
Abstract
Introduction
Non-small cell lung cancer (NSCLC) is the most prevalent and lethal subtype of lung cancer. Most patients are diagnosed at an advanced stage of the disease, resulting in a poor prognosis. Early treatment and clinical intervention for NSCLC following early diagnosis can improve patients’ survival rate. It is of considerable significance to develop a more efficient and precise approach for identifying key genes and clinically pertinent biomarkers in NSCLC to enable its early diagnosis.
Methods
An interpretable two-stage analytical framework integrated with advanced artificial intelligence (AI) technology is proposed to enhance the accuracy of biological gene screening for NSCLC. Firstly, gene-level statistical features derived from the GSE19804,GSE30219 and GSE33532 datasets are standardized and dimensionally reduced via principal component analysis (PCA), which reveals two distinct linear distribution patterns of candidate genes in the PCA projection space. Subsequently, these candidate genes are validated using the TCGA and GEPIA platform by evaluating their differential expression profiles and associations with patient survival outcomes, with the goal of identifying robust predictive biomarkers.
Results
Through AI-driven analytical pipelines, multiple tumor-associated genes are screened and confirmed to be correlated with NSCLC progression. Notably, ADGRD1 (Adhesion G Protein-Coupled Receptor D1) exhibits a close association with pulmonary physiological functions and may serve as a potential biomarker in the initiation and progression of NSCLC.
Conclusion
The proposed method combines unsupervised structural discovery with cross-cohort clinical evidence to prioritize NSCLC biomarkers, providing critical support for early diagnosis, prognostic stratification, and biomarker-guided therapeutic strategies. Furthermore, the study provides technical support for biomarker discovery in other cancer types, and highlights the application value of integrating computational intelligence with oncology research.
Keywords
Introduction
Lung cancer accounts for a substantial proportion of cancer-related morbidity and mortality globally, imposing an enormous burden on public health systems. 1 It is estimated that approximately 787,000 new cases of lung cancer are diagnosed in China each year, with more than 630,000 patients dying from the disease, ranking first globally in both incidence and mortality rates. 2 Among these cases, 85% of lung cancers are non-small cell lung cancer (NSCLC), which mainly includes lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC). 3 As the incidence and mortality rates of NSCLC continue to rise, research on its early screening and prognostic assessment has become a pivotal focus in the field of lung cancer research and several advancements have been made. Early detection and diagnosis of lung cancer can reduce its mortality rate. Currently, early lung cancer detection technologies are evolving toward diversification and non-invasiveness, mainly including traditional imaging detection, non-invasive breath testing technology, liquid biopsy technology, and biomarker detection.4–6 However, with the 5-year overall survival rate remaining between 15% and 20%, the clinical prognosis remains dismal. This poor outcome is primarily attributed to the fact that approximately 70% of patients are diagnosed at an advanced stage, where radical surgical resection is no longer feasible and the efficacy of systemic therapy is significantly limited.7,8
To address this challenge, exploring the key genes involved in the initiation and progression of NSCLC has become imperative. The advancement of high-throughput sequencing technology and the accumulation of large-scale genomic, transcriptomic, and epigenomic datasets have provided robust support for deciphering the complex molecular landscape of NSCLC. Previous studies have identified driver mutations in key oncogenes and tumor suppressor genes, including EGFR, KRAS, TP53, ALK, and ROS1.9–13 These biomarkers are crucial for understanding the pathogenesis and immunotherapeutic responses of NSCLC. Their discovery has ushered in the era of precision oncology and accelerated the development of molecular targeted therapies, such as EGFR tyrosine kinase inhibitors (TKIs), ALK inhibitors, and immune checkpoint inhibitors. 14
However, the tumorigenesis and progression of NSCLC are driven by the complex interplay of multiple factors, including gene mutations, epigenetic modifications, transcriptional dysregulation, and tumor microenvironment perturbations. 15 Although single-gene analysis approaches have demonstrated utility, they fail to fully capture the complexity of intergenic interactions and pathway crosstalk that underpin malignant phenotypes. Therefore, there is an urgent need for comprehensive research strategies that integrate multi-dimensional genomic data to identify reliable biomarkers and key regulatory genes capable of guiding early diagnosis, prognostic evaluation, and personalized treatment. 16 To achieve a more comprehensive identification of NSCLC-related key genes, we have incorporated AI technology—a transformative tool in biomedical research that excels in processing and interpreting high-dimensional, complex biological data. In particular, AI-driven biomarker discovery exerts a remarkably positive effect on lung cancer screening. For example, in the field of non-invasive detection, Binson et al proposed an electronic nose system for analyzing volatile organic compounds (VOCs) in exhaled breath. Combined with machine learning algorithms, this system can effectively distinguish lung cancer patients from those with chronic obstructive pulmonary disease (COPD) and healthy individuals, providing a novel approach for non-invasive early disease screening. 17 Their subsequent research further optimized the system, focusing on lung cancer staging detection and verifying the application potential of this method in assessing disease progression. 18 In the domain of precise subtyping, Dwivedi et al constructed an interpretable AI deep learning framework, which screens clinically meaningful biomarker sets for NSCLC from high-dimensional gene expression data, facilitating the differentiation between LUAD and LUSC subtypes. 19 At the level of prognostic prediction, Fang et al innovatively integrated knowledge graphs with machine learning. By extracting graph embedding features, this integration compensates for the insufficiency of gene panel data and significantly enhances the stability and reliability of survival risk prediction for NSCLC patients. 20 Compared with traditional statistical methods, AI exhibits superior performance in capturing non-linear relationships and hidden patterns within genomic datasets, providing strong support for comprehensive molecular characterization of NSCLC.21–29
