breakthroughs and innovations, biological research is moving into the clinical setting, including ambulatory practices and operating rooms. These biological agents are often received and prepared by pharmacists for patient administration and require distinct protocols and training to minimize unnecessary exposure to those managing the agent and/or cross-contamination with other pharmaceutical agents. This expansion into the clinical setting includes new variables such as patients and health care personnel (HCP) from various departments, patient family members, and visitors. These individuals may or may not be involved in treatments including a biological agent, and therefore new processes need to be developed so that medical research can continue in a safe environment across the organizational continuum.
The use of live biological agents as vectors for gene therapy has promise to revolutionize treatment of cancers and many other diseases. This innovation, however, carries a risk since these agents potentially integrate into the genome of the infected cell (lentivirus and gammaretrovirus vectors) and/or potentially have the ability to replicate and spread beyond the intended host subject. However, most agents used in clinical research are replication incompetent. Therefore, utilization of these agents presents a potential risk to members of the health care team, including physicians, nurses, environmental workers, and pharmacists. Working with these agents in current laboratory research settings does not address the issues involved in patient care sites. There is a need to outline a structure in the clinical setting to ensure clinical research can continue safely.
Case Report
In response to the expansion of biological research into the clinical setting at Northwell, a Biosafety Steering Committee was established. This committee, chaired by the chief quality officer/associate chief medical officer, included representation of the Northwell biosafety officer, principal investigators (PIs), hospital medical directors, members of the preclinical Institutional Biosafety Committee (IBC), employee health services (EHS), nursing, pharmacy, safety, human resources, infection prevention, research compliance, quality, and administration. Project management support was obtained to facilitate the process, identify goals and key milestones for program development, and ensure the completion of key deliverables within the project timeline. Project management assisted with the formation of subcommittees based on members’ areas of expertise. To ensure the process developed was sustainable, this committee initially limited the scope of clinical research to a few selected centers of excellence.
The structure implemented at Northwell is an attempt to integrate those who deliver clinical care with those involved in research to advance medicine and benefit patients. The Biosafety Steering Committee accomplished all goals within 6 months and can serve as a model for other health systems as vector-based research advances over the next decade.
Program Goals and Expansion
Traditionally, biological research is performed within a laboratory setting by scientists who are very familiar with management of the agent’s safety practices defined by the manufacturer of the agent and in accordance with biosafety levels (BSL-1, BSL-2, BSL-3, BSL-4) defined and standardized by the US Department of Health and Human Services (DHHS), Centers for Disease Control and Prevention (CDC), 2 and National Institutes of Health (NIH) 3 to mitigate health risks and reduce infections. These scientists can also use the regulatory environment governing safe practices for biological agents while keeping the environment safe from exposure to harmful agents.
Oversight of biological research in FIMR was governed by the facility’s Biohazard Safety Program. It required risk assessment and mitigation strategies be defined at each touch point (ie, receipt, storage, transport, manipulation, deactivation, and/or disposal) to promote safety. For example, the use of personal protective equipment (PPE) mitigates the risk of transmission—the use of gloves during the handling of the agent reduces agent contact, and the use of face shield minimizes mucous membrane exposure. The use of a transfer device minimizes the use of needles during agent transfer from the manufacturer's container to the syringe and needle with an active safety device, and immediate disposal and destruction of the needle and syringe to minimize a percutaneous exposure. Biological agents in the laboratory setting follow an established protocol with clearly outlined management practices submitted to the IBC prior to the start of a research study to ensure the safe management of biological agents.
After a current state assessment of the Biosafety Program, it was recognized that there was a lack of structure and outlined processes to ensure safe handling of biological agents, so the Steering Committee was tasked with standardizing the definition of a biological agent, identifying any biological agents currently being used within the organization, developing a structure for the inventory and management of agents, developing practice guidelines with approved policies and procedures, establishing an internal IBC responsible for approving all Northwell studies, establishing a standardized process for all research studies, implementing an employee surveillance system, communicating to and educating all appropriate stakeholders, and developing training modules.
