
Abstract
The popular dietary supplements of Ginkgo biloba (Ginkgo) products have been reported to have anti-cancer activities in multiple cellular and animal studies, with the benefits yet to be proven with clinical trials. The mechanisms of action are not clear, forming a barrier to investigation in Gingko-specific benefits to cancer patients, especially when combined with other therapies. Here we reported on the discovery of a novel mechanism by which a Ginkgo golden leaf extract (GGLE) inhibited melanoma cell invasion and angiogenesis. GGLE did not inhibit melanoma cells via direct cytotoxicity. Instead, GGLE significantly inhibited total RNase activities in melanoma cells under both normoxia and hypoxia conditions. The RNase angiogenin was induced twofolds by hypoxia, and the induction was significantly suppressed by GGLE treatment in a dose dependent manner. As a result of angiogenin inhibition, GGLE inhibited melanoma cell migration and invasion in a dose dependent manner. Conditioned media from melanoma cell culture sufficiently induced in vitro angiogenesis in human endothelial cells, whereas the conditioned media of GGLE-treated melanoma cells significantly inhibited this angiogenetic activity. This was accompanied with markedly reduced angiogenin concentrations in the GGLE-treated melanoma cell conditioned media. We concluded that, instead of direct cytotoxicity, GGLE inhibited angiogenin synthesis and secretion by melanoma cells, resulting in inhibition of tumor cell invasion and tumor-induced angiogenesis. This new mechanism opens the door for investigation in GGLE influencing tumor microenvironment, and warrants further investigation and validation in vivo.
Introduction
The tree Ginkgo biloba (Ginkgo), native to China and cultivated around the world, is one of the oldest living tree species and the only living species in the division Ginkgophyta. 1 The seeds and leaves are used as a source of food, and have long been used in Traditional Chinese Medicine (TCM) to treat a number of ailments including respiratory diseases, circulatory disorders, sexual dysfunction, and hearing loss. Today the products of Ginkgo are still popular as a dietary supplement for memory enhancement and for conditions such as dementia, tinnitus, and ocular issues. 2 Many cancer patients use Ginkgo products to treat and prevent cancer, and to reduce chemotherapy-associated toxicities. 3
Ginkgo leaf extract contains flavanols and flavone glycosides, lactone derivatives (ginkgolides), bilobalide, ascorbic acid, catechin, iron-based superoxide, 6-hydroxykinuretic acid, protocatechuic acid, shikimic acid, sterols, and vanillic acid.4,5 The major classes of active ingredients are the ginkgolides and bilobalides (also known as terpenes) and the flavonoids, and upon which the standardization of Ginkgo leaf extract products is established. 6
Many preclinical studies have shown that Ginkgo extracts exhibit anticancer properties,7-9 while few in-human studies have been published. For example, in ovarian cancer and gastric cancer cell lines, Ginkgo extract inhibited cell growth, and sensitized the cancer cells to cis-diaminodichloroplatinum (CDDP) and etoposide.10,11 Ginkgolic acid in the extract of Ginkgo seed coat was reported to inhibit the growth, migration, and invasion of human pancreatic cancer cells, and increase apoptosis. 12 Ginkgo extract also induced apoptosis of estrogen-negative human breast cancer cells and inhibited aromatase in hormone-dependent breast cancer cells.13,14 In a rat mammary tumor model, Ginkgo extract improved efficacy of tamoxifen treatment. 15 Despite positive preclinical results, only a very limited number of clinical studies have been published on Ginkgo extract in cancer treatment, and the results remain inconclusive. Ginkgo extract is found well-tolerated and does not increase the risk of toxicities or interfere the efficacy of anticancer therapies.16-18 Some clinical trials suggested its potential in reducing chemo-induced side effects.19,20 Specific mechanisms of action for Ginkgo extracts are not well understood. This has limited the investigations targeting mechanism-related outcomes of Ginkgo’s anti-cancer effects. Our study here aims to examine whether the extract of Ginkgo biloba golden leaves (GGLE) has inhibitory effects on cancer cells, and to investigate its mechanisms of action, using melanoma cells as a model.
Materials and Methods
Cell Culture and Reagents
Human melanoma cell lines A375 (BRAF mu/mu), SK-MEL-2 (BRAF +/+), and SK-MEL-5 (BRAF mu/+) were used. A normal human skin fibroblast cell line CCD-34SK and a normal human melanocyte line HEMA were used as control cell lines. A human umbilical vein endothelial cell (HUVEC) was used for in vitro angiogenesis assay. All cell lines were purchased from American Type Culture Collections (ATCC). Cells were cultured in recommended media with 10% FBS (F0926, Sigma-Aldrich) and 100 units/mL penicillin/streptomycin (30-001-CI, Corning Life Sciences) in 37°C cell incubator with humidified 5% CO2. HUVEC was used within 10 passages. All other cells were used within 20 passages in our laboratory. GGLE was provided by Natural Source International, Ltd. (New York, NY, USA) as a powder and was prepared in sterile water in 100 mg/mL stock solutions and stored at −20°C in small aliquots and was thawed and diluted in culture media for single use.
Cell Viability Assay
Cell viability was assessed using a 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazo-lium bromide (MTT) assay. Cells (5000/well) in 100 µL complete media were planted into 96-well plate. The next day cells were exposed to serial dilutions of GGLE for 48 hours. Then 20 μL of 5 mg/mL MTT were added into each well, and the plates were incubated in the cell culture incubator for 4 hours. The colorimetric MTT assay assesses relative proliferation, based on the ability of living cells to reduce MTT to formazan. The formazan crystal was dissolved in 150 μL/well dimethyl sulfoxide (DMSO) and color was measured at 570 nm using a microplate reader (BioTek Instruments, Inc.). The 50% inhibitory concentration (IC50) was defined as the concentration of drug that inhibited cell growth by 50% relative to the untreated control.
