
Abstract
Tumor-targeted superantigens (TTSs) have been used to treat a variety of tumors in preclinical studies. The TTS utilizes the powerful T-cell activation strategy by means of staphylococcal enterotoxins (SEs) as superantigens (Sags) to target tumor cells. Monoclonal antibodies and tumor-related ligands have been used as targeting molecules of Sag. In this study, we assessed the antitumor potency of tumor-targeted superantigen (TTS) strategy to design and produce fusion protein as a new antitumor candidate. The third loop (L3) of transforming growth factor α (TGF-α) was genetically conjugated to staphylococcal enterotoxin type B (TGFαL3-SEB), and its in vitro antitumor activity against murine breast cancer cells (A431 cell line) was evaluated. We designed and prepared TGFαL3-SEB chimeric protein and evaluated superantigenic activity, binding property to cancer cells, overexpression of epidermal growth factor receptor (EGFR), and in vitro antitumor activities. Cloning of tgfαl3-seb was confirmed by colony-polymerase chain reaction, enzymatic digestion, and sequencing. The recombinant TGFαL3-SEB fusion protein with molecular weight of 31 kDa was expressed and confirmed by anti-His Western-blot analysis. The TGFαL3-SEB fusion protein attached to A431 cell line with proper affinity and induced dose-dependent cytotoxicity against EGFR-expressing cancer cells in vitro. The TGFαL3-SEB chimeric protein exhibited potent in vitro antitumor activity. Our findings indicated that TGFαL3-SEB may be a promising anticancer candidate in cancer immunotherapy, and further studies are required to explore its potential in vivo therapeutic applications.
Keywords
Introduction
Breast cancer is the most common cancer in women 1 –4 and has been increasing in men. 5 Although advances in screening, surgery, adjuvant radiation, and systemic therapies are in practice, there is no effective cure for patients with advanced stages of the disease. 6 Therefore, the development of novel treatments that can increase the survival of patients with breast cancer is welcome.
Activation of a patients’ immune system is one of the several promising therapeutic methods for controlling progression of cancer because tumor cells often avoid presenting their antigens to T-cells. One of the major goals of tumor immunotherapy is generating tumor-specific T-cells that finally contribute to the eradication of tumors. 7 Advances have been made to understand the role of the host immune responses along with alternative treatments in intervening tumor progression. 8
Superantigens (Sags) are bacterial and viral proteins both share the ability to activate a large number of T lymphocytes. Bacterial Sags bind to the class II major histocompatibility complex (MHC) antigens expressed on antigen-presenting cells outside the peptide-binding groove and consequently bind to the T-cell receptor (TcR) via the variable region of the TcR β-chain. 9 –11 Superantigens are efficient inducers of inflammatory cytokine production and cell-mediated cytotoxicity. 12 –15
Currently purified bacterial products are gaining relevance as new classes of bioactive products to treat and prevent cancer growth and metastasis. 16 Staphylococcal enterotoxins (SEs) are powerful Sags that activate all T-cells expressing a defined set of Vb-TCR, irrespective of their actual antigen specificity. 7 Enterotoxins are produced by Staphylococcus aureus and draw considerable attention as an ideal agent for cancer therapy. 17
Tumor-targeted superantigens (TTS) represent a novel concept for cancer immunotherapy which aim to activate and provoke T lymphocytes to attack tumor cells. These objectives can be achieved by means of fusing bacterial Sags to Fab fragments of tumor-reactive monoclonal antibodies (mAbs) or ligands that bind to receptors that are either uniquely expressed or overexpressed on the target cells relative to normal tissues. This allows a specific delivery system of antitumor agent to the cancer cells. Ligand-targeted therapeutics (LTTs) has advantages to mAbs. Tumor-related ligand is less antigenic than mAbs, plus nonantibody ligands are often readily available, inexpensive to manufacture, and easy to handle 18 and facilitate drug penetration into solid tumors. 19
In this study, we chose the staphylococcal enterotoxin B (SEB) as bacterial Sags which is a potent inducer of cytotoxic T-cell activity and cytokine production in vivo. 7
Variation in the regulation and expression of growth factors and/or their receptors has been correlated with the development and prognosis of malignancies. 20 Generally, the targeted antigen or receptor should have a high density on the surface of the target cells, 18 so we chose the epidermal growth factor receptor (EGFR) as a suitable target for the design of LTTs in breast cancer immunotherapy.
The EGFR is a commonly expressed 170-kDa transmembrane glycoprotein that is a member of the HER tyrosine kinase growth factor receptor family and is involved in signaling pathways affecting cellular growth, proliferation, and differentiation. 20 The EGFR has been implicated in the pathogenesis of multiple human tumors. Overexpression of the receptor has also been noted frequently in breast, bladder, and ovarian tumors as well as in various squamous carcinomas. 21 The EGFR is generally reported as an adverse prognostic marker. 22 –24 Moreover, the degree of EGFR overexpression has been correlated with advanced tumor stage and resistance to standard therapies. High levels of receptor expression have been found in 30% to 40% of carcinomas. 21 The EGFR is encoded by the proto-oncogene, c-erb-B. 25 –27 Activation of this proto-oncogene results in overexpression of EGFR in many human tumors.