Ultimately, the objective was to develop a comprehensive Biosafety Program that would standardize the process for biological research across the organization in both the clinical and preclinical settings. This is inclusive of ensuring adequate containment of biological agents, enabling communication among researchers and health care providers about protocols, and reviewing and monitoring adherence to the research protocol.
System IBC
NIH guidelines require not only committees for protection of research subject rights but also an IBC to provide local review and oversight of nearly all forms of research using biological agents issued a biosafety level (BSL) category. The IBC is responsible for oversight of all aspects of the institution’s program for biological agent use in research conducted at or on behalf of Northwell. The IBC ensures that research involving these agents is conducted in a manner that protects researchers, laboratory personnel, human research subjects, the public, and the environment in compliance with the NIH guidelines, federal and state regulations, and institutional policies.4,5 For Northwell, the IBC also functions as the institutional review entity (IRE) for research considered dual-use research of concern (DURC) 6 ; in other institutions, it may function as a separate committee.
Many hospitals use an external organization, such as the Biological Research Alliance of New York (BRANY), to serve as the IBC for the organization. Northwell had a registered IBC under FIMR for preclinical research, which comprised basic-translational researchers, 2 outside community members, the biosafety officer, and members from EHS and Research Compliance. However, the use of the FIMR IBC was not standardized across the organization, and some PIs sought study approval through BRANY or other external organizations.
It was decided that an enterprise-wide IBC should be created to review, approve, and oversee all biological research (preclinical and clinical) conducted throughout the organization. An internal IBC could provide a centralized, standard process for submission, review, approval, and inventory of agents. It would also promote collaboration between members of the IBC, the biosafety officer, PI, research study team, and site team to outline necessary processes regarding use of the agent in the specified clinical setting.
As a result, a second Northwell IBC for clinical research was established and registered with the NIH Office of Biotechnology Activities (OBA)—Office of Science Policy. This clinical IBC consisted of the majority of members in the preclinical IBC with additional representatives from the institutional review board (IRB), clinical research, infection prevention, safety, and nursing. Meetings for both committees are held monthly unless no applications are scheduled for review.
Relevant program participation documents and forms required to submit a research study application to the IBC are housed on the FIMR website. Completed applications are submitted to the IBC Office, which then notifies the PI regarding approval or the need to submit additional information. The IBC Office and the study PI communicate directly regarding the submission disposition. Consistent with the protection of privacy and proprietary interests, the FIMR website shares meeting information publicly.
The IBC has final authorization for review and modification of the submitted documents. Once IBC approval is granted, an approval letter is sent to the Office of Human Research Protection Program (HRPP). Upon receipt of IBC approval, the HRPP can then grant Northwell institutional approval, which allows research participant enrollment. If a Northwell site is the initial study site and if either the IBC and/or IRB upon registering the study also requests from the NIH Office of Science Policy (OSP) Recombinant DNA Advisory Committee (RAC) review, such NIH OSP determination and possible RAC findings will also be sent to the office of the HRPP.
To date, 3 biological agents have undergone the vigorous review process developed by the Biosafety Steering Committee and have been approved by Northwell’s IBC for use within the clinical setting, and the agents have been administered successfully to patients.
Policies and Procedures
Prescriptive policies and procedures allow for a cohesive, unified approach to managing biological agents across a large organization. Members of the Biosafety Steering Committee collaborated with system resources to develop policies and procedures to standardize daily operational activities and help guide the PI through the process for IBC approval. These documents also provide a reference for HCP roles and responsibilities while performing research with a biological agent.
The first policy, “GR101—Use of Biological Agents in Research (IBC Approved),” defines a biological agent (a biological agent was defined as an agent with recombinant or synthetic nucleic acid molecules; potentially infectious or hazardous agents [bacteria, viruses, protozoans, fungi, prions, other microorganisms, and parasites]; biological toxins; select agents; transgenic animals, invertebrates, and plants; artificial gene transfer; synthetic biology; and DURC), highlights the need for the PI to seek both IBC and IRB approval to work with an agent in a clinical research trial, and outlines mandatory training, auditing, and monitoring. A second policy, “GR-102—Biosafety Requirements for Biological Agent Research,” defines PI responsibilities, including outlining a strict chain of custody for the biological agent, fulfilling regulatory requirements, communicating to appropriate stakeholders, training employees, and ensuring medical surveillance. All required forms are provided as an attachment to the policy.