RNase Activity Detection
RNase activity assay was performed using the RNaseAlert™ QC System Kit (AM1966, Invitrogen™, Thermo Fisher Inc.) according to the manufacturer’s protocol. Activity of secreted RNases was measured in the supernatant culture media, and activity of intracellular RNases was measured in cell lysates. Hypoxic conditions were induced using 100 μM of the hypoxia-mimetic agent Cobalt chloride (CoCl2).21,22
Cells were seeded at 1 × 105 cells per well in a 24-well plate in 1 mL complete growth media. The next day, the media was removed, and cells were washed 3 times with 1 mL PBS. Then 0.5 mL of fresh FBS-free media with or without GGLE was added to each well. The cells were treated for 24 or 48 hours. The supernatant media was collected and centrifuged at 2000g for 10 minutes at 4°C to remove any floating cells and debris in the media, and the supernatant was then used for the RNase activity assay.
After the culture media was removed, 200 μL RNase activity assay cell lysis buffer was added to each well. Each 40 mL of RNase activity assay lysis buffer contains 0.8 mL Tris-HCl pH 8.0 (1M) + 5.48 mL NaCl (1M) + 0.4 mL TritonX-100 + 33.32 mL RNase-free H2O. The cells were then collected by scrapers and lysed on ice for 30 minutes with vortexing every 10 minutes. The cell lysates were then centrifuged at 16 000g for 15 minutes at 4°C. The supernatant was collected for the RNase activity assay. The protein contents in the cell lysates were determined using the Pierce BCA protein assay (23 225, Thermo Scientific) and were used for normalization.
To detect the RNase activity, 80 μL sample was added to each well in a black-wall 96-well plate. Fresh FBS-free media was used as the blank control for supernatant media samples, and fresh cell lysis buffer as blank control for the cell lysates samples. Next, 10 μL 10 × RNase assay buffer was added to each well and then 10 μL of RNase substrate. Immediately after the substrate was added, the plate was put into a plate reader and the first read was performed at 485/525 nm for fluorescence intensity. The plate was kept in the plate reader with 37°C incubation and read every 10 minutes for a total of 60 minutes. The readings were then normalized to the protein contents of each well.
RT-qPCR
Total RNA was extracted from cells using TRIZOL reagents (AM9738, Invitrogen) according to the manufacturer’s protocol. The synthesis of cDNA was carried out with 1 μg of total RNA using OneScript cDNA Synthesis Kit (G234, Applied Biological Materials, Richmond, BC, Canada). cDNA was then diluted 5 times in nuclease-free H2O for RT-qPCR reaction. RT-qPCR was performed using BioRad iQ iCycler detection system with One-Step BrightGreen reagents (MasterMix-S, Applied Biological Materials, Richmond, BC, Canada) according to protocol. Each reaction was carried out in 10 μL volume with 5 μL of 2× BrightGreen qPCR MasterMix, 0.6 μL of forward and reverse primer mix (10 μM), 2 μL of diluted cDNA and 2.4 μL of nuclease-free H2O. All qPCR reactions were run under the following cycling condition according to protocol from the kit: enzyme activation at 95°C for 10 minutes, followed 40 cycles of denaturation (95°C for 15 seconds), annealing/extension (60°C for 60 seconds). Melting curve was detected at 55°C to 95°C with 0.5°C increments. All reaction was carried out in triplicates for each sample with 3 independent experiments. Gene expression was quantified using 2−∆∆Ct method with 18S rRNA as the internal control gene. Primers were designed using PrimeBank (https://pga.mgh.harvard.edu/primerbank/) and the sequences are listed in Supplemental Table S1.
ELISA Assay
Secreted angiogenin was quantified by ELISA assay. Cells were seeded at 1 × 105 cells per well in a 24-well plate in 1 mL complete growth medium. The next day, the medium was removed, and cells were washed 3 times with 1 mL PBS. Then 0.5 mL of fresh FBS-free medium with or without GGLE was added to each well. The cells were treated for 48 hours. The supernatant medium was collected and centrifuged at 2000g for 10 minutes at 4°C to remove any floating cells and debris in the medium. The supernatant was then used for detection of secreted angiogenin using the Human Angiogenin ELISA Kit (Abcam, Cat ab219629) following the manufacturer’s protocol. Briefly, samples were diluted 2 to 5 times in Sample Diluent NS (provided in the kit), and 50 μL of diluted samples were added to wells pre-coated with anti-angiogenin antibody. Then 50 μL of Antibody Cocktail was added to each well and incubated at room temperature for 1 hour. The Antibody Cocktail contains human angiogenin capture antibody (primary antibody), and human angiogenin detector antibody (secondary antibody) provided in the kit. After incubation, wells were aspirated and washed 3 times with 350 μL Wash Buffer provided in the kit, and then 100 μL TMB Development Solution was added to each well and color was developed for 7.5 minutes. Stop Solution (100 μL/well) was added and OD was immediately read at 450 nm. Concentrations of angiogenin was quantified using a standard curve and normalized to the protein contents of the cells.