The EGFR is composed of an extracellular ligand-binding region that is the ligand-binding site for both the epidermal growth factor (EGF) and transforming growth factor α (TGF-α), 20 a lipophilic transmembrane anchor sequence, and an intracellular domain characterized by an adenosine triphosphate (ATP)-dependent tyrosine kinase. 25,28,29 Human transforming growth factor α (hTGF-α) is a native ligand, co-overexpressed with its receptor EGFR in many human tumors. 30 When ligand binds, receptor dimerization occurs and the intracellular tyrosine kinase domain triggers subsequent signaling pathways involved in cell growth regulations and survival. The hTGF-α consists of 3 loops, the third of which (TGFαL3) retains the binding ability to EGFR. 20 Ligand–receptor internalization occurs upon binding of the intact ligand to its receptor; however, the Sag functions extracellularly and its function is independent of its cellular uptake. In this study, TGF-α was selected as a ligand to block induced ligand–receptor internalization, and SEB Sag was fused to the third loop of TGF-α (TGFαL3). This will not only retain the binding ability to EGFR but also prevent the ligand/receptor internalization. Moreover, TGFαL3 is presumably less antigenic compared to mAbs; thereby, it has a longer half-life in vivo. These interesting properties of TGFαL3 make it an attractive targeting molecule for the Sags toward tumors. 30
Here, we designed and constructed TGFαL3-SEB chimeric protein, and its binding property to EGFR-expressing cancer cells was determined in vitro. Also, its antitumor activity on 4T1 murine breast cancer cell line was evaluated. The 4T1 cells grow as adherent epithelial cells in vitro and are used as a model of murine estrogen-nonresponsive mammary carcinoma cells. When injected into Balb/c mice, 4T1 cells multiplied rapidly resulting in highly metastatic tumors and served as an animal model for human stage IV breast cancer. 31,32 Interestingly, we found that the TGFαL3-SEB fusion protein could bind to EGFR-expressing tumor cells with proper affinity and exhibited an apparent inhibitory effect on the growth of 4T1 tumor cells in vitro.
Material and Method
Material and Reagent
Restriction endonucleases and silica-based DNA gel extraction kits were obtained from Fermentas Thermo Fisher Scientific, Inc. (Waltham, Massachusetts). Nickel-nitrilotriacetic acid (Ni-NTA) column, polymerase chain reaction (PCR) purification kit, and DNA extraction kit were purchased from Qiagen GmbH (Germany), Metabion (Germany), and Bioneer (Korea), respectively. Deoxynucleoside triphosphate was obtained from Fermentas. The 10× thermophilic buffer, pfu DNA polymerase, and MgSO4 were purchased from Promega (Germany).
Nitrocellulose membrane and diaminobenzidine were purchased from Schleicher (USA) and Sigma (Germany), respectively.
Oligonucleotides used as primers for PCRs and tgfα-linker sequence were synthesized by Bioneer and ShineGene Molecular Biotech, Inc (China), respectively.
Bacterial Strains and Cell Culture
Staphylococcus aureus was isolated from clinical samples. Escherichia coli strain TOP10 was used for plasmid propagation and cloning and E coli strain BL21 (DE3) as a host for the production of fusion proteins. pET-28a+ was used as expression vector, supplied by Novagen Inc (Madison, Wisconsin).
A431 human epidermal carcinoma cell line (NCBI C204) and 4T1 mouse breast cancer cell line (NCBI C604) were purchased from Pasteur Institute of Iran and were maintained in Dulbecco's Modified Eagle medium (Gibco; Life Technology, Maryland) and Roswell Park Memorial Institute medium (RPMI) 1640 (Gibco; Life Technology), respectively. The media were supplemented with 10% fetal bovine serum (Gibco; Life Technology), 100 units/mL penicillin, and 100 mg/mL streptomycin.
Mouse spleen cells, freshly separated from healthy BALB/c mice, were grown in RPMI cell culture medium.
Optimization of the PCR for Detection of seb-Positive S aureus Strain
In this study, 200 clinical specimens were screened for detecting seb-positive S aureus isolates. All biochemically tested S aureus were confirmed by PCR using specific primers for S aureus thermonuclease (nuc gene), then PCR was performed to screen for seb-positive strains. Staphylococcus aureus isolates were grown in 10 mL brain heart infusion broth at 37°C overnight under aerobic conditions. Bacterial cells were collected and DNA was extracted with DNA extraction kit based on kit instructions. The amplification reaction was performed in a final volume of 25 μL, containing 1 μL genomic DNA, 0.5 μL Taq polymerase (5 unit/μL), 1 μL each of the primers (10 ρmol/μL), 2 μL each of 2.5 mmol/L deoxy ATP, deoxythymidine triphosphate, deoxycytidine triphosphate, and deoxyguanosine triphosphate, 2.5 μL of 10× PCR buffer (50 mmol/L KCl), 10 mmol/L Tris-HCl (pH 8.3 at 25°C), and 1.5 μL MgCl2 (50 mmol/L). Primer sequences and PCR programs used for detection of nuc and seb are shown in Table 1. The amplified products were analyzed by electrophoresis with a 1% agarose gel followed by ethidium bromide staining and ultraviolet transilluminator visualization. The PCR products were verified by sequencing.
Characteristics of Published Primers, Nucleotide Sequences, and PCR Programs.

Abbreviations: F, forward; PCR, polymerase chain reaction; R, reverse.
a1, 30 times (94°C, 1 minute; 55°C, 1 minute; 72°C, 1 minute); 2, 30 times (94°C, 1 minute; 64°C, 45 seconds; 72°C, 1 minute); and 3, 35 times (94°C, 1 minute; 63°C, 1 minute; 72°C, 1 minute).
