
Abstract
Introduction
HER2-positive breast cancer, accounting for 15%-20% of cases, remains challenging due to therapy resistance and immunosuppressive “cold tumor” microenvironments. Current strategies combining immunotherapy with chemotherapy or radiotherapy often face toxicity limitations. To address this, we developed HER2-targeted nanoliposomes co-delivering viral peptides and the STING agonist diABZI, aiming to convert cold tumors into immunologically active “hot tumors” by enhancing antigen spreading and immune recognition.
Methods
Viral peptides with high Human Leukocyte Antigen-A2 affinity were selected using NetMHCpan-4.1/4.0 and incorporated into nanoliposomes via thin-film dispersion. Trastuzumab F(ab’)2 fragments were conjugated for HER2-specific targeting. Nanoliposomes were characterized for size, stability, encapsulation efficiency (HPLC), and in vitro release. Immune efficacy was assessed via ELISPOT, flow cytometry (CD3+/CD8+/NK cells), and TCR β sequencing in HER2+ SK-BR-3 and HER2− MCF-7 cells. Cytotoxicity and cellular uptake were evaluated using CCK-8 assays and fluorescence imaging.
Results
The nanoliposomes exhibited uniform size (∼70 nm), stability (5% size variation over 25 days), and high encapsulation efficiency (75.5% for peptides). Targeted delivery to SK-BR-3 cells peaked at 60 µL (P < .05), with sustained release of peptides (52% at 48 h) and diABZI (46.2%). In HER2+ cells, nanoliposomes synergistically enhanced IFN-γ (2.5-fold, P < .01) and granzyme B (3-fold, P < .05) secretion, overcoming antagonism seen with free agents. Flow cytometry revealed dominant CD8+ T-cell infiltration (50.9% vs 0.67% in controls) and expanded NK/NKT populations. TCR β sequencing showed increased clonotype diversity (60,915 vs 57 574 clones) and reduced clonal dominance, indicating broadened antigen recognition.
Conclusion
Our HER2-targeted nanoliposomes effectively reprogrammed cold tumors by dual activation of innate (STING pathway) and adaptive (viral peptide-driven TCR diversity) immunity. The platform demonstrated robust targeting, safety, and immune activation, offering a promising strategy to overcome immunotherapy resistance. Future studies will validate in vivo efficacy and explore adaptations for other cold tumors via alternative targeting ligands.
Introduction
Breast cancer remains one of the most common cancers worldwide, with HER2-positive cases accounting for 15-20% of all diagnoses. 1 The introduction of HER2-targeted therapies, such as trastuzumab, has significantly improved the survival rates of these patients However, despite these advancements, many patients still develop resistance to various breast cancer treatments, including targeted therapies and immunotherapies, which reduces long-term outcomes for this group of patients. 2 Additionally, tumor subgroups known as “cold tumors” respond poorly to immunotherapies due to low immunogenicity, weak neoantigen expression, and an inhibitory tumor microenvironment. 3 These challenges highlight the need for novel strategies, particularly for tumors resistant to immunotherapy, that can induce neoantigen spreading, enhance immune recognition, and improve the immune system's ability to attack tumor cells. One promising approach to overcoming these limitations is combining traditional cancer treatments, such as radiotherapy and chemotherapy, with immunotherapy. This combination has shown improved outcomes by expanding tumor antigen epitopes. 4 Radiotherapy and chemotherapy induce tumor cell death, activating the cGAS-STING pathway,5,6 a key immune signaling mechanism that can enhance the immune system's recognition of hidden tumor antigens and increase the immunogenicity of weak neoantigens 7 However, despite the success of combination therapies, they often come with significant side effects, underscoring the need for safer, more targeted methods that can enhance immune responses without introducing additional toxicity.
To address these issues, our research focuses on developing a novel nanoliposome-based therapy targeting HER2-positive breast cancer, 8 aimed at transforming cold tumors into “hot tumors” that are more likely to respond strongly to immunotherapy. 9 This strategy leverages viral peptides as dominant antigens to induce a stronger immune response than typical tumor neoantigens, activating the cGAS-STING pathway with a STING agonist. This leads to a robust immune response against the viral peptides within tumor cells, triggering the expansion of neoantigen epitopes and enhancing the immunogenicity of tumor antigens. Specifically, we incorporated the trastuzumab F(ab’) 2 fragment into the nanoliposome design to increase specificity for HER2-positive cancer cells, 8 ensuring that the viral peptides and STING agonist are directly delivered to the tumor cells. Given the rising incidence of breast cancer and its increasing recognition as the most common cancer globally, 10 there is an urgent need for novel therapeutic strategies. Our nanoliposome-based approach not only has the potential to improve outcomes for HER2-positive patients but also offers broader applications in transforming cold tumors into immunotherapy-responsive tumors. This study evaluates the immune efficacy of these targeted nanoliposomes and explores their potential as a novel cancer vaccine platform capable of enhancing the immune microenvironment and boosting anti-tumor activity.
Material and Methods
Selection of Viral Peptides
Using NetMHCpan-4.1 and NetMHCIIpan-4.0 software, viral peptides with high affinity for the Human Leukocyte Antigen (HLA) alleles of SK-BR-3 cells were identified. Peptides ranked < 2% were classified as weak binders (WBs) and those < 0.5% as strong binders (SBs). Five strong-binding peptides were selected and synthesized by Jinyu Chemical Co., Ltd, for subsequent liposome preparation.
Determination of Optimal diABZI Dose via TNF-α Measurement in PBMC Cultures
To determine the optimal dose of the STING agonist diABZI, the level of TNF-α, a cytokine involved in inflammation and tumor suppression, was measured. PBMCs were cultured in IL-2 medium (RPMI 1640 with 10% FBS, antibiotics, 60 µg/L IL-2, and β-mercaptoethanol) at 37°C and 5% CO2, and lymphocytes were stimulated with varying concentrations of diABZI. After 3 days, TNF-α levels were assessed via Enzyme-Linked Immunosorbent Assay (ELISA) to establish the effective dose.
F(ab’)2 Fragment Preparation
F(ab’)2 fragments were prepared via the Pierce™ F(ab’)2 Preparation Kit (Thermo Fisher Scientific Inc.). The protein concentration was measured at 280 nm with an ultramicro spectrophotometer. To determine the connection efficiency of F(ab’)2 to liposomes, the fragments were coupled to the liposome surface. The sample was then centrifuged in a 0.5 mL ultrafiltration tube (100 kD) at 5000 rpm for 5 min, and this process was repeated three times. The filtrate was analyzed to determine the free antibody concentration. The connection efficiency was calculated by subtracting the free antibody concentration from the total concentration of added antibody, dividing the result by the total concentration of added antibody, and then multiplying by 100 to express it as a percentage.