Research Study Application
The Northwell Biosafety Program includes 3 documents that must be filed with the IBC:
Risk Assessment—completed by a multidisciplinary team to assess risk throughout the entire chain of custody of the biological agent Standard Operating Procedures (SOPs)—developed based on the Risk Assessment IBC Registration Form—completed by the PI of the study
Risk Assessment
As defined in the NIH guidelines, “Risk assessment is ultimately a subjective process. The PI must make an initial risk assessment based on the risk group of an agent” (NIH 2016, Biosafety, Fifth Edition). Northwell developed the Biological Agent Risk Assessment, which is a form used for both clinical and preclinical research with biological agents, including select agents and toxins and DURC agents. The risk assessment helps to develop standards for working with an agent from receipt to disposal. The sections and key features are shown in Table 1.
Risk Assessment Contents.
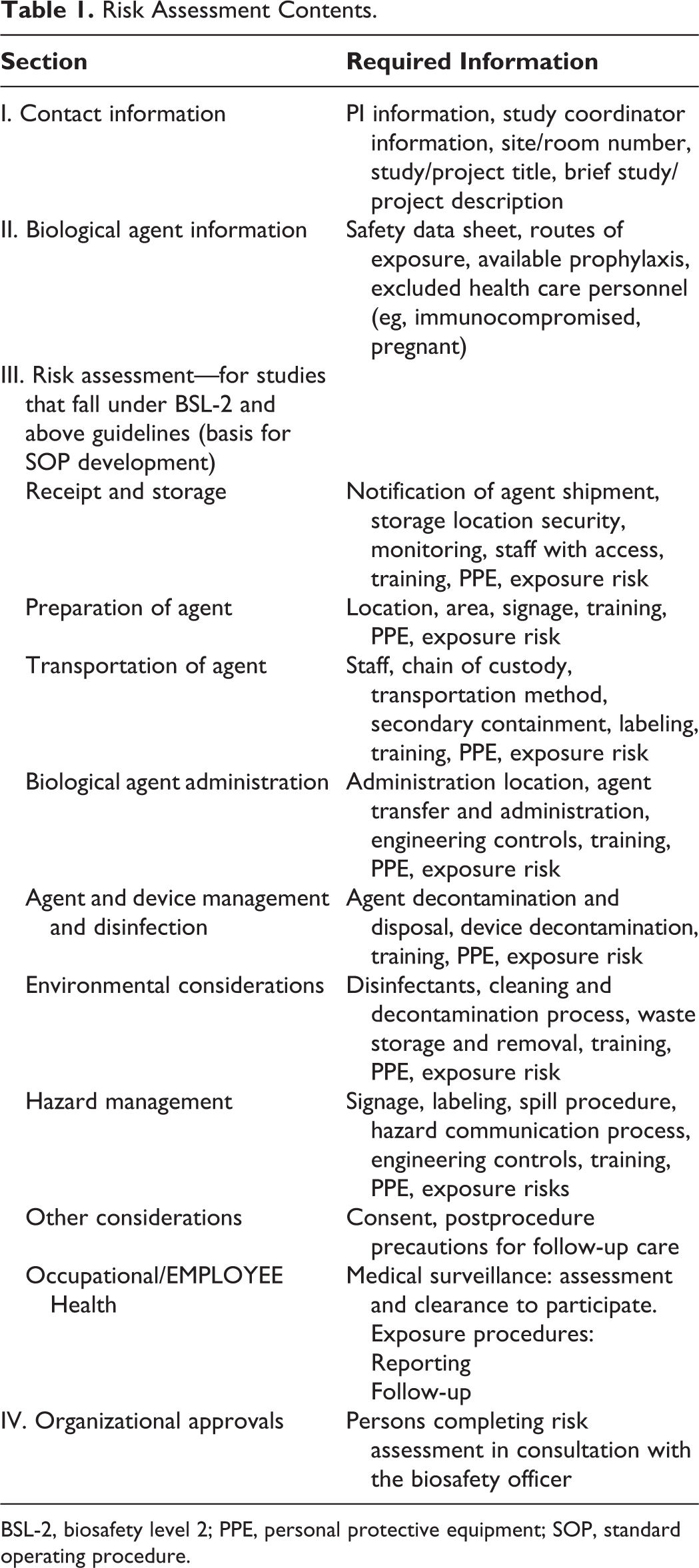
BSL-2, biosafety level 2; PPE, personal protective equipment; SOP, standard operating procedure.