Design and Construction of Chimeric Gene
To design TGFαL3-SEB chimeric construct, we chose 17 amino acids from the N-terminus of third loop of transforming growth factor α (TGFαL3). This sequence was reported to be involved in binding to the receptor, EGFR. 33 The 239 amino acids of the full length of mature SEB (accession: KC428707.1, http://www.ncbi.nlm.nih.gov/nuccore/KC428707.1) were selected and fused to the C-terminus of the chimeric fusion protein. The 2 fragments were bridged by a linker consisting of 8 amino acids (GGSGSGGG). The amino acid composition of TGFαL3 and SEB sequences was computed using bioinformatics databases and tools (Figure 1).

Schematic model of TGFαL3-SEB fusion protein. This model shows the construction of TGFαL3 and SEB bound together by the GGSGSGGG linker. TGFαL3-SEB indicates third loop of transforming growth factor α genetically conjugated to staphylococcal enterotoxin type B.
Cloning of seb and tgfαl3-seb Genes
The 720-bp DNA sequence encoding the mature SEB from seb-positive S aureus was amplified by PCR using specific primers that included 5′ EcoRI and 3′ HindIII restriction enzyme sites, respectively, to facilitate cloning (Table 1). For PCR amplification, the reaction mixture (30 µL) contained 1 μL of each of the primers (10 pmol/μL), 1 μL of deoxynucleotide triphosphate mixture (2.5 mmol/L each), 3.0 µL of 10× PCR buffer, 2.5 µL MgSO4 (25 mmol/L), 1 µL pfu DNA polymerase (5 units/μL), and 19.5 µL distilled water. Finally, 1 µL DNA preparation was added to each 0.2-mL reaction tube. The tubes were subjected to thermal cycling (Eppendorf Mastercycler gradient) using the program shown in Table 1. The amplified product was cleaved with EcoRI and HindIII, bound into similarly digested pET28a to produce recombinant vector pET28a:: seb. DNA sequence encoding GGSGSGGG linker and third loop of TGFαL3 were obtained from Uniprot Knowledgebase database using accession no. P01135 and synthesized by ShineGene Molecular Biotech, Inc with 5′ BamHI and 3′ EcoRI restriction enzyme sites, respectively. Then this 100-pb DNA sequence was cleaved with BamHI and EcoRI and ligated into similarly digested pET28a:: seb to produce recombinant vector pET28a:: tgfαl3-seb. These 2 constructs were transformed into E coli TOP10 competent cells and cultured onto Luria–Bertani (LB) medium, containing 100 mg/mL ampicillin at 37°C. The PCR-selected clones were confirmed by restriction digestion and DNA sequencing.
Expression Optimization and Purification of TGFαL3-SEB Fusion Protein and SEB
Expression host E coli BL21 (DE3) was transformed with the isolated recombinant constructs SEB and TGFαL3-SEB. The individual clones were grown at 37°C in LB broth, containing 50 µg/mL kanamycin. For expression of TGFαL3-SEB fusion protein , analysis of the expression level in different hours (2, 4, 6, and overnight), temperature (37°C and 30°C), and isopropyl-b-
Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis and Western Blot Analysis
The recombinant proteins were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot. The SDS-PAGE was carried out using 12.5% gels. Recombinant proteins were solubilized in reducing sample buffer. Polyacrylamide gels were stained by Coomassie blue G-250 to detect the protein. Gels were blotted onto a 0.45-μm nitrocellulose membrane, and then nonspecific binding sites were blocked using 5% bovine serum albumin (BSA) dissolved in 0.1 mol/L PBS plus 0.05% Tween 20 (PBS-T) for 1 hour at 22°C. The membrane was then incubated with anti-His antibody (diluted 1:1000 in PBS-T) for 1 hour at room temperature. After 5 washes in PBS-T, the membranes were incubated for 1 hour at room temperature in PBS-T containing anti-rabbit-immunoglobulin G horseradish peroxidase (HRP)-conjugated secondary antibody (Abcam, Tokyo, Japan). The membranes were washed 3 times in PBS-T and twice in PBS and then stained using 1 mg/mL 3,3′diaminobenzidine in PBS with 0.04% H2O2 and 8.3% methanol.
Tumor Cell Binding Assay
Binding analysis of TGFαL3-SEB to EGFR on the cancer cells was carried out by cell enzyme-linked immunosorbant assay (ELISA) assay. The 1.0 × 104A431 cells were seeded per well into 96-well plates overnight. The cells were then washed with PBS and fixed with 10% neutral formaldehyde (0.1 mol/L PBS, 10% formaldehyde, pH 7.4) at room temperature for 1 hour. After washing, the fixed cells were blocked with blocking solution (0.5% BSA in PBS) for 2 hours and incubated with different concentrations of TGFαL3-SEB and SEB (0.1, 1.0, 10, 100, and 1000 ng/µL) at 37°C for 1 hour. After being washed 5 times with PBS-T (pH 7.4), the cells were incubated with antihexahistidine HRP-conjugated antibody (1:1500) at room temperature for 1.5 hours and washed as described previously. Finally, the color was developed by tetramethylbenzidine solution with 1.5% H2O2, after adding stop solution (H2SO4) and the absorbance was read at 450 nm using a ELISA plate reader.