Preparation of Nano-Liposomes Containing Viral Peptides and STING Agonists
Nanoliposomes containing viral peptides and the STING agonist diABZI were prepared via a thin film-ultrasonic dispersion method. A lipid mixture of hydrogenated soybean phospholipid, DSPE-mPEG2000, and DSPE-PEG2000-Mal (40 mg) was dissolved in ethanol, followed by vacuum evaporation at 45 °C for 10 min to form a uniform lipid film. Cholesterol (10 mg) dissolved in chloroform (3 mL) was added, and further evaporation removed the solvent, leaving a lipid film. A solution of viral peptide (5 mg) and diABZI in PBS (5 mL) was then added, and rotary evaporation at 60 °C for 30 min detached the lipid gel, yielding a milky white suspension. This mixture was sonicated (150 W, 1 s/1 s pulse) for 5 min, forming a semitransparent suspension, which was extruded through 400 nm and 100 nm polycarbonate membranes 20 times to produce liposomes with uniform particle sizes.
Conjugation of F(ab’)2 to Nano-Liposomes
F(ab’)2 fragments were thiolated by reacting with 2-iminothiolane at a 1:10 molar ratio under gentle agitation for 1 h at room temperature, followed by dialysis to remove excess reagent. The thiolated F(ab’)2 was then conjugated to nanoliposomes containing viral peptides and STING agonists (1:50 molar ratio) through agitation for 3 h. After dialysis to remove unbound F(ab’)2, the resulting purified liposomes, abbreviated as LPs (viral peptide + diABZI), were obtained. Similarly, F(ab’)2-conjugated liposomes containing only viral peptides (LPs [viral peptides]) and empty liposomes (LPs [empty]) were prepared.
Characterization of Nanoliposomes
The characterization of the nanoliposomes involved assessing their morphology, particle size, zeta potential, and encapsulation efficiency. Morphological analysis was conducted by applying the liposome samples onto a copper grid, which was subsequently stained with 2% phosphotungstic acid, and images were captured via transmission electron microscopy. The particle size, polydispersity index (PDI), and zeta potential were measured via nanoparticle size and zeta potential analysis, as these parameters are critical for evaluating colloidal stability and dispersion, with the zeta potential indicating electrostatic repulsion between particles. The encapsulation efficiency was determined via high-performance liquid chromatography (HPLC). A standard curve for peptide quantification was generated using known peptide concentrations (0.05 to 0.25 mg/mL). The total peptide content in the liposomes was measured by rupturing the liposomes with methanol and analyzing the sample via HPLC, while the free peptide content was quantified after ultrafiltration and HPLC analysis. The encapsulation efficiency was calculated by subtracting the free peptide content from the total peptide content, providing a measure of the liposome's drug-loading capacity.
Physicochemical Stability and in Vitro Release Characterization of Nanoliposomes
The stability of nanoliposomes was evaluated by storing freshly prepared samples in amber vials at 4 °C for 25 days. Aliquots (0.5 mL) were collected at Days 0, 5, 10, 15, 20, and 25 to assess physical stability via dynamic light scattering (DLS) for particle size, PDI, and zeta potential. For in vitro drug release studies, 1 mL of nanoliposomal suspension was loaded into a dialysis bag (MWCO: 8-14 kDa) and immersed in 100 mL of release medium (0.01 M PBS, pH 7.4, 0.5% Tween-80) under physiological conditions (37 °C, 100 ± 5 rpm). At predetermined intervals (0.5-48 h), 1.0 mL of medium was sampled and replaced with pre-warmed medium to maintain sink conditions. The antigenic peptide concentration was quantified using a bicinchoninic acid (BCA) assay, while diABZI was analyzed by UV-Vis spectroscopy at 300 nm. Free drug concentrations were calculated using pre-validated standard curves for each compound. Cumulative release rates were determined via a dynamic mass-balance model to correct for sampling dilution effects. All samples were filtered (0.22 μm hydrophobic membrane) and stored at −80 °C prior to analysis.
Cytotoxicity/Cellular Uptake Assessment of Nanoliposomes
The cells were thawed at −80 °C in a 37 °C water bath, transferred to a centrifuge tube with fresh medium, and washed through centrifugation. They were cultured in medium containing 90% DMEM, 10% FBS, and 1% penicillin/streptomycin, with color changes indicating the need for medium replacement. Upon reaching approximately 80% confluency, the cells were digested with trypsin, passaged into new vessels, and cryopreserved in freezing medium for future experiments. The cytotoxicity of the liposomal formulations was assessed via the CCK-8 assay, where cells were seeded into 96-well plates and treated with various concentrations of liposomal drugs. The absorbance at 450 nm was measured to determine cell viability. For the cellular uptake experiment, fluorescently labeled liposomes were prepared by adding a lipophilic dye and incubating in the dark, followed by ultrafiltration to remove excess dye. Various concentrations of these labeled liposomes were then introduced into the cells and incubated for 2, 12, or 24 h, after which the fluorescence intensity was quantified via a fluorescence microscope and a microplate reader.
Immunological Evaluation of Liposomal Formulations in Breast Cancer Cells
SK-BR-3 and MCF-7 cells were seeded in a 96-well plate at a density of 3 × 10³ cells per well. On the second day, the liposomal drugs and 100 ng/mL IFN-γ were added to the culture medium, which was replaced 24 h later to remove residual liposomes. Human peripheral blood mononuclear cells (hPBMCs) were obtained from Miaoshun Biotechnology Co., Ltd (Shanghai), thawed, and cultured in fresh medium. On Day 3, the liposome-treated tumor cells were cocultured with healthy PBMCs for three days. The immune effects were assessed via ELISA to quantify the levels of cytokines such as TNF-α and IFN-γ in the supernatants, with IFN-γ indicating immune activation. Additionally, the ELISPOT assay was used to measure the frequency of IFN-γ secretion from T cells at a concentration of 1 × 105 cells per well. Finally, flow cytometry was performed at Hangzhou Normal University Hospital to characterize TILs via the CD3, CD4, and CD8 markers.