Western Blot
Western blots were performed to detect angiogenin levels in the cell culture media and in cell lysate. After GGLE treatments, cell culture media was collected and lyophilized dry, and then re-dissolved in water. Bovine serum albumin (23209, Thermo Scientific) 10 μg/mL was added to each sample as an internal control. Cell lysate was collected in Pierce RIPA buffer (89901, Thermo Scientific) with protease and phosphatase inhibitor cocktails (P8340, P5726, P0044, Sigma-Aldrich) and protein concentrations determined using the Pierce BCA protein assay (23225, Thermo Scientific). Samples were then run in 12% SDS-PAGE gel and transferred onto 0.2 mm PVDF membranes (ISEQ00010, MilliporeSigma). Immuno-blotting was performed as routine. Rabbit polyclonal anti-angiogenin (PA5-34422, Thermo Scientific), mouse monoclonal anti-albumin (A11133, Thermo Scientific) and anti-β-actin (ab8227, Abcam) were used at 1:1000. HRP-linked anti-rabbit (7074S, Cell Signaling) or anti-mouse (7076S, Cell Signaling) secondary antibodies were used at 1:5000. Bands were then developed by using Pierce ECL plus reagents (32132, Thermo Scientific).
Matrigel Migration/Invasion
Boyden chamber trans-well cell culture inserts (Corning Life Science, 353097) were coated or non-coated with 1 mg/mL (0.1%) of Matrigel (Corning Life Sciences, 356237). Cells were seeded at 5 × 104 cells/insert in FBS-free medium. Inserts were then placed into 24-well tissue culture plate with culture medium containing 10% FBS. Treatments were present in the medium in both the inserts and the wells. Hypoxia was induced by adding 100 μM Cobalt chloride to the culture media (CoCl2) as previously described.21,22 After 48 hours treatment, inserts were taken out from the wells, and cells inside the inserts were removed by cotton swap. Migrated/invaded cells on the bottom side of the insert membrane were fixed in 4% formaldehyde for 10 minutes, and then stained by 0.5% crystal violet solution for 10 minutes. Inserts were then washed in water and air dried. Pictures of the whole insert were taken under light microscopy. Total number of cells in each insert were counted using ImageJ software.
Tube Formation
SK-Mel-2 cells (5000/well in 96-well plate) were treated with GGLE (1 and 2 mg/mL) under both nomoxia and hypoxia conditions for 48 hours. Conditioned media was collected and immediately used or frozen at −20°C until used for the tube formation assay. In vitro angiogenesis was assessed using an Angiogenesis Assay Kit (in Vitro) (Abcam, cat. # ab204726). Briefly, HUVEC cells under 10 passages were grown into 90% confluence, harvested and resuspend in HUVEC medium supplemented with Bovine brain extract (0.2%), rhEGF (5 ng/mL), hydrocortisone (1 μg/mL), heparin (0.75 U/mL), l-glutamine (10 mM), ascorbic acid (50 μg/mL), and 2% FBS. Black-wall 96-well plate were coated with Extracellular Matrix Gel provide in the kit. After the gels was solidified, HUVEC cells were seeded into the wells at 3 × 104/well in 100 μL volume. The conditioned SK-Mel-2 media (100 μL/well) was added. Suramin (30 μM) was used as an inhibitor control, and melanoma medium (DMEM + 10% FBS) supplemented with 50 ng/mL of angiogenin was used as another control. The cells were incubated in the cell culture incubator for 18 hours, and then stained with the fluorescent dye provided in the kit. The endothelial tube formation was then analyzed under fluorescent microscope.
Data Analysis
All data represent mean ± SD of at least 3 independent experiments each done in triplicates. Comparisons among multiple groups were carried out by one-way ANOVA with Bonferroni and Holm Multiple Comparison with pairs relative to control group simultaneously compared. Comparisons between 2 groups were done by student t-tests.
Results
GGLE Inhibited Total Activities of Secreted RNases by Melanoma Cells
To examine the direct cytotoxic effects of GGLE, melanoma cells were exposed to serial concentrations of GGLE and cell viability was assessed after 48 hours. Because mutations in the BRAF gene represent ~50% of the melanoma cases and influence clinical outcomes, we used melanoma cells lines harboring homozygous BRAF mutations (A375), heterozygous mutations (SK-Mel-5), or wild type BRAF (SK-Mel-2). A normal skin fibroblast cell line (CCD-34SK) and a normal melanocyte cell line (HEMA) were subjected to the same treatments. At 48 hours treatment, GGLE did not exhibit potent cytotoxicities to the tested melanoma cell lines, until high concentrations were used, regardless of the cells’ BRAF status (Figure 1). The IC50 values were around 3 to 4 mg/mL. The cytotoxic effects of GGLE toward normal cells and toward cancerous cells were not well-separated. The data indicated that the anti-tumor effects of GGLE was not likely due to direct cytotoxicity.

Cytotoxicity of GGLE to melanoma cells and normal cells. Melanoma cells A375, SK-Mel-5, and SK-Mel-2 were exposed to 0 to 5 mg/mL of GGLE. Cell viability was assessed after 48 hours. A normal skin fibroblast cell line CCD-34SL and a normal melanocyte cell line HEMA were subjected to the same treatment. BRAF mutation status of the melanoma cells was noted on the right. IC50 values stand for GGLE concentrations needed to reduce 50% of cell viability compared to untreated controls. Data represents mean ± SD of 3 to 6 experiments each done in triplicates.