The risk assessment is completed by the PI with support of the biosafety officer and a multidisciplinary team of onsite experts. This team can include, but is not limited to, system representatives from EHS, safety, infection prevention, nursing, pharmacy, and site representatives from quality, safety, nursing, medical director, and pharmacy. Together, this team completes the risk assessment by outlining the workflow and chain of custody, providing a list of staff members that must receive appropriate training and enrollment in medical surveillance, and determining if the research study can be safely operationalized at the designated site. Adherence to outlined standards of practice to ensure safe management is the responsibility of the PI with oversight from the biosafety officer and site quality through periodic audits.
After the completion of the risk assessment, SOPs are generated for handling the specified agent from receipt through disposal. In addition, medical surveillance must be implemented and completed training documented to submit the research study application, study protocol, PI’s brochure, and informed consent to the IBC for review.
Employee Safety Program
Occupational risk assessment
The occupational health risk assessment begins with the NIH guidelines for research involving recombinant or synthetic nucleic acid molecules. The guidelines classify biological agents (viral, bacteria, fungal, parasitic, and others) that are known to infect humans. This risk group classification system is based on the seriousness of known human disease.
One important observation when referencing these guidelines is that the risk group assignment is based on risk to a healthy adult human. The occupational physician needs to consider that individuals may have increased susceptibility to biological agents. Preexisting diseases, medications, compromised immunity, pregnancy, and/or breastfeeding are all risks that must be considered in the individual risk assessment.
A second important observation is that the agents considered in the NIH guidelines are the naturally occurring forms of genera and species. Genetically mutated and/or recombined species are not considered. Therefore, it is the role of the occupational physician to conduct an independent risk assessment based on the virulence and pathogenicity of the altered biologic agent. The guidelines recommend a 2-level assessment: “The first involves a consideration of the risk groups of the source(s) of the sequences and the second involves an assessment of the functions that may be encoded by these sequences (e.g., virulence or transmissibility)” (IBC Protocol).
Health Hazard Identification
The manufacturer (sponsor) information on the biologic agent is often an important source of hazard information. In addition, the hazard information available for the parent virus (eg, adenovirus, lentivirus, gammaretrovirus) also provides important information on the risks of the biologic agent vector. The occupational health professional should be familiar with the research protocols, sponsor education materials, and safety data information made available by the manufacturer. The sponsor will provide the investigator’s brochure that will offer valuable information, including the name of the agent, manufacturer contact information, and a general description. Important characteristics such as replication competence, pathogenicity, mode of transmission, and communicability may also be provided. Essential to the occupational health professional are manufacturer recommendations for first aid/treatment, post-exposure testing and prophylaxis, and any recommendations for pre- and post-exposure immunization.
Post-exposure prophylaxis treatment using antimicrobials and antivirals should be considered by the occupational physician prior to any potential exposure. Post-exposure treatment requires comprehensive risk benefit discussion with a well-educated worker. Agreement and consensus on prophylactic treatment rest on employee input and understanding. Consultation with a knowledgeable infectious disease expert may also be helpful in making treatment decisions.