RNA Extraction, TCR β Chain Sequencing, and Bioinformatics Analysis
Total RNA was extracted from peripheral blood mononuclear cells (PBMCs). RNA concentration was measured using spectrophotometry, and integrity was assessed via agarose gel electrophoresis. For TCR β chain sequencing, a 5′ rapid amplification of cDNA ends (RACE) protocol was employed with the TCR profiling system (ImmuQuad Biotech, Hangzhou, China). High-throughput sequencing was conducted on an Illumina NovaSeq® system in PE150 mode. Raw sequencing data were filtered, quality controlled, and merged before alignment to the ImMunoGeneTics (IMGT) database. Subsequent bioinformatics analyses were conducted using the immunarch package in R, 11 including quantifying the frequencies of CDR3 gene and amino acid sequences, assessing the formation of V, D, J, and C segments, determining the length distribution of the CDR3 region, and evaluating sequence diversity and clonality across samples.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism v5.0. Statistical significance was evaluated using a one-way analysis of variance with a Bonferroni post-test (*** P < 0.001, ** P < .01, * P < .05, ns = not significant (P > .05)). To present the results and findings clearly, visual representations in the form of plots and graphs were generated using the ggplot2 package in R. The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Results
Viral Peptide Screening
To identify viral peptides capable of specifically targeting HER2-positive breast cancer cells, we performed HLA typing on SK-BR-3 cells. As shown in Table 1, the cells predominantly expressed the HLA-A*02:01 allele. We subsequently employed the bioinformatics tools NetMHCpan-4.1 and NetMHCIIpan-4.0 to predict viral peptides with high binding affinity to the HLA-A2 molecule via a comprehensive viral peptide database. On the basis of the prediction results, five nonapeptides with strong binding affinities were selected, as detailed in Table 2. These peptides originated from human papillomavirus, human T-lymphotropic virus, and influenza virus.
HLA Genotyping of SK-BR-3 Cells.

Abbreviation: HLA, Human Leukocyte Antigen.
Binding Affinity Predictions Using NetMHCpan-4.1 and NetMHCpan-4.0.

DiABZI Dose-Dependently Enhanced TNF-α Production in PBMCs
To evaluate the immunomodulatory effects of diABZI, a stimulator of interferon genes (STING) agonist, we measured tumor necrosis factor-alpha (TNF-α) production in peripheral blood mononuclear cells (PBMCs) treated with various concentrations of diABZI. As shown in Figure 1, diABZI induced a dose-dependent increase in TNF-α secretion by PBMCs. Logistic regression analysis revealed a strong positive correlation between the diABZI concentration and TNF-α production (R²=0.95989). Notably, even at low concentrations (0.78-1.5 µM), diABZI elicited a significant immune response. Given these findings and to minimize potential cytotoxic effects and experimental interference, we selected a diABZI concentration of 1 µM for subsequent experiments.

Dose Response Curves Show TNF-α Secretion (pg/mL) with Increasing diABZI Concentrations (0-50 μM). TNF-α Production was Significantly Increased Even at low diABZI Concentrations (0.78-1.5 μM). the Curves Showed Saturation at Higher Doses, Indicating That TNF-α Secretion Reached a Plateau Around 40 μM.
Nanoliposome Characterization
The particle size distribution of nanoliposomes coated with antigenic peptide and STING agonist was determined by dynamic light scattering (DLS) particle size analyzer. The measurement results (Figure 2) showed that the average particle size of the nanoliposomes was about 70 nm, and the PDI was 0.181, indicating a narrow and uniform particle size distribution. The measured zeta potential was about −9.18 mV, indicating a moderate negative surface charge, which can lead to strong electrostatic repulsion between particles, thereby preventing aggregation and promoting stable dispersion in solution. HPLC analysis was performed to further characterize the nanoliposome formulation. It was observed that different concentrations of antigenic peptide produced a stable solvent baseline and a clear drug peak at about 7 min, confirming that the antigenic peptide can be reliably detected under the established experimental conditions. The clear peak indicates minimal interference and supports the accuracy of peptide quantification. The standard curve was generated by plotting the peak area (A) of the standard solution against the peptide concentration (C) (Figure 3). Linear regression analysis yielded the equation Y = 11576X − 40.93 with an R2 of 0.9996, indicating a good linear correlation between peptide concentration and peak area in the concentration range of 0.05-0.25 mg/mL. This high linearity confirms the reliability of the HPLC method in accurately quantifying the antigenic peptides in the nanoliposomal formulation. For a sample volume of 500 µL, the total peptide content was determined to be 131.45 µg, while the free peptide content was 32.233 µg, with an encapsulation efficiency of 75.5%. This high encapsulation efficiency highlights the effectiveness of the nanoliposomal formulation in retaining the antigenic peptide within the lipid bilayer, which is critical for its controlled release and targeted delivery.
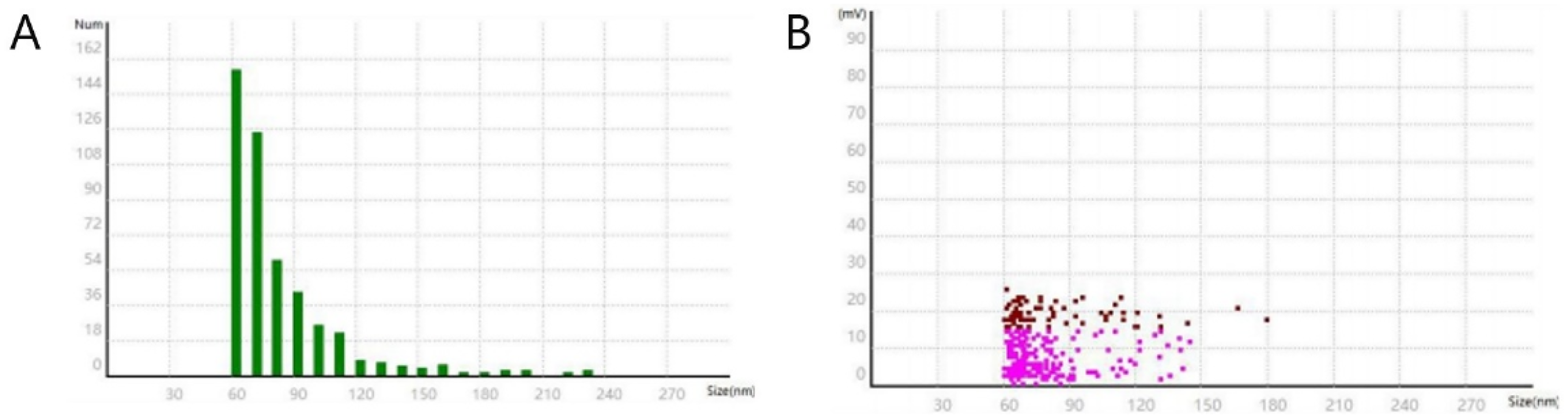
2A Liposome Drug Particle Size Measurement, Average Particle Size: 70 (nm), Polydispersity Index: 0.181, Median Particle Size: 69 (nm), D90: 96 (nm), D10: 60 (nm), D90:D10: 1.6, Largest Particle Size: 223 (nm), Smallest Particle Size: 57 (nm). 2B Liposome Drug Potential Measurement, Zeta Potential: −9.18 (mV), Zeta Potential Variance: 7.050, Median Zeta Potential: −6.42 (mV), Zeta Potential D90: −20.64 (mV), Zeta Potential D10: −2.28 (mV).