Instead, at sub-cytotoxic concentrations of 1 and 2 mg/mL, GGLE significantly inhibited total RNase activities in melanoma cells. The RNases secretion was found different in the BRAF-mutated cells A375 and the BRAF wild type cells SK-Mel-2. The secreted RNases activity was not detectable in the culture media with A375 cells (Figure 2A), and GGLE treatment only minimally reduced RNase activity in the A375 cell lysate (Figure 2B). On the other hand, SK-Mel-2 cells secreted high levels of RNases, and GGLE markedly reduced the RNases activity in the culture medium (Figure 2C), whereas the RNase activity in SK-Mel-2 cell lysate was not affected (Figure 2D).

Secreted and intracellular RNase activities of melanoma cells treated with GGLE. A375 and SK-Mel-2 cells were treated with 0, 1, and 2 mg/mL of GGLE for 48 hours. Supernatant medium and cell lysate were separately collected and detected for RNase activity, and the results were normalized to cell protein amounts, shown as fluorescence intensity per μg of cell protein. (A) RNase activity in culture medium of A375 cells. (B) RNase activity in cell lysate of A375 cells. (C) RNase activity in culture medium of SK-Mel-2 cells. (D) RNase activity in cell lysate of SK-Mel-2 cells. Data represents mean ± SD of 3 experiments each done in triplicates. *P < .05, **P < .01, and ***P < .001 by one-way ANOVA with Bonferroni and Holm Multiple Comparison with pairs relative to control group simultaneously compared.
Hypoxic conditions were mimicked using the hypoxia-mimetic agent Cobalt chloride (CoCl2, 100 μM).21,22 The hypoxic condition did not influence the RNase activity in A375 cells, as data showed that the activity in culture media was still undetectable and the activity in the cell lysate did not change (Supplemental Figure S1). Under the hypoxic condition, SK-Mel-2 cells markedly increased secreted RNase activity compared to the normoxia condition. GGLE treatment was able to reduce the secreted RNase activity of SK-Mel-2 cells under both normoxia and hypoxia (Figure 3).

Secreted RNase activities of SK-Mel-2 cells treated with GGLE under normoxia and hypoxia. SK-Mel-2 cells were treated with GGLE for 48 hours. Supernatant medium was collected and detected for RNase activity, and the results were normalized to cell protein amounts. Hypoxic condition was induced by using 100 μM of the hypoxia-mimetic agent CoCl2. Data represents mean ± SD of 3 experiments each done in triplicates.
To determine whether GGLE directly interfered with RNases, we added GGLE to purified recombinant RNase A, as well as to DMEM medium containing 10% fetal bovine serum (FBS). The direct addition of GGLE did not influence the RNase activities in either purified RNase A solution or in FBS. This indicated that GGLE did not interact with the RNases directly, but rather the inhibition was on a cellular level (Figure 4).

Direct influences of GGLE to RNases. GGLE (2 mg/mL) was added to RnaseA solution (100 U/mL) or DMEM media containing 10% FBS, or pure water. The RNase activities were detected for 60 minutes at 37°C. Data represents mean ± SD of 3 experiments each done in triplicates.
GGLE Inhibited RNases Expression by Melanoma Cells
In addition to their conventional ribonuclease enzymatic function, RNases have diverse biological functions that are implicated in cancers.23,24 To investigate the implications of the changes in secreted RNases on melanoma cells induced by GGLE, we determined which RNases were influenced by the GGLE treatment. We examined the mRNA expression of 14 RNases known to have important functions related to tumors (Supplemental Table S2).23,24