Splenocyte Proliferation Assay
Freshly isolated splenocytes from the spleen of healthy BALB/c mice were seeded into 96-well plates at a density of 1 × 105 cells per well in the presence of 1.0, 2.0, and 6.0 μg/mL of TGFαL3-SEB and SEB, 20 μg/mL phytohemagglutinin (PHA) as a positive control and PBS as negative control. The treated cells were cultured at 37°C for 72 hours, then 50 μL sodium 2,3,-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)-carbonyl]-2H-tetrazolium (XTT), 1 mg/mL, 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide was added and incubated at 37°C for 4 hours. Finally, the OD450 was measured on an ELISA plate reader (Biorad, USA). The relative proliferating activity of the fusion protein was calculated by 100% × OD450 of the TGFαL3-SEB fusion protein/OD450 of the SEB.
In Vitro Tumor Cell Growth Inhibition Assay
Mouse breast cancer 4T1 cells as target cells were seeded into a 96-well plate at a density of 3.0 × 104 cells/well. Following an overnight incubation period, mouse spleen cells, freshly separated from healthy BALB/c mice, were cocultured at an effector-to-target (E:T) cell ratio of 1:1, 10:1, 30:1, and 100:1, respectively, in triplicate to a total volume of 100 μL in the presence of 5 ng/μL TGFαL3-SEB. The plates were incubated at 37°C and 5% CO2 for 72 hours. Tumor cell viability was determined using the XTT assay. The data were reported as the percentage of tumor cell growth inhibition (TCGI%), which was calculated as follows:
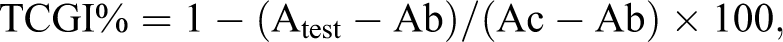
where Atest is the absorbance of tumor cells grown in the presence of the effector cells and Sags, Ab is the absorbance of the medium only, and Ac is the absorbance of the tumor cells grown in the medium.
Upon determination of the E–T ratio causing 50% TCGI, 4T1 cells were cocultured with fresh mice splenocytes at defined E–T ratio in the presence of various concentrations of Sags (0.05, 0.5, 5, 50, and 200 ng/µL). Cells were cultured at 37°C in 5% CO2 for 72 hours. The remaining viable tumor cells were determined using the XTT assay. The data were reported as the TCGI% as described previously.
Statistical Analysis
All experiments were performed in triplicate and results were presented as mean ± standard deviation. Statistical comparisons were made by Student t test or analysis of variance. The accepted level of significance was P value <.05.
Results
Detection of seb-Positive S aureus Strain From Clinical Samples
To confirm the biochemical identification of S aureus isolates, a conserved region of the thermonuclease gene (nuc gene) of S aureus was amplified by PCR. The PCR amplification of the nuc gene of S aureus yielded the expected product of 318 bp (Figure 2A). Staphylococcus aureus ATCC 25923 and Staphylococcus saprophyticus ATCC 49453 were used as positive and negative controls, respectively. All the isolates were confirmed as S aureus. To detect the seb-positive S aureus strains PCR was performed using specific primers for seb gene that encodes S aureus enterotoxin type B (Figure 2B). Staphylococcus aureus ATCC 14485 and nontoxigenic S aureus strain ATCC 6538 were used as positive and negative controls, respectively. The PCR amplification of the seb gene yielded the expected product of 495 bp. The 17 seb-positive S aureus were detected among the confirmed S aureus isolates. After sequencing, we aligned their sequences and 1 seb-positive S aureus was selected as the representative of the 17 sequences for cloning purpose (data not shown).

A, PCR detection of S aureus nuc gene. Agarose gel electrophoresis of the nuc gene of S aureus, M: 100+ bp DNA ladder, C+: S aureus ATCC 25923, C−: S saprophyticus ATCC 49453, and lanes 1-3: nuc+ S aureus strains. B, The PCR detection of staphylococcal enterotoxin type B gene. Agarose gel electrophoresis of PCR product using primers for detection of the seb gene of Staphylococcus aureus, M: 100+ bp DNA ladder, C+: seb+ Staphylococcus aureus ATCC 14485, C−: nontoxigenic Staphylococcus aureus strain ATCC 6538 was used as negative control, and seb-positive S aureus that was selected for cloning purpose was showed in lane 3. PCR indicates polymerase chain reaction.
Cloning, Expression, and Characterization of Purified of Proteins
Full-length region of mature SEB gene (seb) was amplified by PCR from seb-positive S aureus clinical isolates. As expected, yielded amplicons of 720 bp was observed on agarose gel electrophoresis (Figure 3A). The amplified product bound into similarly digested pET28a to produce pET28a:: seb recombinant vector. The cloned pET28a:: seb construct was transformed to BL21 (DE3) expression system and recombinant His-tagged SEB protein of ∼28 kDa was purified by Ni-NTA column (Figure 4A) and confirmed by Western blot analysis (Figure 4B). Construction of pET28a:: tgfαl3-seb was done by inserting 100 pb synthetic DNA sequence encoding third loop of TGFαL3 and GGSGSGGG linker in 5′ of pET28a:: seb recombinant vector. This chimeric construct was transformed into E coli BL21 (DE3) for expressing TGFαL3-SEB fusion proteins. Protein of interest with the expected molecular weight of ∼31 kDa was detected in the total cell pellets. After expression optimization (induction time, temperature, and IPTG concentration), the soluble phase in the buffer contained 80% SEB and 50% TGFαL3-SEB. The optimal TGFαL3-SEB native expression was induced with 0.5 mmol/L IPTG concentration at 37°C. The arrow in Figure 3B indicates the optimal native expression of TGFαL3-SEB fusion protein. Soluble TGFαL3-SEB fusion protein was purified from the supernatant of sonicated bacterial pellets through a Ni-NTA affinity column. Coomassie blue staining after SDS-PAGE separation showed that both SEB and TGFαL3-SEB were purified up to 95% homogeneity (Figure 4A). Finally, the identity of SEB and TGFαL3-SEB fusion protein was confirmed by Western blotting using the anti-His antibody (Figure 4B). The optimal purification of SEB and TGFαL3-SEB is shown in Figure 4A, lanes 1 and 4.