Standard Curve of Peptide Quantification by HPLC.
Stability Assessment of Nanoliposomes
The stability of nanoliposomes encapsulating antigenic peptides and STING agonists was systematically evaluated over 25 days under controlled storage conditions. Dynamic light scattering (DLS) and zeta potential measurements revealed that the particle size remained consistent (Figure 4), with minimal fluctuations between 67.0 nm (Day 25) and 72.9 nm (Day 20), representing a modest increase of approximately 5% from the initial size of 68.6 nm (Day 0). The PDI, a critical indicator of size homogeneity, remained below 0.2 throughout the study (range: 0.123-0.222), demonstrating a narrow and stable size distribution despite transient variability (PDI = 0.222 on Day 15). Zeta potential measurements further confirmed colloidal stability, with surface charges maintaining a negative range between −9.3 mV (Day 0 and 25) and −8.1 mV (Day 20), indicative of sufficient electrostatic repulsion to prevent aggregation. Notably, the zeta potential D90:D10 ratio (spanning −20.64 mV to −2.28 mV) and particle size D90:D10 ratio (1.6-fold difference) underscored uniform surface charge and size profiles. These data collectively validate the robust physical stability of the nanoliposomal formulation, ensuring consistent performance for therapeutic applications during the tested storage period.

Stability Evaluation of HER2-Targeted Nanoliposomes Over 25 Days. (4A) Particle Size (Yellow Line) and Zeta Potential (Blue Line) Profiles During Storage. Particle Size Fluctuated Minimally Between 67.0 nm and 72.9 nm (5% Variation), While Zeta Potential Stabilized Between −9.3 mV and −8.1 mV, Confirming Colloidal Stability. (4B) Storage Time-Dependent Changes in Polydispersity Index (PDI) of HER2-Targeted Nanoliposomes. PDI Remained Below 0.2 (Range: 0.123-0.222), Indicating Consistent Particle Size Homogeneity.
Drug Release Profiling Under Simulated Physiological Conditions
The in vitro release kinetics of antigenic peptide- and diABZI-loaded nanoliposomes were evaluated under simulated physiological conditions over 48 h. All reported release data represent mean values from three independent replicates (n = 3). Both formulations exhibited sustained release profiles, with cumulative release increasing over the incubation period. For antigenic peptide-loaded nanoliposomes, 5.3% of the encapsulated peptide was released within the first 0.5 h, followed by a gradual escalation to 52.0% at 48 h (Figure 5). In contrast, diABZI demonstrated slower initial release kinetics, with 2.0% released at 0.5 h, and reached 51.8% cumulative release by 48 h, reflecting its distinct diffusion-controlled mechanism. The biphasic release pattern—characterized by an initial burst phase (attributed to surface-associated drug fractions) and subsequent sustained release (from core-encapsulated drug)—aligns with typical nanoliposomal behavior. Notably, antigenic peptide displayed faster overall release kinetics compared to diABZI (Figure 5), likely due to differences in molecular size, hydrophobicity, and interactions with the lipid bilayer, even though their cumulative release at 48 h was similar. These results underscore the tunable release properties of the nanoliposomal platform, which can be optimized for therapeutic payloads requiring either rapid bioavailability or prolonged action.
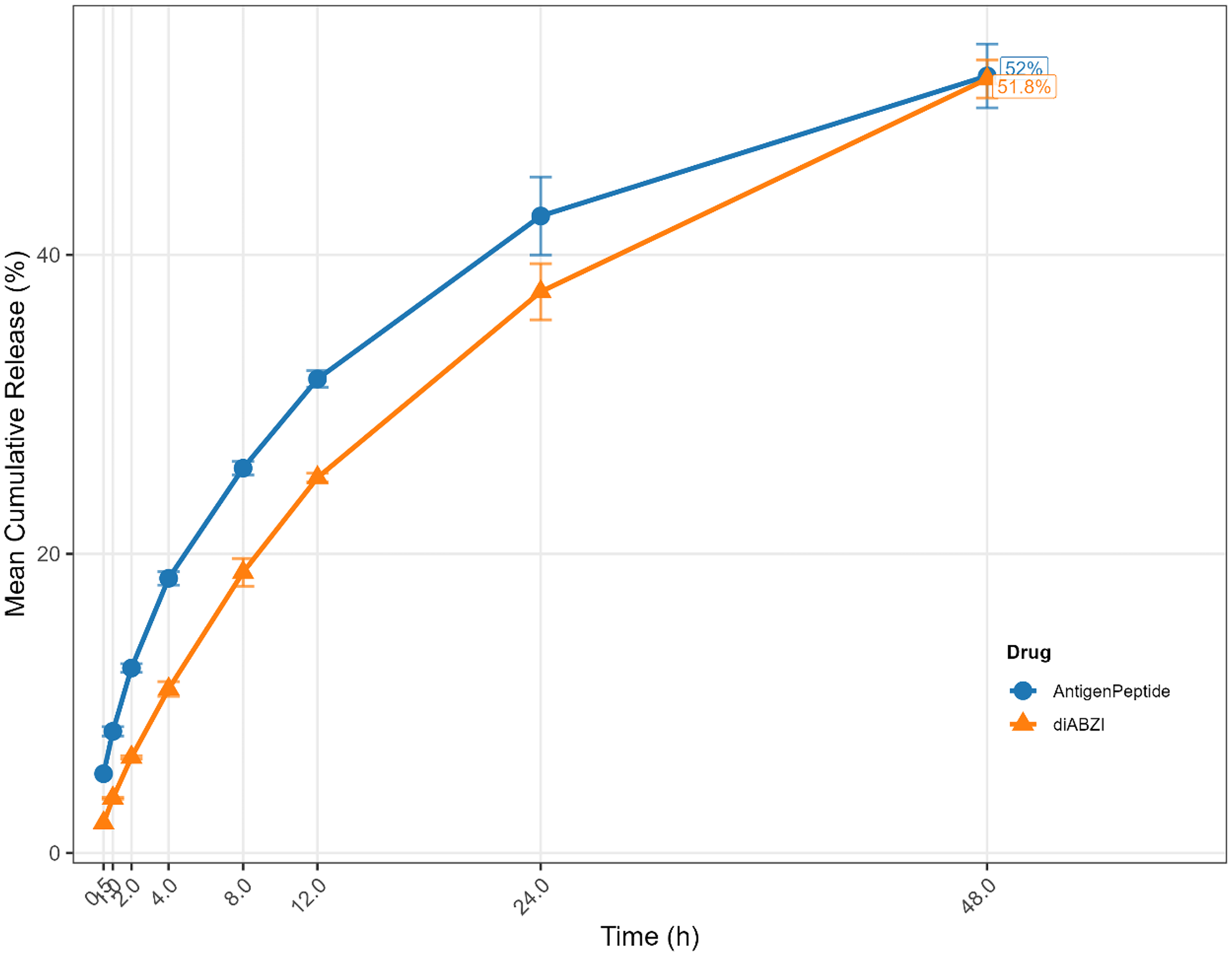
In Vitro Cumulative Release Profiles of Antigenic Peptide and diABZI from HER2-Targeted Nanoliposomes Under Simulated Physiological Conditions. Antigenic Peptide (Blue Line) Exhibited More Rapid Release Kinetics, with a Mean Cumulative Release of 5.3% at 0.5 h and Reaching 52.0% at 48 h. DiABZI (Orange Line) Showed a Slower Initial Release (2.0% at 0.5 h), Attaining a Mean Cumulative Release of 51.8% at 48 h. Both Formulations Demonstrated Sustained Release Patterns Over the 48-h Period, Consistent with Diffusion-Controlled Mechanisms. All Data Points Represent the Mean of Three Independent Replicates (n = 3); Error Bars Indicate Standard Deviation.