The RT-qPCR results showed that GGLE treatment decreased the mRNA levels of 7 out of the 14 RNases examined, under normoxia culture conditions (Figure 5A). Five RNases had no change in mRNA levels or the changes were not GGLE concentration-dependent. Two RNases had increased mRNA levels after GGLE treatment. The increased RNases were IRE1a and FEN1, which were related to stress responses. The RNases that had decreased mRNA expression were mainly related to the canonical RNA decay activities.
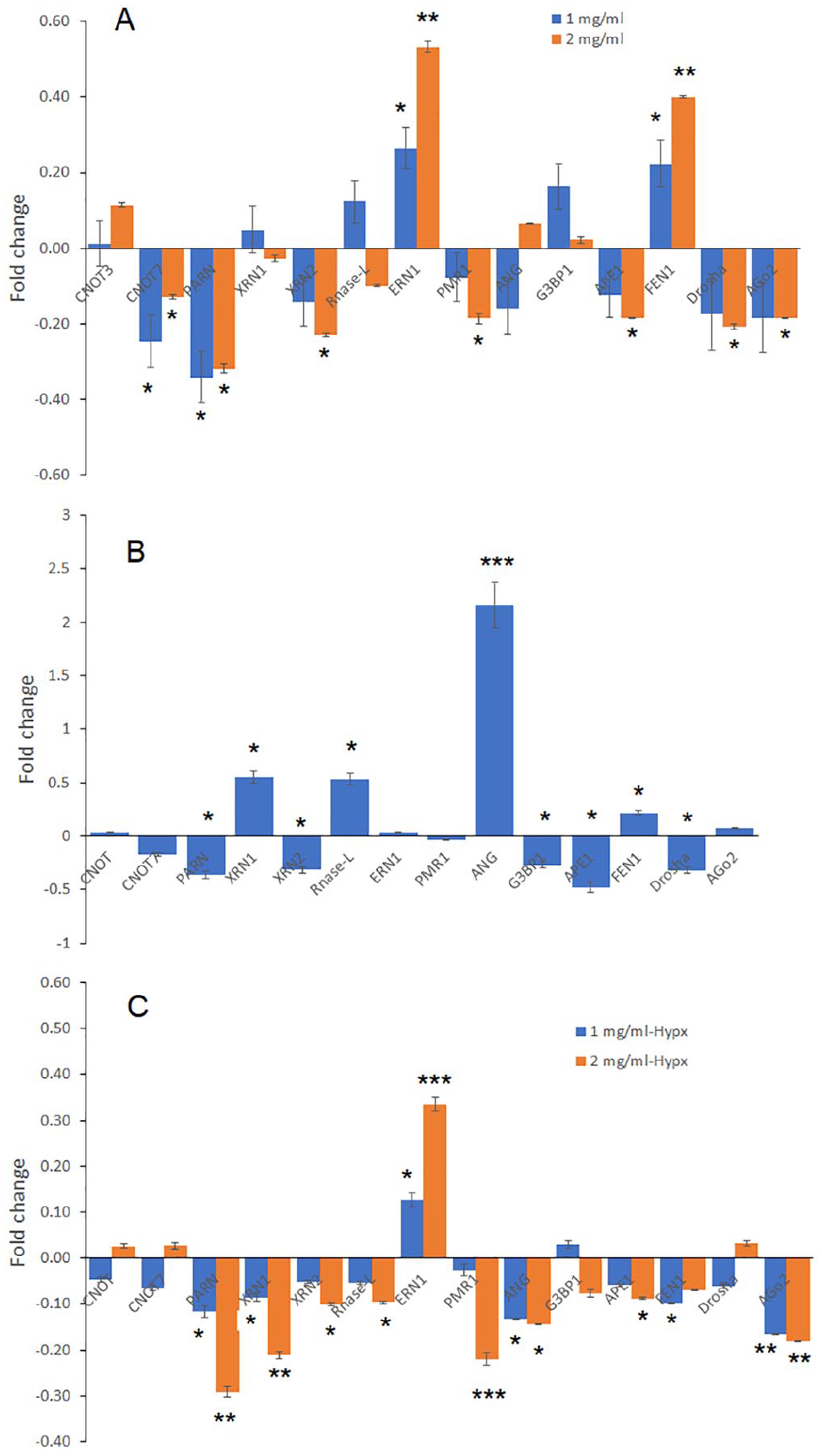
Changes in mRNA levels of 14 RNases in melanoma cells by GGLE treatment. SK-Mel-2 cells were treated with GGLE for 48 hours. Cell lysates were collected and RT-qPCR was done to examine the changes of 14 RNase genes implicated in cancer (Supplemental Table S2, see Supplemental Table S1 for primer sequences). Fold changes are relative to untreated cells, displayed as 2−ΔΔCt − 1. 18S rRNA was used as internal control. (A) Changes under normoxia by GGLE treatment. (B) Changes under hypoxia without GGLE treatment, relative to normoxia. (C). Changes under hypoxia by GGLE treatment. Data represents mean ± SD of 2 experiments each done in triplicates. *P < .05, **P < .01, and ***P < .001 by one-way ANOVA with Bonferroni and Holm Multiple Comparison with pairs relative to control group simultaneously compared.
We then examined the mRNAs of these RNases under hypoxic culture condition, a condition commonly found in cancer microenvironment. Hypoxia moderately influenced the mRNA levels of most of the RNases tested, with some increased and others decreased compared to normoxic culture condition (Figure 5B). One of the RNases, namely angiogenin, had the most significant increase in its mRNA level compared to normoxia condition (Figure 5B). The GGLE treatment decreased the mRNAs of 9 RNases including angiogenin, with the other 4 unchanged and 1 increased (Figure 5C). The increased RNase was IRE1a, the stress response gene. Taken together, the data indicated that GGLE stimulated a stress response in melanoma cells, and overall decreased RNases expression in melanoma cells. The decrease was more profound under hypoxic condition.
GGLE Decreased Angiogenin Levels Secreted by Melanoma Cells
One of the RNases, angiogenin, had the most profound increase of expression under hypoxia, and its mRNA levels were decreased by GGLE treatment (Figure 5B and C). Angiogenin has been reported as a secreted factor to promote angiogenesis. 25 We detected the protein level of angiogenin secreted by melanoma cells. SK-Mel-2 cells were cultured under normoxia and hypoxia, and were treated with GGLE. Normal melanocytes (HEMA cells) were subjected to the same conditions. Supernatant media was collected and ELISA assay was used to detect the amount of angiogenin. Data showed that the culture medium of SK-Mel-2 cells had much higher angiogenin concentrations than that of HEMA cells, under both normoxia and hypoxia conditions. GGLE treatment significantly reduced secreted angiogenin, in both SK-Mel-2 cells and HEMA cells, and under both normoxia and hypoxia (Figure 6A). The inhibition was more profound in cancer cells than in normal cells.

Reduction of angiogenin levels by GGLE treatment. Melanoma cells SK-Mel-2 or normal melanocyte (HEMA) were treated by GGLE for 48 hours, under either normoxia or hypoxia. (A) Supernatant medium was collected, and ELISA assays were used to detect angiogenin concentrations in the medium. Data represents mean ± SD of 2 experiments each done in triplicates. *P < .05 by one-way ANOVA with Bonferroni and Holm Multiple Comparison with pairs relative to control group simultaneously compared. (B) Supernatant medium was lyophilized and re-dissolved in water for western blotting. Albumin (10 μg/mL) was added as an internal control. Total cell lysates were also used for western blotting. Band intensity was normalized to β-actin for the cell lysate, and to albumin for the culture medium. Data represents mean ± SD of 2 experiments. *P < .05 by student t-tests with treated group relative to control.