A, PCR amplification of seb gene. Agarose gel electrophoresis of PCR product using primers for cloning of the seb gene of Staphylococcus aureus; lane 1: negative control and lane 2: mature staphylococcal enterotoxin type B gene (seb). M: 100+ bp DNA ladder. B, Expression optimization of TGFαL3-SEB fusion protein. Optimization of TGFαL3-SEB expression was done in different concentrations of IPTG and temperature. Lane 1: supernatant of bacteria was induced with 1 mmol/L (final) IPTG concentration at 37°C, lane 2: pelet of bacteria were induced with 1 mmol/L IPTG concentration at 37°C, lane 3: supernatant of bacteria were induced with 0.8 mmol/L IPTG concentration at 37°C, lane 4: pelet of bacteria were induced with 0.8 mmol/L IPTG concentration at 37°C, lane 5: supernatant of bacteria were induced with 0.5 mmol/L IPTG concentration at 37°C, lane 6: pelet of bacteria were induced with 0.5 mmol/L IPTG concentration at 37°C, lane 7: supernatant of bacteria were induced with 1 mmol/L IPTG concentration at 30°C, lane 8: pelet of bacteria were induced with 1 mmol/L IPTG concentration at 30°C, lane 9: supernatant of bacteria were induced with 0.8 mmol/L IPTG concentration at 30°C, and lane 10: pelet of bacteria were induced with 0.8 mmol/L IPTG concentration at 30°C. Arrow indicates the best native expression of TGFαL3-SEB fusion protein. IPTG indicates isopropyl-b-

Purification and characterization of SEB and TGFαL3-SEB fusion protein. A, The best purified bands of TGFαL3-SEB (lane 1) and SEB (lane 1) by Ni-NTA column . B, Western blot analysis of TGFαL3-SEB fusion protein. Lane 1:, Uninduced BL21; Lane 2: TGFαL3-SEB fusion protein; Lane 3: SEB protein as a control, M and L: protein marker. TGFαL3-SEB indicates third loop of transforming growth factor α was genetically conjugated to staphylococcal enterotoxin type B.
TGFαL3-SEB Fusion Protein Can Bind Efficiently to EGFR-Expressing Cells
The A431 cells were used as in vitro cell culture model to investigate the binding properties of the fusion proteins to EGRF. The A431 cells are derived from human epidermoid carcinoma with high levels of EGFR expression. After overnight culture in 96-well plates, the cells were incubated with different concentrations of 6His-tagged TGFαL3-SEB or SEB (ranging from 0.1 ng/μL to 1 μg/μL). The cells were then incubated with HRP-conjugated antihexahistidine antibody. Unlike SEB, TGFαL3-SEB fusion protein bound to A431 EGFR-expressing cells with a significant affinity (Figure 5; P < .05). The binding affinity of SEB was like the negative control, PBS.

Binding of fusion proteins to EGFR by cell ELISA. A431 cells were seeded in a 96-well plate and then fixed with 10% neutral formaldehyde at room temperature for 1 hour. The bindings of cells to various concentrations (0.1-1000 ng/mL) of fusion proteins were detected by cell ELSIA. The results showed that TGFαL3-SEB fusion protein as a ligand-targeted superantigen unlike SEB bind to A431 EGFR-expressing cells with a significant affinity (P < .05), whereas SEB did not bind to A431 cells and it had binding affinity in compare to PBS as a negative control. Each point shows the mean ± standard deviation (SD) of triplicate samples. EGFR indicates epidermal growth factor receptor; ELISA, enzyme-linked immunosorbant assay; TGFαL3-SEB, third loop of transforming growth factor α was genetically conjugated to staphylococcal enterotoxin type B.
Third Loop of TGF-SEB Maintains Its Superantigenic Properties
To investigate whether the fusion of TGFα-L3 to SEB can affect superantigenic activity of this protein, we compared the cell proliferation upon exposure to either TGFαL3-SEB or SEB by XTT assay. Mice splenocytes were incubated with TGFαL3-SEB or SEB or PHA at 37°C for 72 hours. As shown in Figure 6, TGFαL3-SEB maintained 95.5% of the proliferative activity of SEB at 1 µg/mL concentration of the proteins (Figure 6). We also compared fusion protein superantigenic activity in higher concentrations (2 and 6 µm/mL) of proteins to investigate whether the superantigenic activity of TGFαL3-SEB was maintained in higher concentration of this protein. Results indicated that TGFαL3-SEB maintained 91.2% and 84.9% of the proliferative activity at 2 and 6 µg/mL TGFαL3-SEB concentrations, respectively (data not shown). As a control, PHA induced strong proliferation of splenocytes. Our results showed that fusing of TGFαL3 in N-terminal of the TGFαL3-SEB construct has no significant effects on either MHC binding or its superantigenic activity.