Medical clearance and medical surveillance
At Northwell, a comprehensive occupational health program has been implemented to identify any health issues that may make an individual employee more susceptible to illness in the event of an accidental exposure. All health professionals are required to receive Occupational Safety and Health Administration (OSHA) bloodborne pathogen training upon hire and thereafter annually. 7 Most biological agents being used in the clinical setting do not pose any higher risk than blood and/or body fluids, but in the spirit of transparency and safety, personnel awareness, assessment of risk, education, and option to opt out were incorporated into the Biosafety Program.
First, all personnel participating in biologic agent research are required to enroll in the Medical Surveillance Program. This includes completing a medical questionnaire with an occupational health professional prior to working with any biological agents. This questionnaire focuses on eliciting any health information that may adversely affect the employee’s immune system. Medications, uncontrolled diabetes, HIV infection, current chemotherapy, or chronic use of steroids are all specifically questioned. Pregnancy and breastfeeding are also factors that should be discussed during the medical clearance examination. Presence of any of these health conditions should not be an automatic disqualifier for working with biological agents, but it does indicate the need for a more individualized risk assessment.
During the completion of this questionnaire, employees are educated on routes of exposure, required PPE, the availability of post-exposure prophylaxis, and need to report the exposure to EHS and EHS’s obligation to report to the exposure to the IBC, and they are provided the opportunity to have a confidential dialogue with a knowledgeable health care provider. Ultimately, the decision of whether or not the employee is cleared to work with the biological agent is frequently met by consensus between the employee and the medical provider.
In addition to the medical clearance screening, a medical surveillance consent form is required for all at-risk employees. After employees have been medically cleared and given the opportunity to ask questions, employees are asked to sign a consent/declination form to participate in the medical surveillance program. This allows the EHS department to track employees who might have been exposed to a biological agent.
Employee Training and Communication
Communication, education, and training of employees are crucial components to ensure shared knowledge about biosafety and to foster adherence to safety standards. Considerations in developing a meaningful education strategy for employees include role-specific education based on the risk assessment, timing of education to study implementation, and general safety concerns related to embarking on a new clinical practice.
Communication
Before engaging in any education strategies, a general message regarding the organization’s participation in clinical studies involving biological agents was distributed via email and presented to employees at the selected sites where these studies would take place. This communication ensured safety processes were in place to protect employees and patients, directed employees where to go for more information, and touted the exciting possibilities that this research provides for patients who have exhausted or are not qualified for other life-saving options.
Training and education program design
A crucial first step is the determination of which employees require training and education. Subsequently, it was necessary to identify what type of education was appropriate for identified groups. Using the completed risk assessment and associated SOPs, a team including the study PI, study coordinator, and IBC members partnered to identify departments and individuals deemed essential for participation in education.
A multilevel approach was designed to provide comprehensive and appropriate biosafety education for employees. A blended education process was established via face-to-face sessions and online support through an enterprise-wide learning management system (iLearn). Three tiers of education were designed, and individuals and groups were designated to a specific education plan.
Tier 1 education is required for any employee identified as working in areas along the chain of the custody of the biologic agent. For example, this would include employees who work in operating rooms and may see a biohazard sign posted. Tier 1 education provides foundational knowledge about biosafety, including an explanation of gene therapy treatment, biosafety levels, biosafety signage, and safety measures in place for patients, employees, and the environment.
Tier 2 education is required for all employees in the immediate area of the study but not directly handling the agent. For example, this includes employees in the outpatient practice where the clinical trial is being performed, but they do not work directly with the clinical trial. Tier 2 education includes the tier 1 education module and advanced biosafety information such as principles of biosafety in the clinical setting, risk group definitions, standard precautions, safety equipment, waste and spill management, and injury/illness reporting.
Tier 3 education is required for all employees directly involved in the clinical trial and handling of the agent. Tier 3 education includes tier 1 and tier 2 education modules, but it is agent specific and therefore not standardized across all clinical trials. A template to develop protocol-specific training was created to ensure key training items were addressed. This includes information about the biological agent, pathogenicity, modes of transmissions, signs and symptoms of exposure, special warnings, recommended precautions, emergency procedures, and other resources.