The objective of this study is to establish a reliable computational framework for molecular diagnosis of NSCLC and biomarker discovery by integrating AI approaches with the analysis of public gene expression datasets (GSE19804/GSE30219/ GSE33532/TCGA and GEPIA), and constructing a gene classification and prediction model. This study proposes an AI-based method that combines unsupervised structural discovery with cross-database validation to prioritize the screening of NSCLC biomarkers. After applying the proposed method, multiple NSCLC-related genes identified in previous studies are validated; additionally, ADGRD1—a gene previously understudied in the context of NSCLC—is newly identified. ADGRD1 is one of the group V members of the adhesion G protein-coupled receptor family. Current studies have shown that ADGRD1 is highly expressed in acute myeloid leukemia (AML) and is closely associated with shorter survival time in patients. Furthermore, studies have found that ADGRD1 is highly expressed in the hypoxic regions of malignant gliomas and is regulated by the HIF-αtranscription factor, thereby promoting the occurrence and progression of gliomas. These studies suggest that ADGRD1 functions as an oncogene. ADGRD1 participates in tumor cell differentiation, proliferation, invasion, infiltration and tumor growth. It is abnormally expressed in glioblastoma, oral squamous cell carcinoma, and other tumors, and correlates with the prognosis of oral squamous cell carcinoma patients. Additionally, ADGRD1 is highly expressed in acute myeloid leukemia and closely associated with patients’ survival time. 30 We conduct an in-depth investigation into the correlation between ADGRD1 expression in NSCLC and patient prognosis. ADGRD1 is lowly expressed in lung cancer and linked to patients’ poor prognosis, indicating that ADGRD1 may exert a tumor suppressor role in lung cancer. Therefore, exploring the causes of ADGRD1 downregulation in lung cancer is essential, and its regulatory mechanism may provide a crucial basis for lung cancer treatment targeting ADGRD1. 31 This discovery is expected to advance the development of early diagnostic strategies and personalized treatment regimens, ultimately improving clinical outcomes. Furthermore, this study provides technical support for biomarker discovery in other cancer types, highlighting the value of integrating computational intelligence with oncology research.
Data Sources and Method
Data Sources
This study integrates public gene expression datasets to conduct a comprehensive analysis of the molecular characteristics associated with NSCLC. The data are primarily obtained from the following two sources:
This data integration feature ensures that the gene information used in the study is both comprehensive and reliable, so it was employed as the validation dataset.
AI-Based Processing Method
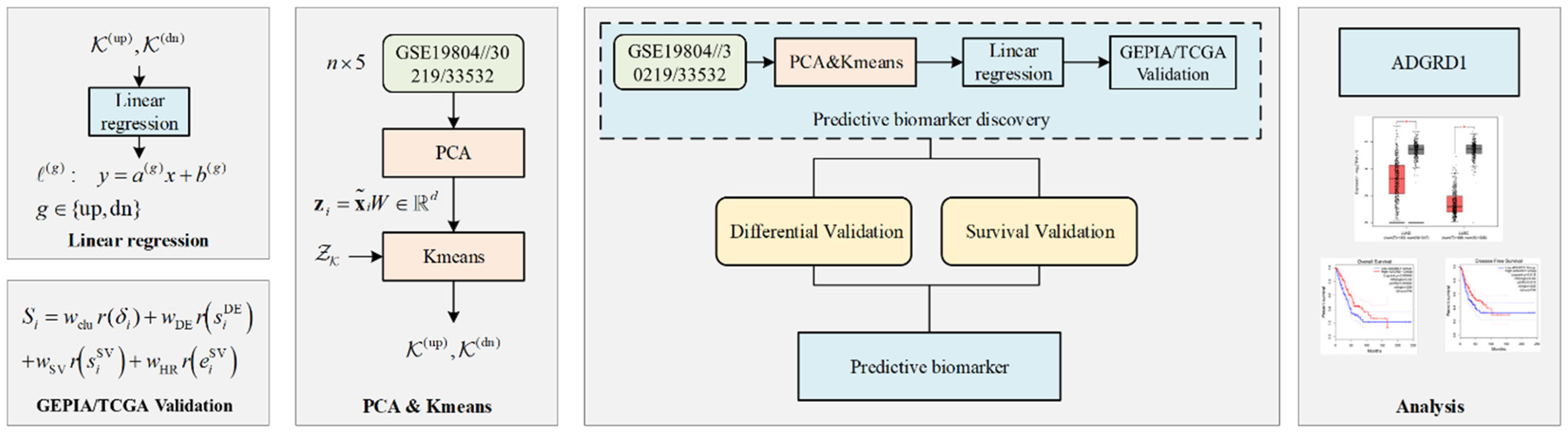
In this study, GSE19804//30219/33532 datasets were used as the core discovery datasets. By combining AI technology with bioinformatics analysis, a two-stage “screening-verification” research framework was constructed. The overall research followed the logical sequence of “AI model-based candidate gene screening → GEPIA platform-based validation → predictive biomarker identification”.32,33 The ultimate goal was to identify disease-related biomarkers with clinical predictive value, and the specific protocol is as follows:
A. Discovery Stage: AI Model-Based Candidate Gene Screening First, preprocessing was performed on GSE19804/30219/33532 datasets, including data normalization, missing value imputation, and quality control of the gene expression matrix, to ensure reliable input for the predictive model. A linear regression model based on artificial intelligence was employed to construct predictive models using gene expression levels as input features. Clinical endpoints, such as disease stage and prognostic status, were clearly defined as indicators of patient outcomes: disease stage was represented as categorical variables reflecting tumor progression, while prognostic status (eg, overall survival or disease-free survival) was quantified using patient survival time and event status, and summarized through statistical measures such as log-rank test p-values and Cox proportional hazards model hazard ratios (HRs). These clinical endpoints were integrated into the model as additional input features, enabling the linear regression model to simultaneously capture the relationships between gene expression and patient outcomes. This model was chosen because it can effectively handle continuous gene expression data while maintaining interpretability, allowing the regression coefficients to directly reflect the contribution of each gene to the prediction of clinical endpoints, and it is stable to train on a dataset with a moderate sample size, avoiding overfitting. To identify the most informative genes contributing to the prediction, feature selection was performed using principal component analysis (PCA), initially narrowing down the set of candidate genes for subsequent analysis. B. Verification Stage: GEPIA Platform-Based Dual Validation This stage utilized GEPIA (Gene Expression Profiling Interactive Analysis)—an online tool built on The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) databases—to perform differential expression verification and survival association verification of the candidate genes. C. Confirmation Stage: Predictive Biomarker Establishment By synthesizing the results of the above two stages, genes that simultaneously met the criteria of “high importance in the AI model, significant differential expression in GEPIA, and significant association with survival in GEPIA” were screened. These genes were ultimately established as biomarkers with clinical predictive value, providing core targets for subsequent mechanism research or the construction of diagnosis/prognosis models. The workflow is shown in Figure 1.
The framework of predictive biomarker discovery.
Data Preprocessing
A variety of statistical indicator features were extracted from the original GSE19804 dataset, including log fold change (