Comparison of superantigenic activity of TGFαL3-SEB and SEB proteins. Freshly isolated splenocytes were seeded in each well in a 96-well plate at a density of 1.0 × 105 cells/well. The cells were then incubated with 1.0 μg/mL TGFαL3-SEB fusion protein, 1.0 μg/mL SEB, and 20 μg/mL phytohemagglutinin (PHA) as a positive and PBS as a negative control at 37°C for 72 hours, and then XTT solution was dispensed to each well and the optical density at 450 nm was measured. As shown in the figure, TGFαL3-SEB maintained 95.5% of the proliferative activity of SEB at 1 µg/mL concentration of proteins. Each column shows the mean ± standard deviation (SD) of triplicate samples. TGFαL3-SEB indicates third loop of transforming growth factor α was genetically conjugated to staphylococcal enterotoxin type B.
Third Loop of TGF-SEB Fusion Protein Can Inhibit Tumor Cell Growth In Vitro
We investigated whether TGFαL3-SEB is able to bridge between tumor cells and immune effecter cells leading to the lysis of breast cancer cells. 4T1 cells, as target cells, were initially cocultured with increasing numbers of activated spleen cells as effector cells in the presence of either 5 ng/μL TGFαL3-SEB or SEB. As shown in Figure 7A, 50% inhibition of tumor cell growth by TGFαL3-SEB was achieved when the E–T ratio was around 30:1. Under this E–T ratio, SEB exerted more potent inhibitory effect than TGFαL3-SEB, whereas from the ratios of 30:1 to 300:1, the 2 curves overlapped and there was no difference between SEB and TGFαL3-SEB in TCGI% (Figure 7A). The E–T ratio of 30:1 was used as the 50% TCGI in the subsequent treatments. The 4T1 cells were cocultured with activated spleen cells at 30:1 E–T ratio in the presence of increasing concentrations of either TGFαL3-SEB or SEB. As shown in Figure 7B, 50% inhibition of tumor cell growth by TGFαL3-SEB and SEB was achieved at 0.05 ng/µL of both proteins. Upon 5 ng/µL treatment, SEB showed much higher inhibitory effect than TGFαL3-SEB. Higher concentrations of TGFαL3-SEB showed higher cytotoxicity compared with the higher concentrations of SEB in a dose-dependent manner (Figure 7B). Also, as shown in Figure 7B, 100% inhibition of tumor cell growth by TGFαL3-SEB was achieved at 200 ng/µL of the fusion protein.
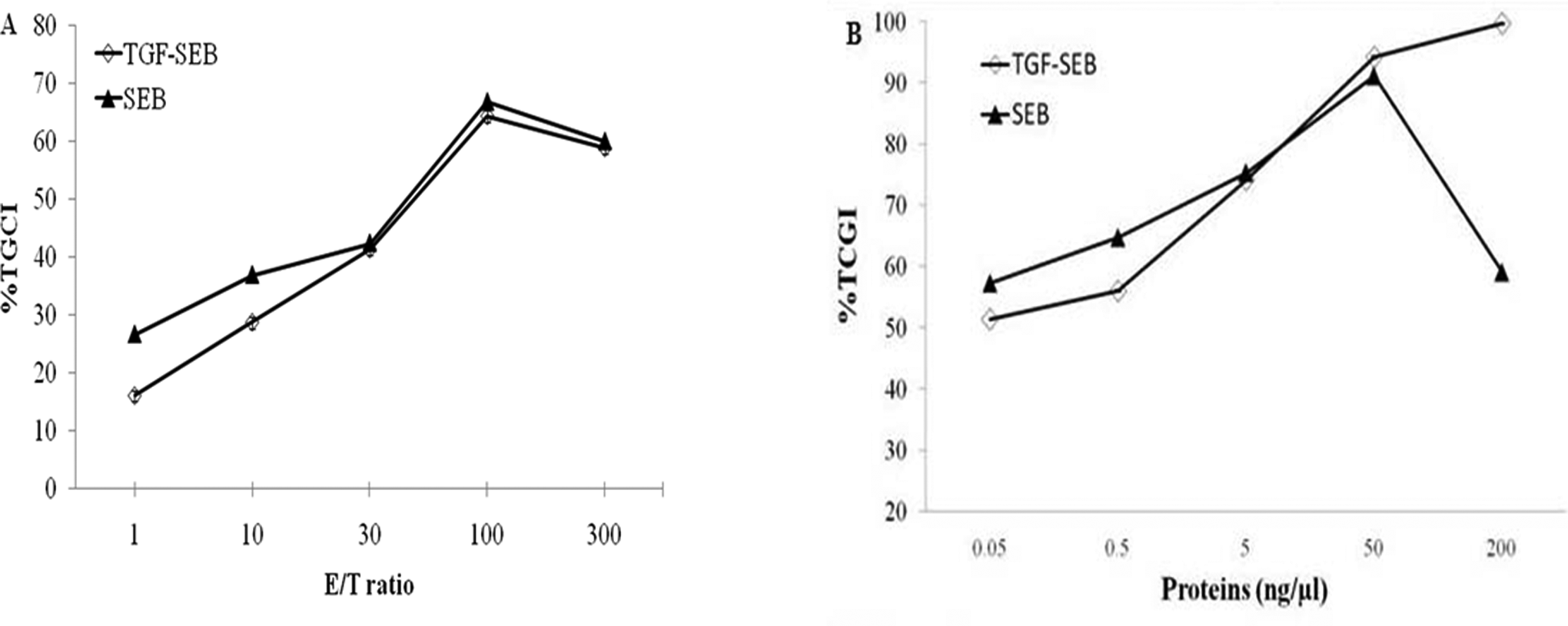
TGFαL3-SEB inhibits tumor growth in vitro. A, 4T1 cells were seeded into a 96-well plate at a density of 2.5 × 104 cells/well. Following an overnight incubation period, various ratios of effector cells cocultured with 4T1 cells as a target cells were in the presence of 5 ng/μL rSEB(diamond) or TGFαL3-SEB (black triangle). Viable tumor cells were determined using an 2,3,-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)-carbonyl]-2H-tetrazolium (XTT) assay, and the data were given as a percentage of tumor cell growth inhibition (TCGI). As shown in the figure, 50% inhibition of tumor cell growth by TGFαL3-SEB was achieved when the effector–target (E–T) ratio was around 30:1. B, 4T1 cells were plated in a 96-well plate at a density of 2.5 × 104 cells/well, followed by treatment with the indicated concentrations of TGFαL3-SEB (black triangle) and rSEB (diamond) at an E–T ratio of 30:1 in vitro. Our result indicated 50% inhibition of tumor cell growth by TGFαL3-SEB and SEB were achieved in 0.05 ng/µL of both proteins. The data were given as the percentage of tumor cell growth inhibition (%TCGI), which was described previously. rSED indicates recombinant staphylococcal enterotoxin type B; TGFαL3-SEB, third loop of transforming growth factor α was genetically conjugated to staphylococcal enterotoxin type B.