Cytotoxicity Assessment of Nanoliposomes in SK-BR-3 and MCF-7 Cells
The cytotoxicity of blank liposomes and nanoliposomes (containing viral peptides and diABZI) was evaluated using the Cell Counting Kit-8 (CCK8) assay in SK-BR-3 and MCF-7 cell lines across a range of concentrations, as depicted in Figure 5. For blank liposomes, the optical density (OD) values in SK-BR-3 cells (Figure 6A) did not exhibit a significant decrease across the tested concentrations, and no statistically significant differences were observed between the treatment groups. Similarly, in MCF-7 cells treated with blank liposomes (Figure 6B), the OD values remained consistent across the concentration range, with no significant inter-group variations, indicating a lack of notable cytotoxicity.

Cytotoxicity Assessment of Blank Liposomes (A and B) and Nanoliposomes Containing Viral Peptides and diABZI (C and D) in SK-BR-3 (A and C) and MCF-7 (B and D) Cells Using the CCK8 Assay. Optical Density (OD) Values at 450 nm are Plotted Against Drug Concentration. Statistical Significance is Indicated by Asterisks (*P < .05, **P < .01, ***P < .001, ****P < .0001).
The treatment with nanoliposomes containing viral peptides and diABZI resulted in significant differences in OD values. For SK-BR-3 cells (Figure 6C), statistical analysis revealed significant variations among the treatment groups (F = 72.91, P < .0001), with doses of 10, 15, 20, 25, and 30 µL leading to significantly higher OD values compared to the control group (P < .01), suggesting no substantial cytotoxic effects at these concentrations. Likewise, in MCF-7 cells treated with the same nanoliposomes (Figure 6D), the OD values significantly differed across the groups (F = 43.60, P < .0001), with doses of 5, 10, 15, 20, 25, and 30 µL resulting in significantly elevated OD values compared to the control group (P < .001). Overall, within the tested concentration range (0-30 µL), neither the blank liposomes nor the nanoliposomal formulation induced significant cytotoxic effects on either cell line.
Optimized Cellular Uptake and Targeting Efficiency of HER2-Targeted Nanoliposomes in Breast Cancer Cells
The effects of targeted and nontargeted liposomes on SK-BR-3 and MCF-7 cells were evaluated by fluorescence intensity measurements at 2, 12, and 24 h (Figure 7). Figure 7B shows that the fluorescence intensity at 24 h was significantly higher than that at 2 and 12 h, indicating that 24 h is the optimal incubation period. In SK-BR-3 cells, 60 µL of targeted liposomes produced significantly higher fluorescence than 30 µL or 120 µL (P < .05). Fluorescence increased between 30 µL and 60 µL but decreased at 120 µL, indicating that higher doses may reduce targeting efficiency due to nonspecific interactions with the Her2 receptor. In contrast, MCF-7 cells, which lack significant Her2 expression, showed consistent dose-dependent fluorescence increases at higher doses without inhibition.

(A) Fluorescence Microscopy Images of MCF-7 and SK-BR-3 Cells Treated with Fluorescently Labeled Liposomal Drugs at Increasing Concentrations (0, 15, 30, and 60 µL). Green Fluorescence Indicates Liposomal Drug Uptake, with Fluorescence Intensity Positively Correlated to Drug Concentration. (B) Time-Dependent Changes in Fluorescence Intensity (2, 12, and 24 h) in SK-BR-3 and MCF-7 Cells Under Varying Liposomal Drug Concentrations. Both Cell Lines Exhibited Progressive Fluorescence Enhancement, Reaching Maximal Intensity at 24 h. (C) Concentration-Response Analysis of Fluorescence Intensity in SK-BR-3 and MCF-7 Cells Treated with Liposomal Drugs (0, 15, 30, 60, and 120 μL). Fluorescence Intensity Increased Dose-Dependently in Both Cell Types, with SK-BR-3 Cells Demonstrating Higher Intensity at Intermediate Concentrations (15-60 μL). (D) Fluorescence Intensity Comparison Between Targeted and non-Targeted Liposomes in SK-BR-3 and MCF-7 Cells. Targeted Liposomes Showed Superior Uptake at 15 and 60 μL Concentrations in SK-BR-3 Cells (P < .05), Whereas MCF-7 Cells Exhibited Uniform Responses Across Treatments.
Comparison with nontargeted liposomes (Figure 7C) confirmed these findings. In SK-BR-3 cells, targeted liposomes produced significantly higher fluorescence than nontargeted liposomes at 15 µL and 60 µL (P < .05), but fluorescence was significantly lower at 120 µL (P < .01), supporting the conclusion that excessive doses compromise targeting efficiency. In contrast, MCF-7 cells showed stronger fluorescence of targeted liposomes at all doses (P < .05), indicating no loss of targeting at high concentrations.