Delivery and Documentation
Education sessions are held onsite where studies are performed, led by the study coordinator and the system biosafety officer using a slideshow presentation. Tier 1 education programs lasted approximately 10 minutes. Tier 2 programs lasted approximately 20 minutes. A full tier 3 session lasted approximately 60 minutes. During these sessions, attendees had the opportunity to ask questions and have an open discussion with the study coordinator and biosafety officer. Information was supplemented with site visits and other preparatory meetings.
Documentation of education related to biosafety and agent-specific education is necessary prior to the beginning of a study. This process confirms employees directly involved in the clinical trial have been appropriately trained in the protocols. Attendance at in-person training sessions is transcribed into iLearn, which enables administrators to include notes on the employee’s individual education transcript. Another advantage of iLearn is the ability to track and report employee compliance with appropriate biosafety education. A third advantage is the capacity to upload the education module presented in the face-to-face session to attendees, which offers access to this reference information beyond the time spent at the training session.
Clinical Research Study Process
To memorialize the expanded Northwell Biosafety Program process, the Biosafety Steering Committee documented a standardized, reproducible workflow to support the handling of biological agents within the clinical environment. Experts from the Steering Committee and their respective departments collaborated to develop the comprehensive and robust process shown in Figure 1. To ensure the safety of Northwell’s patients and employees, completion of the detailed risk assessment was incorporated into the process map, and checks and balances were built in to ensure the study could be put on hold if proven unsafe at any time.
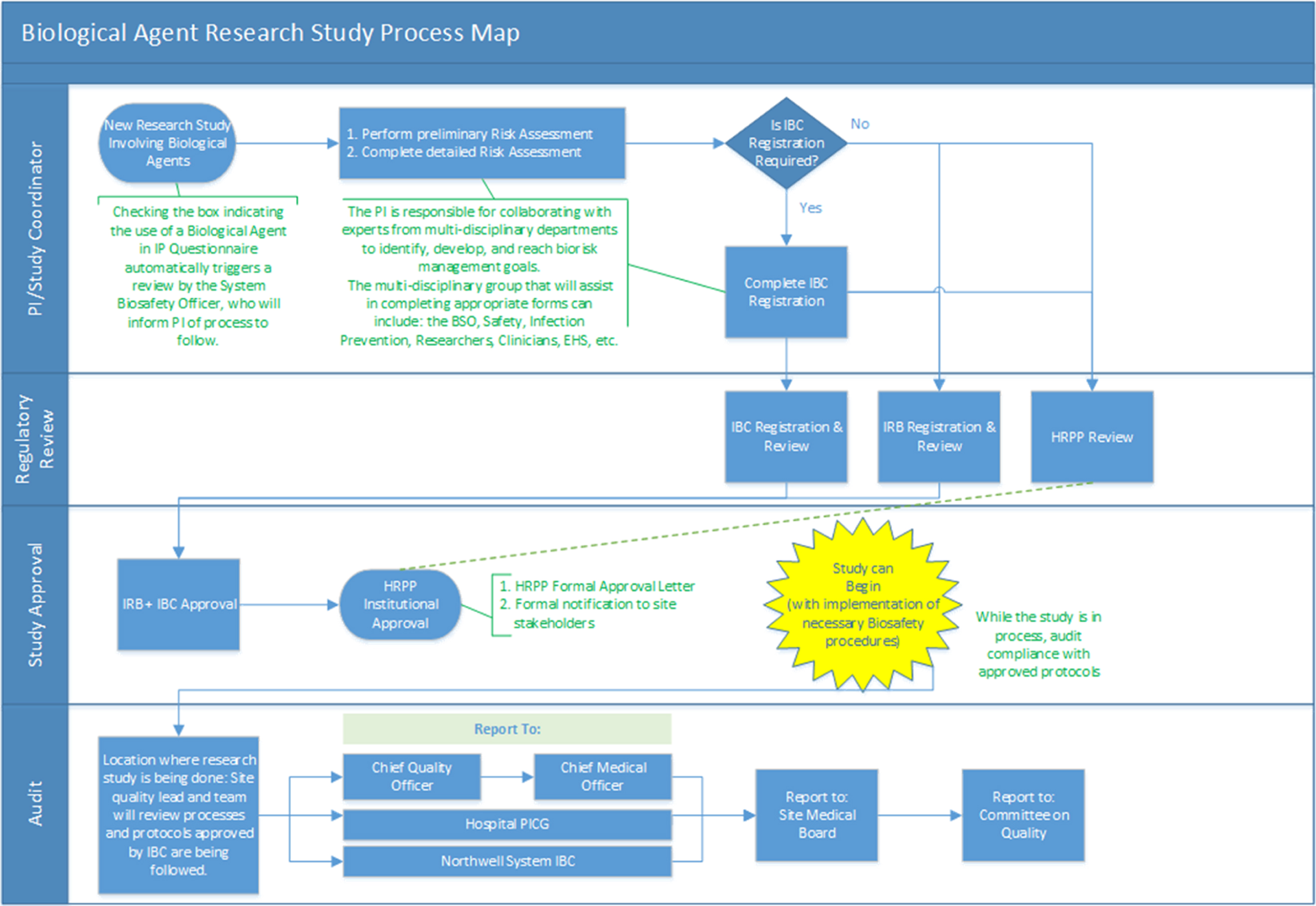
Biological agent research study process map. BSO, Biosafety Officer; EHS, employee health services; HRPP, Human Research Protection Program; IBC, Institutional Biosafety Committee; IRB, institutional review board; PI, principal investigator; PICG, Performance Improvement Coordinating Group.
The detailed process map outlines the necessary steps and approvals for each stakeholder prior to the approval of a research study. Four essential phases were defined to initiate a new research study: PI/study coordinator submissions; regulatory review and study approval by the IRB, IBC, and HRPP; and audit led by site quality leadership. This process was developed in conjunction with preclinical and clinical researchers to ensure applicability to both the clinical and preclinical areas of research across the system.