We also investigated the effect of SEB and TGFαL3-SEB fusion protein on murine splenocyte by adding 5 ng/µL proteins on increasing number of splenocytes ranging from 25,000 to 7,500,000 cells. Our results indicated that maximum proliferative effect obtained in the presence of 750,000 of murine splenocytes was 30 times more than 4T1 target cells (30:1 E–T ratio; data not shown). In higher number of splenocytes (2,500,000 and 7,500,000), the cells began to die and XTT optical density decreased presumably because of the abnormal confluency of the cells in the wells and lack of nutrients.
Discussion
T-cells have multifarious antitumor effects via releasing tumor-suppressive cytokines (interferon γ [IFN-γ] and tumor necrosis factor α [TNF-α]) or cytotoxic effector molecules such as perforin, 22,25 –27 but generally the number of tumor-specific T-cells is insufficient to mediate potent antitumor responses against progressive tumor growth. 34
Superantigens can activate a large number of T-cells irrespective of their antigen specificity resulting in a massive release of cytokines from T-cells and monocytes. They enhance the antitumor activity of the immune system and prevent tumor growth and metastasis. 7 According to our data, Sags can be used to enhance specific antigen responses. However, in vivo administration of intact Sags at sufficient therapeutic amounts could produce unwanted cytotoxic effects on normal cells. This is the main problem associated with most of the anticancer agents because of their nonspecific activities. 35 Therefore, anticancer chemotherapeutics are often given at suboptimal doses, resulting in the failure of therapy, often accompanied by the development of drug resistance and tumor metastasis. 18
For decades, several approaches have been employed for specific delivery of drugs to the cancer cells and subsequently improving the selective toxicity of anticancer therapeutics. One such therapeutic method includes the delivery of antineoplastic drugs to cancer cells by conjugating the drugs to the antigen-binding molecules (mAbs) or receptor-binding ligands that are either uniquely expressed or overexpressed on the target cancer cells relative to normal tissues. This allows a specific delivery of drugs to the cancer cells. Ligand-targeted therapeutics (LTTs) have advantages to mAbs. Tumor-related ligands are less antigenic than mAbs, are often readily available and inexpensive, and are easy to handle 18 and facilitate drug penetration into solid tumors. 19 Over the past few years, mAb-targeted Sags have been applied to several types of tumors; however, human antimouse antibody responses reduced circulating half-lives of these targeting molecules in vivo, while native ligands such as SAgs have gained limited application in targeting tumors due to induction of ligand–receptor internalization. This prevents SAgs from being presented to the surface of the tumor cells and subsequently prevents activation of T-cells.