Figure 7D reinforces these results by comparing fluorescence intensity in both cell lines after 24 h. In SK-BR-3 cells, targeted liposomes produced significantly higher fluorescence than nontargeted liposomes at 15 µL and 60 µL (P < .05), but fluorescence was lower at 120 µL (P < .01), confirming reduced targeting efficiency at high doses. However, MCF-7 cells showed stronger fluorescence of targeted liposomes at all doses (P < .05) and was not affected by increasing doses. These findings suggest that while targeted liposomes are effective at enhancing drug delivery in SK-BR-3 cells at moderate doses, higher doses may reduce specificity due to competitive inhibition by free antibody fragments. A dose limit of 60 µL is recommended for optimal targeting in SK-BR-3 cells to prevent off-target effects.
Nanoliposome-Mediated Immune Activation Overcomes Drug Antagonism in HER2-Positive Breast Cancer Cells
Our ELISPOT studies delineated HER2-targeted nanoliposome-mediated resolution of drug antagonism in combinatorial immunotherapy for breast cancer models(Figure 8).

ELISPOT Images of IFN-γ and Granzyme B Production in SK-BR-3 and MCF-7 Cells. Cells Were Treated with Control, Viral Peptide + diABZI, Liposomes (Control), or Liposomes (Viral Peptide + diABZI). Representative ELISPOT Wells Showing Spot-Forming Cells (SFCs) for IFN-γ and Granzyme B are Depicted for Each Treatment Group in Both Cell Lines.
In HER2-overexpressing SK-BR-3 cells, HER2-directed nanoliposomes co-encapsulating viral peptide and diABZI (STING agonist) markedly enhanced IFN-γ secretion compared to equivalent free agents (P < .01; Figure 9A). Notably, the unconjugated combination of viral peptide and diABZI paradoxically suppressed IFN-γ responses by over half relative to liposomal delivery (P < .001; Figure 9A), indicating HER2-dependent interference in free drug interactions. In contrast, HER2-negative MCF-7 cells exhibited robust IFN-γ activation with combinatorial nanoliposomes, achieving responses threefold above baseline (P < .001; Figure 9B). These differential outcomes underscore the critical role of HER2-targeted delivery in bypassing receptor-mediated signaling conflicts, as HER2-dense SK-BR-3 cells experience competition between viral peptide-driven antigen presentation and diABZI-activated STING pathways when drugs are unformulated. Liposomal co-delivery spatially synchronized these processes within endosomal compartments, resolving kinetic discordance inherent to free drug combinations.

Combinatorial Liposomal Therapy Enhances IFN-γ Secretion in HER2-Positive (SK-BR-3) and HER2-Negative (MCF-7) Cells. (A) SK-BR-3 Cells Treated with LPs (Viral Peptide + diABZI) Showed a Significantly Higher IFN-γ+ Response Versus Free Agents (P < .01). Free Combination Therapy Significantly Reduced the Response (P < .001). (B) MCF-7 Cells Exhibited Maximal IFN-γ Activation with Combinatorial LPs (P < .001 vs Control). Data are Normalized to 105 Cells; Error Bars Represent the Standard Error of the Mean (SEM) (n = 3). Statistical Analysis was Performed Using one-way ANOVA with Tukey's post-hoc Test.
Cell-type-specific cytotoxic responses further validated this mechanism. While SK-BR-3 cells showed modest granzyme B elevation with combinatorial nanoliposomes (P < .05; Figure 10A), MCF-7 cells demonstrated fourfold-enhanced granzyme B secretion (P < .001; Figure 10B), reflecting HER2-independent susceptibility to STING-driven innate immunity. This divergence highlights how HER2 receptor density dictates delivery system requirements: SK-BR-3 cells necessitated receptor-targeted nanoliposomes to circumvent HER2-mediated interference with cytosolic signaling crosstalk, whereas MCF-7 cells broadly responded to combinatorial immunotherapy regardless of targeting.

Cell-Type-Specific Granzyme B Induction Under Combinatorial Therapies. (A) SK-BR-3 Cells Showed Moderately Elevated Granzyme B+ Levels with LPs (P < .05 vs Control). (B) MCF-7 Cells Achieved a Significantly Higher Granzyme B Secretion with Combinatorial LPs (P < .001 vs control). Statistical Analysis was Performed Using One-Way ANOVA with Tukey's post-Hoc Test.
Collectively, HER2-directed nanoliposomes eliminated antagonistic interactions observed with free drug combinations by ensuring temporally coordinated intracellular release. The inverse efficacy profiles between formulated and unformulated therapies emphasize that pharmacokinetic harmonization through receptor-targeted delivery is indispensable for overcoming biological barriers in HER2-positive tumor models.
Immune Cell Analysis by Flow Cytometry
In our study, we investigated the immunomodulatory effects of lipopeptides (LPs) [viral peptide + diABZI] on SKBR3 cells, a human breast cancer cell line. The experimental design included two distinct groups to assess these effects through flow cytometry analysis. Group A consisted of SKBR3 cells cocultured with peripheral blood mononuclear cells (PBMCs) for three days and served as the control group. Group B included SKBR3 cells treated with LPs (viral peptide + diABZI), followed by three days of coculture with PBMCs, prior to flow cytometry analysis.
The flow cytometry results, as illustrated in Figure 11, highlighted notable differences in immune cell dynamics between the two groups. Group A, the control, presented a CD3+ T-cell prevalence of 78.63% among lymphocytes. Conversely, in Group B, where the cells were treated with the lipopeptide formulation, there was a significant increase in CD3+ T cells, accounting for 90.85% of the lymphocytes. This increase suggests that an augmented T-cell response is potentially triggered by LP treatment. Additionally, the percentage of CD8 + cytotoxic T cells in Group B increased to 50.9%, which was markedly greater than the 0.67% observed in Group A, indicating pronounced cytotoxic immune activation. Conversely, the percentage of helper CD4+ T cells decreased from 31.02% in Group A to 18.39% in Group B, indicating a strategic shift toward cytotoxic T-cell responses over helper T-cell responses in the presence of LP. Natural killer (NK) cell activity also displayed significant alterations, with increases in both the NK and NKT-like cell populations in Group B. These changes underscore an enhanced innate immune response, which is crucial for the initial defense against potentially transformed cells harboring malignancies.