To confirm the reduction in secreted angiogenin, western blots were used as a complementary method to detect angiogenin expression. SK-Mel-2 cells were treated with 2 mg/mL GGLE for 48 hours and total cell lysate was used for western blotting. For the secreted angiogenin, cell culture medium was collected, lyophilized dry and re-dissolved in water for western blotting. The results showed that the angiogenin levels in the cell lysate did not change. The angiogenin levels in the culture media were significantly reduced by GGLE treatment, under both normoxia and hypoxia conditions (Figure 6B).
GGLE Inhibited Melanoma Cells Migration and Invasion, and Melanoma-Induced Angiogenesis
Angiogenin has been reported to be involved in tumor angiogenesis and cancer cell migration and invasion. 25 We postulated that the GGLE-inhibited angiogenin expression had important functional outcomes in melanoma cells. The migration and invasion of melanoma cells were assessed by Matrigel un-coated and coated Boyden Chambers, with the uncoated indicating migration ability of the cells and the coated assessing invasion ability. Data showed that GGLE significantly inhibited both migration and invasion of SK-Mel-2 cells, under either normoxia or hypoxia condition (Figure 7A–D). For comparison, A375 cells which had minimal secreted RNase activity (Supplemental Figure S1) were exposed to the same treatment conditions. GGLE did not influence migration and invasion of A375 cells (Supplemental Figure S2).
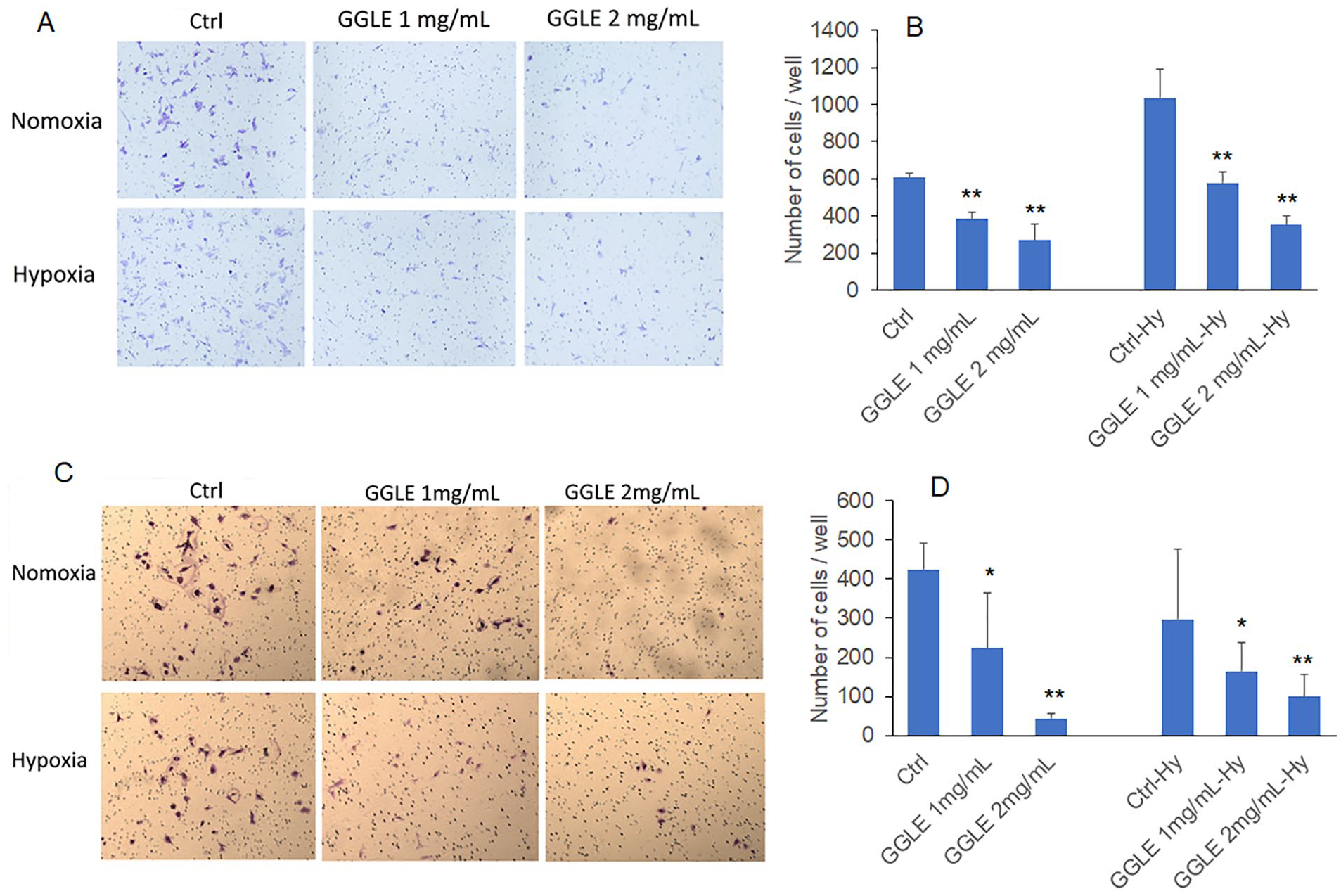
Matrigel migration/invasion of SK-Mel-2 cells treated with GGLE: (A and B) indicating migration as the Boyden chamber membrane was not coated with Matrigel. A shows representative images of cells migrated through the membrane, (B) is quantification of number of cells per well that migrated through the membrane, and (C and D) indicating invasion as the membrane was coated with Matrigel. Data represents mean ± SD of 2 to 4 experiments each done in triplicates. *P < .05 by one-way ANOVA with BonfeBroni and Holm Multiple Comparison with pairs relative to control group simultaneously compared.