In 2010, Xu et al suggested that the third loop of hTGF-α (TGFαL3) retained the binding ability to EGFR. The TGFαL3 is a native ligand to its receptor, EGFR, in many human tumors. The binding of the ligand to EGFR was relatively weaker than mAbs but was sufficient enough to bring SAgs toward EGFR-overexpressing tumor cells. 30 They fused TGFαL3 to mutant staphylococcal enterotoxin A (SEA; SEAD227A, introduced by Holzer). 36 Attempts to define the relationship between the structures of SEs and their various biologic activities have focused primarily on the activation of T lymphocytes. These studies indicated that more than 1 region of the toxin is involved in T-cell activation. 37
The Sag SEA binds to MHC class II molecules at 2 sites on either side of the peptide groove. Two separate but cooperative interactions to the human class II molecule human leukocyte antigen (HLA) DR1 were detected. The first high-affinity interaction to the DR1 β chain is mediated by a zinc atom coordinated by H187, H225, and D227 in SEA and H81 in the polymorphic DR1 β chain. The second low affinity site is to the DR1 β chain analogous to SEB binding and is mediated by residue F47 in SEA. Both binding sites on SEA are required for maximal activity. Thus, unlike SEB, SEA requires 2 separate binding sites for optimal activity, which may allow it to stabilize SEA interaction with TcRs as well as to activate the antigen-presenting cell by cross-linking MHC class II. 38 SEAD227A point mutant was constructed by Holzer in order to decrease MHC class II affinity and therefore prevent unwanted binding of the Sag SEA to MHC class II molecules. This mutation (SEAD227A) resulted in an approximately 3-log reduction in affinity to HLA-DR, and consequently T-cell stimulating activity was 4- to 5-log less effective than native SEA when activation of resting T-cells was assayed in terms of blast formation, expression of cell surface activation markers, and cytokine release (interleukin 2, IFN-γ, and TNF-α). Furthermore, lysis of MHC class II-positive cells was reduced approximately 1000-fold, and stimulation of T-cells required 4- to 5-log higher concentrations of SEAD227A when compared to the wild-type SEA. Overall, according to their findings, the potency of the mutant SEAD227A in T-cell activation was severely reduced when compared to the wild-type Sag. 38 In the case of SEB, several studies have been done to detect the key fragment of SEB structure essential to T-cell proliferation. Binek et al revealed that a 17-kDa carboxy-terminal fragment of SEB was crucial for T-cell proliferation. 39 Buelow et al indicated that residues 1 to 138 were sufficient to promote T-cell proliferation. Deletion of the first 30 amino-terminal residues of SEB eliminated activation of at least 1 VP type (V beta 3.1), while polyclonal T cells still responded to this molecule. 40 On the other hand, analysis of site-directed mutants of SEB confirmed the importance of specific amino-terminal and central regions of SEs in MHC class II and VP interactions. In general, deletions of amino-terminal and carboxy-terminal residues of SEA and SEB reduced their stability and mitogenic activity. 37
The present study aimed to evaluate the antitumor activity of the recombinant TGFαL3-SEB in vitro using the third loop of TGF-α to deliver whole SEB molecule to 4T1 murin breast cancer cell line. The potency of TGFαL3-SEB was examined in a T-cell activation assay. Based on 40 independent studies, the mean percentage of EGFR expression on breast cancer cells is 45% (range 14%-91%). 22 Since expression of EGFR is highly associated with tumor invasion and metastasis, we used the EGFR-overexpressing 4T1 breast adenocarcinoma mouse cell line 41 in our in vitro study. The 4T1 mammary carcinoma cell line was originally isolated by Miller and coworkers at the Karmanos Cancer Institute. 42,43 This cell line is a widely studied model for basal-like breast cancer with metastatic potential. 31,44,45 We used the 4T1 breast adenocarcinoma mouse cell line as a model system because tumor growth and metastatic spread of 4T1 cells mimic very closely to human breast cancer and has the advantage of being able to be transplanted into immunocompetent recipients. 46 This cell line is spontaneously capable of metastasis to several organs including lungs, liver, and brain as well as bone. 31,47 –49
We included controls (4T1 + media, 4T1 + SEB, and 4T1 + TGFαL3SEB) in TCGI assay to rule out direct cytotoxic effect of SEB and TGFαL3-SEB fusion proteins on 4T1 cell line. No cytolytic effect was detected when 4T1 target cells were incubated with the proteins only.
Xu et al examined the effects of TGFαL3-SEAD227A fusion protein on A431 and B16 cancer cells. The A431 cells contained as many as 2.0 × 106 EGF receptors per cell. The EGFR gene was indeed amplified 15- to 20-fold in A431 cell line, 50 while murine melanoma B16 cells expressed lower EGFR compared to that found in A431 cells. For this reason, they used A431 cell line in their TCGI assay and found that TGFαL3-SEAD227A fusion protein inhibits the cell growth much more effectively in a dose-dependent manner compared to SEA. However, in the case of B16 cell line, SEA exerted more potent inhibitory effects than TGFαL3-SEAD227A at concentrations below 5 ng/μL. Interestingly, TGFαL3-SEAD227A showed considerable dose-dependent inhibition effect at concentrations higher than 5 ng/μL compared to SEA. 24 Our results were consistent with Xu et al when 4T1 cell line was used instead of B16 cell line. Expression of EGFR in 4T1 and B16 cell lines is equally low compared to A431 cell line. In TCGI assay using 4T1 cell line, SEB showed higher inhibition effect than TGFαL3-SEB at concentrations below 5 ng/μL, but at concentrations above 5 ng/μL, TGFαL3-SEB exerted remarkable dose-dependent inhibition effect.
This evidence suggests that the ligand density is high enough at concentrations higher than 5 ng/µL TGFαL3-Sag fusion protein to target Sag toward cancer cells (such as 4T1 and B16 cell lines) that express lower surface EGFR than A431. Also, as shown in Figure 7B, 100% inhibition of tumor cell growth by TGFαL3-SEB was achieved in 200 ng/µL of fusion protein, but in Xu et al study, 500 ng/µL of TGFαL3-SEAD227A caused 70% TCGI. This may be due to mutations in the SEA protein and subsequent reduction in its antiproliferative properties.
Conclusions
Our results indicated that TGFαL3-SEB fusion protein is capable of targeting EGFR-expressing cancer cells. Therefore, it may be a promising anticancer candidate for immunotherapy on EGFR-expressing tumors, but further efforts are needed to explore this potential therapeutic strategy.
Footnotes
Authors’ Note
All authors certify that this manuscript has not been published in whole or in part nor is it being considered for publication elsewhere.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from the applied microbiology research center of Baqiyatallah University of Medical Sciences.