Immune Cell Profiles in SKBR3 Cell Cocultures. A (Control): SKBR3 Cells Were Cocultured with PBMCs for Three Days Without Treatment. the Immune Cell Profile Revealed 78.63% CD3+ T Cells, of Which 30.67% Were CD8 + Cytotoxic T Cells and 31.02% Were CD4 + Helper T Cells. Additionally, 6.44% of Cells Were CD19+ B Cells, and a Total of 5.38% Were CD56+ NK Cells (Comprising 1.56% NK Cells and 3.82% NKT-Like Cells). B (Treatment): SKBR3 Cells Were Pretreated with Lipopeptides (Viral Peptide + diABZI) and Then Cocultured with PBMCs for Three Days. the Immune Cell Profile Showed 90.85% CD3+ T Cells, with 50.9% CD8 + Cytotoxic T Cells and 18.39% CD4 + Helper T Cells. the Proportion of CD19+ B Cells was 5.35%, and the Total CD56+ NK Cells Increased to 7.07% (Comprising 1.92% NK Cells and 5.15% NKT-Like Cells).
These results suggest that treatment with LPs (viral peptide + diABZI) in SKBR3 cells not only increases the immune system's cytotoxic capabilities but also potentially increases the efficacy of the delivered therapeutic agents. The observed shifts in immune cell distributions indicate pronounced activation of both innate and adaptive immune responses, which is pivotal for the successful implementation of nanoliposome-based drug delivery systems in cancer therapy. These findings demonstrate the potential of LPs to modulate immune responses favorably, enhancing cytotoxic functions while minimally impacting B-cell proportions.
LPs Enhances TCR β CDR3 Repertoire Diversity and Alters Clonality Compared to Controls
Basic analysis and clonality assessment of TCR β CDR3 repertoires were conducted to compare the diversity and distribution of clonotypes in the Control and LPs (viral peptide + diABZI) groups. The Control group contained 57 574 unique clonotypes, while the LPs group had 60 915 (Figure 12A). Both groups exhibited a similar CDR3 length distribution, with a peak at 15 amino acids (Figure 12B). Clonotype abundance analysis revealed lower diversity in the Control group, with a sharp decline in clonotype numbers as abundance increased. In contrast, the LPs-treated group showed a broader range of abundance, indicating greater diversity at lower clonotype frequencies, consistent with a trend toward an activated immune response (Figure 12C).

Analysis of T Cell Repertoire Diversity and Clonotype Profiles. (A) Number of Unique Clonotypes for 2 Samples. (B) Shows the Length Distribution of CDR3. (C) Presents the Clonotype Abundance Distribution. (D) Compares the Top Clonal Proportion and the Difference of Top Clonal Proportion with Specific Indices Between Control and LPs (Viral Peptide + diABZI). (E) Examines the Rare Clonal Proportion and the Difference of Rare Clonal Proportion with Specific Count Between Control and LPs (Viral Peptide + diABZI). (F) Provides a Statistical Analysis of the V Gene Family Usage in the Control and LPs (Viral Peptide + diABZI). (G) Displays the Chao1 Diversity Estimator and D50 Diversity Index Values. (H) Offers True Diversity Index Values and Inverse Simpson Index Values. (I) Depicts Rarefaction Curves for Control and LPs (Viral Peptide + diABZI), with the Interpolated and Extrapolated Regions Denoted bySolid and Dashed Lines, Respectively. (J) Features Representative Donut Charts of TRB CDR3 Clones, Illustrating Clonal Expansion in Control and LPs (Viral Peptide + diABZI). The Fan-Shaped Area Indicates the Corresponding Clonal Frequency, where Radians of 1, 2+, and 3 + Represent the Total Frequency of CDR3 with 1, 2, and 3 or More Reads, Respectively. Q1–Q5 in the Second Layer Represents the Clonal Frequencies of T Cells per 20\% (from High to Low). Amino Acid Sequences are Shown for the Top Five Clones in the CDR3 Repertoires, with their Frequencies Indicated by the Radian of the Shape. The Larger the Radian, the Higher the Frequency of Expansion of a Specific TCR Clone. The Areas Labeled “1,” “2+,” and “3+” Represent the Summed Frequencies of T Cell Clones that had One, More than Two, and More than Three Sequence Reads, Respectively. The Proportion of “1” and “2+” Serves as an Important Indicator of T Cell Diversity.
Further clonality analysis revealed that the LPs group contained more rare clones, while high-frequency clones dominated the Control group (>50% of total clones) (Figure 12D-E). TRBV gene segment usage was broadly similar between groups, though certain gene segments, such as TRBV12-3 and TRBV5-1, were more prevalent in the LPs group, while TRBV29-1 was more abundant in the Control group (Figure 12F). This suggests that LPs stimulation induces selective changes in TRBV gene usage, potentially reflecting antigen-driven alterations in the T-cell receptor repertoire.
To further evaluate the repertoire diversity, the TCR β CDR3 repertoires of the Control and LPS (Viral Peptide + diABZI) groups were analyzed using a combination of Chao1, D50, true diversity index, and Inverse Simpson index (Figure 12G-H). Although statistical significance was not reached (P > .05), all four diversity indices consistently showed a directional increase in the LPS group compared to the Control group. This lack of significance may reflect limitations in sample size or the magnitude of biological effect, but the concordant trends across multiple metrics suggest that LPs stimulation enhances clonotype diversity. Rarefaction plots (Figure 12I) further supported this observation: while the control group plateaued at larger sample sizes, the LPS group exhibited a continued increase in estimated clonotype diversity, indicating a broader yet unexhausted repertoire.
These findings collectively imply that LPs treatment promotes a shift toward a more diverse TCRβ repertoire, characterized by reduced clonal dominance and expansion of low-frequency clones. While the absence of statistical significance in some indices warrants caution in interpretation, the consistency of trends across complementary analytical approaches (abundance curves, clonality distribution, and rarefaction dynamics) aligns with the hypothesis that LPs stimulation broadens the T-cell response.
Discussion
Our research addresses the significant challenges posed by “cold” tumors, which are characterized by a scarcity of neoantigens, low immunogenicity, and resistance to conventional immunotherapies. By developing a novel nanoliposome formulation containing viral peptides and the STING agonist diABZI, our goal is to induce antigen epitope spreading in tumor cells, enhance immune recognition, and stimulate a more effective immune response against these tumors. The targeted liposomes we developed have shown great potential in promoting the expansion of tumor neoantigen epitopes, increasing their immunogenicity, and improving the immune microenvironment. However, despite the encouraging results, several aspects of this therapeutic strategy require further investigation and refinement to achieve clinical applicability.