In vitro angiogenesis was examined using the human umbilical vein endothelial cell (HUVEC) tube formation assay. SK-Mel-2 cells were cultured under normoxia and hypoxia, and were treated with GGLE for 48 hours. Conditioned media was taken and added to HUVEC cells seeded on an extracellular matrix. The tubes were stained with a fluorescent dye and quantified under a fluorescent microscope. The data showed that the conditioned medium without GGLE treatment mimicked the fresh HUVEC medium, and fresh melanoma media with angiogenin supplementation. GGLE-treated conditioned medium significantly reduced the total number of tubes formed by HUVEC (Figure 8).
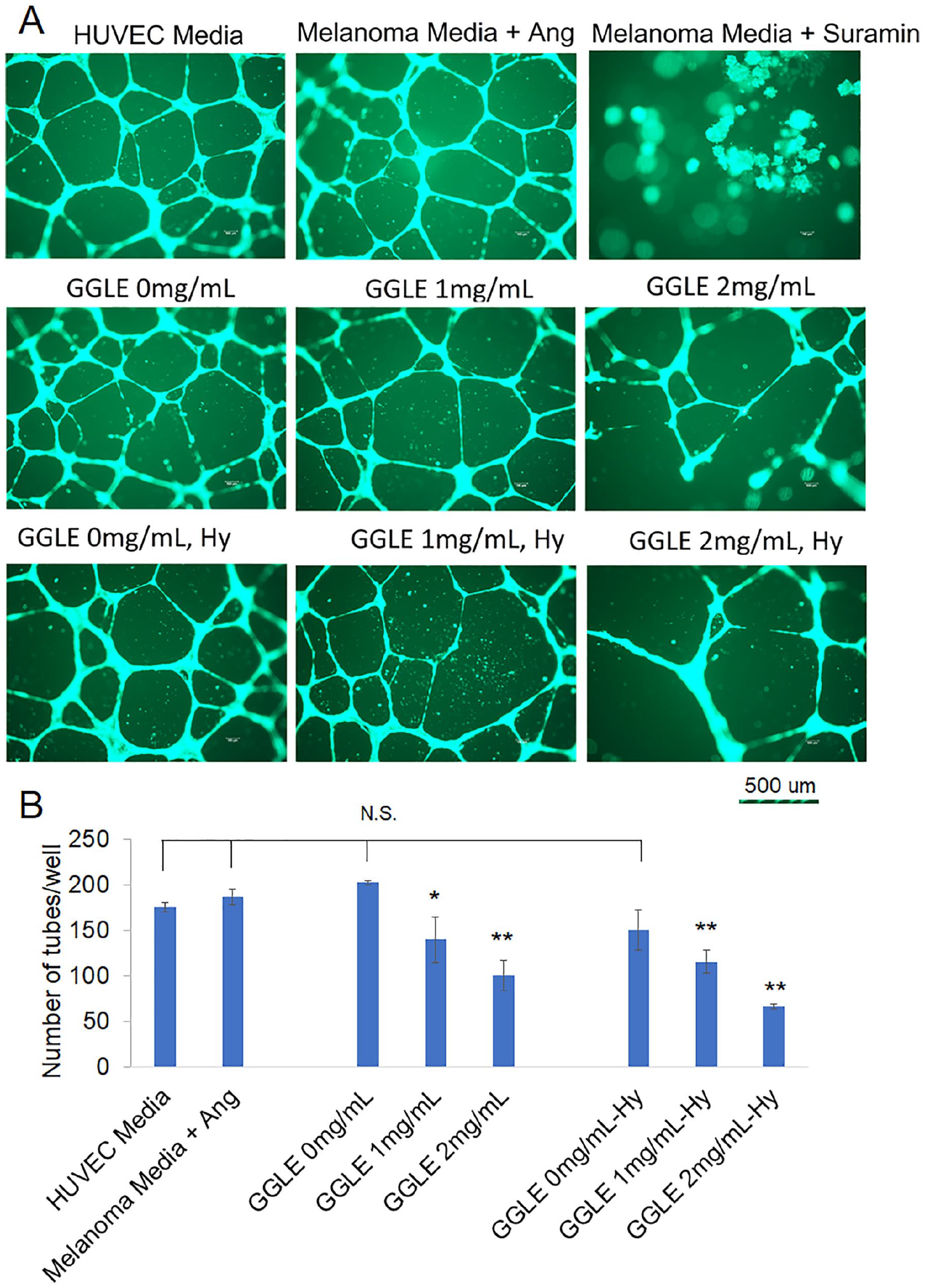
Tube formation of human endothelial cells treated by melanoma cell conditioned-media with and without GGLE treatment. Human umbilical vein endothelial cells (HUVEC) within 10 passages were seeded into Extracellular Matrix Gel and treated with conditioned medium as indicated. Tube formation was examined under fluorescent microscope after 18 hours. (A) Representative images. (B) Total number of tubes formed per well. HUVEC medium indicates medium used for HUVEC culture with all supplements and growth factors indicated in Materials and Methods. Melanoma medium indicates DMEM medium without FBS. Ang, angiogenin 50 ng/mL. Suramin (30 µM) was an inhibitor control. All GGLE treatments indicate conditioned DMEM medium of SK-Mel-2 cells treated with GGLE. Hy, hypoxia. *P < .05; **, P < .01 by one-way ANOVA with Bonferroni and Holm Multiple Comparison with pairs relative to control group simultaneously compared.