An additional level of oversight was built into the process through an innovative, thorough audit process, which ensures the approved research protocols are still in place. Studies can be done over a multiyear period at various locations across Northwell. As Northwell comprises numerous sites, site quality leads are considered the experts of their respective sites’ nuances and therefore have been tasked with auditing IBC-approved studies. A tripartite reporting structure was developed to ensure communication of results to system leadership, hospital leadership, and back to the system IBC. Ultimately, each individual hospital presents audit results at both the local and the system levels. This ensures that throughout the course of the research study, all approved protocols remain in place for the safety of all involved.
Conclusion
Northwell established an IBC and developed a comprehensive approach that goes beyond protocol approval by the IBC. The multidisciplinary team reviewed all aspects of the introduction of these agents to a hospital or ambulatory environment. This included the chain of custody of the study agent from delivery of the agent to appropriate disposal after use. The training program for staff promotes safe handling throughout the workflow, as well as appropriate steps for spill response and decontamination. Such spills must be reported to the biosafety officer, who in turn reports such incidents to the IBC for reporting of a potential biocontainment issue to the NIH. In addition, the long-term risk of exposure to these agents is unknown, so a surveillance system was developed to monitor employees. Finally, a robust oversight and audit process by trained quality nurses was instituted to ensure protocol and policy compliance.
The establishment of a defined process has allowed Northwell’s clinicians to outline processes to promote safety, attain approval for use, and offer their patients research therapies that could potentially improve their quality of life. The policies and processes developed at Northwell are applicable to other health care facilities and systems. We propose that this level of attention to detail and oversight is necessary to proactively prevent potential unnecessary exposures or downstream issues, as well as ensure appropriate reporting of exposures to the IBC and other research entities such as the NIH when indicated.
Footnotes
Acknowledgments
The authors thank the following members of the Biosafety Steering Committee, who were critical to the program development and the final review of this article: Michelle Aparicio, John Boockvar, Christina Brennan, Donna Drummond, Stephen Frattini, Paul Kaufman, Victoria Maffea, Steve Marzo, Catherine Milares-Sipinl ,Kaie Ojamaa, Andrea Restifo, Andrew Schulz, Richard Schwarz, Chantal Weinhold, Maureen White.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.