The liposomal formulation was meticulously designed using a cholesterol-to-lipid ratio of 1:4 and a drug-to-lipid ratio of 1:8 and was fabricated through the thin film dispersion method. This design ensures the production of nanoscale, uniform, and stable particles with a consistent size distribution, which is crucial for effective drug delivery. The surface of the liposomes was covalently conjugated to the HER2 monoclonal antibody F(ab’)2 fragment, allowing precise targeting of HER2-positive breast cancer cells. 12 This targeting capability not only optimizes drug delivery to the tumor site but also minimizes off-target effects and systemic toxicity, 13 which is a major challenge with traditional chemotherapy and combination treatments. This targeted approach reflects the shift in cancer therapy toward precision medicine, where maximizing efficacy while reducing adverse effects is paramount. 14
Our findings demonstrated that these engineered liposomes successfully induced the expansion of antigen epitopes, a key mechanism for enhancing the immune recognition of tumor cells. Through immunological assays such as ELISPOT and flow cytometry, we observed elevated levels of IFN-γ- and granzyme B-positive cells, which are indicative of heightened cytotoxic immune activity. Meanwhile, TCR sequencing revealed a significant increase in the number of activated TCR clone types after the nanoliposomes bound to the tumor, indicating that the targeted nanoliposomes induced substantial antigen epitope spreading. Additionally, we detected increases in B lymphocytes, NK cells, and NKT-like cells, further supporting the enhanced immune response. These results indicate that using viral peptides as dominant antigens, combined with the activation of the cGAS-STING pathway mediated by a STING agonist, generated a strong immune response specifically targeting the viral peptides within tumor cells. This, in turn, activated the expansion of tumor neoantigen epitopes, leading to the exposure and formation of more neoantigens, which enhanced their immunogenicity. This approach effectively stimulates both innate and adaptive immune responses. This dual activation could play a critical role in overcoming the immunosuppressive microenvironment of cold tumors.
Despite the strong preclinical data, there are several limitations to our study that warrant discussion. First, the experiments were conducted in cell line models, which, while useful for preliminary assessments, cannot fully capture the complexity of in vivo tumor–immune interactions. 15 Future studies must prioritize testing in animal models not only to evaluate the efficacy of liposomes in a more complex biological environment but also to assess their pharmacokinetics, biodistribution, and potential off-target effects. Animal studies will also be critical for determining whether the immune responses observed in vitro translate to significant tumor regression and improved survival rates. 16
Additionally, while our study focused on HER2-positive breast cancer, it is important to consider the broader applicability of this liposomal formulation to other cancer types, particularly those with cold tumor characteristics. Investigating the potential to modify the targeting mechanisms of different tumor-specific markers could expand the versatility of this therapeutic strategy. For example, targeting other antigens expressed in immunotherapy-resistant cancers, such as PD-L1 or Epidermal Growth Factor Receptor (EGFR), could open new avenues for treating cancers with low immunogenicity. 17
Another important consideration is the interaction between the liposomal components and the immune system. Although our study demonstrated the immunostimulatory potential of the STING agonist diABZI, further research is needed to explore potential immune system-related side effects, such as excessive inflammation or autoimmunity, which could limit the clinical application of this approach. Understanding the long-term effects of STING activation and how to fine-tune the immune response will be crucial for optimizing this therapy. 18 Additionally, identifying the optimal dose of STING agonists and viral peptides for achieving a therapeutic balance between efficacy and safety is essential.
Finally, future research must explore patient-derived tumor models to validate the translational potential of our findings. The incorporation of such models would enable a more accurate assessment of the effectiveness of therapy in the complex, heterogeneous TME found in actual patients. 19 Furthermore, analyzing the impact of this therapy on the immune landscape of patient-derived tumors, such as changes in immune cell infiltration and the tumor immune profile, could provide critical insights into how cold tumors are reprogrammed into hot tumors in a clinical context. 20
In conclusion, our novel liposomal formulation incorporating viral peptides and the STING agonist diABZI represents a promising strategy for overcoming the challenges associated with cold-induced tumors by enhancing immune recognition and response. While our preclinical findings provide a strong foundation, further research involving animal models, patient-derived tumor cells, and a deeper understanding of liposome–immune interactions will be critical to translate this approach into a viable clinical therapy. By addressing these challenges and expanding our research, we hope to contribute to the development of more effective and safer cancer immunotherapies capable of transforming the treatment landscape for patients with immunotherapy-resistant tumors.
Conclusion
Our study presents a HER2-targeted nanoliposome platform co-delivering viral peptides and the STING agonist diABZI to reprogram immunosuppressive “cold” tumors into immunologically active states. Through precise HER2 targeting via trastuzumab-derived F(ab’)2 fragments, the formulation achieved tumor-specific uptake with peak fluorescence intensity at 60 µL in HER2-positive SK-BR-3 cells, while minimizing off-target effects in HER2-negative models. By combining viral peptides as dominant neoantigen surrogates with diABZI-mediated STING pathway activation, the platform synergistically enhanced both innate and adaptive immunity. This dual mechanism was evidenced by 2.5-fold elevated IFN-γ secretion (P < .01), 3-fold granzyme B induction (P < .05), and broadened TCR β repertoire diversity characterized by reduced clonal dominance—all indicative of enhanced tumor antigen recognition.
Notably, the nanoliposomes demonstrated robust stability (5% particle size variation over 25 days) and high encapsulation efficiency (75.5% for viral peptides), with sustained drug release profiles (52% cumulative release at 48 h) under physiological conditions. Flow cytometry further revealed a dominant cytotoxic immune response in HER2+ tumors, where CD8+ T cells accounted for 50.9% of infiltrating lymphocytes (vs 0.67% in controls), alongside expanded NK/NKT cell populations, collectively overcoming microenvironment immunosuppression.
Despite these advances, clinical translation requires addressing critical limitations. First, in vivo validation of pharmacokinetics and tumor regression in animal models is essential to confirm therapeutic efficacy beyond cell-line studies. Second, optimizing the dosing balance between STING-mediated immunogenicity and inflammation risks remains crucial for safety. Finally, expanding this platform's applicability to other cold tumors—through alternative targeting ligands such as PD-L1 or EGFR—could unlock broader therapeutic potential. Our study establishes a blueprint for precision immunotherapy against resistant cancers, bridging nanomedicine design with immune microenvironment remodeling to transform the treatment landscape.
Footnotes
Abbreviations
Author Contributions
QS contributed to the study design, experiments, and writing of the initial draft. YZ and DLZ collected the data and helped perform some of the experiments. LLY and SYZ assisted in the experimental design and helped with the data collection. CXH contributed to the study coordination, technical issues and revision of the manuscript. All the authors read and approved the final manuscript. All authors reviewed the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article:
Development of HER2-Targeted Nanoliposomes Loaded with Novel Dominant Neoantigen Peptides for Gastric Cancer and Investigation of Their Anti-Tumor Immune Effects, Hangzhou Science and Technology Development Program Project, (grant number A20251602, 202004A21).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.