Discussion
Ginkgo products are popular dietary supplements used for a variety of reasons including anti-cancer purposes.2,3 Pre-clinical studies supported the antitumor effects of Ginkgo extracts, mostly focusing on cancer cell proliferation and/or apoptosis. Few clinical investigations have been carried out and most were single-arm small scale trials or observational studies focusing on protection of toxic side effects or cognitive functions when patients were going through chemotherapy.16-20 There lacks a full understanding on the effectiveness and mechanisms of Ginkgo’s antitumor actions.
Our data here indicated that it was unlikely that GGLE inhibited melanoma cells through direct cytotoxicity or proliferation inhibition. Rather, the secreted RNase activity was inhibited. Especially, secreted angiogenin, a known growth factor and angiogenetic factor for cancer was reduced. As an outcome, melanoma cell migration and invasion were inhibited, at sub-cytotoxic concentrations of GGLE. Tumor-induced angiogenesis was also inhibited. This data revealed the novel mechanism of Ginkgo on inhibiting secretive factors by cancer cells influencing migration/invasion and tumor-induced angiogenesis, and implicated Ginkgo’s potential in influencing the tumor microenvironment. These effects and novel mechanisms warrant further in-depth investigation.
Accumulating evidence has shown that various types of RNases play important roles in tumorigenesis and tumor progress. Angiogenin (ANG), a secreted ribonuclease specifically found in vertebrates, has been extensively reported for its angiogenic activity, as well as ability to regulate cell proliferation, survival, migration, invasion, and differentiation. 25 Moreover, the human ribonuclease inhibitor (RI), which is an endogenous cytoplasmic protein that degrades RNAases, was found to interact with angiogenin (ANG) and suppressed ANG activities and suppressed PI3K/AKT/mTOR signaling pathway. Up-regulation of RI inhibited melanoma proliferation and enhanced apoptosis through ILK/PI3K/AKt pathways, 26 and down-regulation of RI enhanced metastasis of bladder cancer. 27 The ability of Ginkgo extract in downregulating angiogenin resulted in inhibition of migration/invasion and in vitro angiogenesis here. Modulating RNases activities by Ginkgo extracts, including angiogenin, could mediate important anticancer functions.
There are also some limitations of the current work. First, the biological significance and implication of the reduction of RNase activity by Ginkgo are not fully understood. Some early studies demonstrated that serum of patients with various types of cancers possessed elevated RNase activities relative to that of healthy persons.28-31 Moreover, treatment with a Ginkgo golden leaf extract significantly reduced the RNase activity in the plasma of cancer patients to levels close to that of healthy persons. 28 As RNases have diverse cellular and physiological functions, 23 the implication of the elevated RNase activities in cancer patients and their reduction by Ginkgo extract needs further understanding. Our data focused on angiogenin. There are other RNases influenced, including those upregulated or downregulated that also deserve investigation. Second, the mechanism of how GGLE inhibited angiogenin activity is not clear. Our data demonstrated reduction in mRNA and secreted protein of angiogenin. The reduction in the protein appeared to be in a larger degree than the reduction of mRNAs. It is possible GGLE inhibited the translation and/or secretion of angiogenin, in addition to transcription. The detailed mechanism needs further investigation. Third, skin fibroblasts are known to secret angiogenin. 32 The effect of GGLE on angiogenin and other growth factors from cancer associated fibroblasts (CAFs) needs to be investigated. Also, in the tube formation assay, we cannot completely exclude the possibility that the residues of GGLE in the medium directly work on the endothelial cells to reduce tube formation, independent of melanoma-secreted angiogenin. However, considering that the GGLE was exposed to the melanoma cells for 48 hours prior to being added to the HUVEC cells, and when added to the HUVEC cells there was a dilution factor (1:1), the residue GGLE would be much lower than what was used to treat the melanoma cells. If the inhibition of angiogenesis was from the residue GGLE on endothelial cells, it would indicate an outcome in favor of the GGLE effects on anti-angiogenesis. Whether GGLE would influence endothelial cells in angiogenesis is also an interesting question worth investigating, the same as with CAFs.
Conclusions
Taken together, data reported here revealed a novel mechanism, showing that Ginkgo inhibited melanoma cell-secreted angiogenin, leading to inhibition of tumor cell invasion and tumor-induced angiogenesis. This new mechanism sets a basis for investigation in Ginkgo extracts influencing tumor growth, metastasis and tumor microenvironment, and warrants further investigation and validation in vitro and in vivo.
Supplemental Material
sj-docx-1-ict-10.1177_15347354221134513 – Supplemental material for Ginkgo biloba Golden Leaf Extract (GGLE) Inhibits Melanoma Cell Invasion and Angiogenesis Through Inhibition of Angiogenin
Supplemental material, sj-docx-1-ict-10.1177_15347354221134513 for Ginkgo biloba Golden Leaf Extract (GGLE) Inhibits Melanoma Cell Invasion and Angiogenesis Through Inhibition of Angiogenin by Ping Chen, Tao Wang and Qi Chen in Integrative Cancer Therapies
Footnotes
Acknowledgements
We thank the Maison Beljanski (New York, NY, USA, formerly Natural Source International Ltd.) for providing the Ginkgo golden leaf extract (GGLE).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by the Beljanski Foundation with a research grant. The Beljanski Foundation has no influence on the study design, the experiments performance, data collection and analysis, and manuscript preparation.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.