To ensure data comparability and avoid bias caused by scale differences, all features were normalized using Z-score standardization
34
:

Missing values were handled by imputation using the k-nearest neighbors (k-NN) algorithm to maintain dataset integrity. 35
Principal Component Analysis (PCA)
Let the Z-score–standardized data matrix be

Eigenvalue decomposition was performed, and the top d eigenvectors associated with the largest eigenvalues are retained to form:


The dimensional representation of a gene i is obtained by:

Unsupervised Clustering (K-Means)
In the PCA space

We select K by grid-searching

The value of K corresponding to the largest

A larger

Results of principal component analysis and linear regression.
On the PCA plane, known biomarkers were dichotomized and fitted with weighted linear regressions separately. The perpendicular distance to each fitted line was used as a similarity metric to screen potential candidates. Let the standardized, PCA-reduced coordinates be
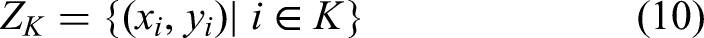
Run K-means with K = 2 on

To identify genes most consistent with these two linear relationships, define the near-line sets using the (Euclidean) perpendicular distance to each line. For all samples compute:
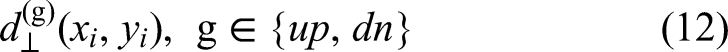
Take the q-th quantile (eg, q = 15%) as the threshold

Assign each sample in

The final set of near-line candidates is:

The set N is aligned across databases with GEPIA and incorporated into the composite scoring, yielding a list of candidate biomarkers that conforms to the two linear patterns and carries stronger statistical support.
It should be noted that the four-dimensional (4D) feature vectors constructed from statistical indicators (
Cross-Database Validation with GEPIA
Based on GEPIA (Gene Expression Profiling Interactive Analysis), we quantified candidate selection using a differential-expression score and a survival score.
Differential expression (DE) score can be denoted as:

Using the gene's log-rank value and hazard ratio (HR), the survival score can be denoted as:

Given three classes of measures—cluster representativeness

Then we composite score as below:

Genes are ranked in descending order of

To ensure the reliability and specificity of candidate gene screening, the following threshold criteria were applied for the dual validation in this study:
Differential Expression Validation Threshold:
Consistent with the differential gene screening criteria of the GSE19804/GSE30219/GSE33532 discovery dataset (| The absolute value of
Survival Association Validation Threshold:
Genes were determined to be significantly associated with patient survival outcomes if they satisfied the following statistical criteria in GEPIA: (1) For both overall survival (OS) and disease-free survival (DFS), the log-rank test P-value was less than .05, meaning the survival curve difference between the high-expression and low-expression groups of the gene was statistically significant; (2) The hazard ratio (HR) calculated by the Cox proportional hazards model was not equal to 1. Among them, HR < 1 indicated that high gene expression was a protective factor for patient survival (reducing the risk of death or recurrence), while HR > 1 indicated that high gene expression was a risk factor.
Only genes that simultaneously met the above two sets of validation thresholds and had high feature importance in the AI model were included in the final list of predictive biomarkers, ensuring that the screened genes not only had significant molecular-level expression differences but also closely correlated with clinical prognosis, providing reliable targets for subsequent translational research.
AI Model and Training Details
A linear regression model based on artificial intelligence (AI) was employed to predict clinical phenotypes from gene expression data. The input features to the model were derived from standardized 4D statistical vectors for each gene, which included log fold change, p-values, adjusted p-values, and t-statistics. These feature vectors were first reduced via principal component analysis (PCA), retaining the top principal components that captured the majority of variance. Subsequently, K-means clustering was applied in the PCA space to identify gene clusters, and near-line filtering based on perpendicular distance to fitted lines was used to select candidate genes for modeling.
The linear regression model was trained using ordinary least squares (OLS) to minimize the mean squared error (MSE) between predicted and observed clinical outcomes (eg, disease stage, prognostic status). To enhance robustness and avoid overfitting, the model incorporated L2 regularization (ridge regression), with the optimal regularization parameter selected via 5-fold cross-validation. The training process involved randomly partitioning the dataset into 5 folds, iteratively using 4 folds for training and 1 fold for validation, and averaging the performance metrics across folds.
Model evaluation metrics included mean squared error (MSE), coefficient of determination (R2), and Pearson correlation coefficients between predicted and observed clinical phenotypes. Feature importance was assessed directly from the regression coefficients, which quantified the contribution of each gene to the predicted clinical outcome. Genes with higher absolute coefficients and those meeting dual validation criteria from GEPIA (differential expression and survival correlation) were prioritized as candidate biomarkers.
This workflow ensures full reproducibility of the AI modeling process, provides interpretable results for biological interpretation, and allows integration of both statistical and clinical information in a transparent manner.
Results
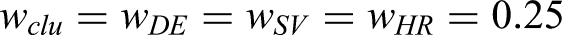
The findings were validated using Formulas 19 and 20 combined with the GEPIA database, and partial scoring results for key genes are presented in Figure 3.

Results of candidate biomarker screening.
Among the highest-scoring candidate genes, many have been previously confirmed to be associated with lung cancer36–42; in contrast, research on ADGRD1 (Adhesion G Protein-Coupled Receptor D1) remains relatively limited. Therefore, a focused analysis of this gene was conducted. In terms of differential expression, ADGRD1 expression levels were significantly lower in lung cancer tissues but higher in normal lung tissues (Figure 4), suggesting that this gene may play a critical role in NSCLC pathogenesis.

ADGRD1 expression in GEPIA.
Subsequently, the association between ADGRD1 expression in lung adenocarcinoma (LUAD) and patient survival rates was analyzed using the GEPIA database (Figure 5). The results showed that patients in the high ADGRD1 expression group had a significantly better prognosis than those in the low expression group. For overall survival (OS), the log-rank test yielded a p-value of .00048; the Cox proportional hazards model showed a hazard ratio (HR) of 0.59 (p = .00056), indicating a 41% reduction in the risk of death in the high-expression group. For disease-free survival (DFS), the log-rank test resulted in a p-value of .018, with an HR of 0.69 (p = .018), corresponding to a 31% decrease in the risk of recurrence/progression. Collectively, these data confirm that high ADGRD1 expression serves as a protective prognostic biomarker for NSCLC.

Prognostic relevance of ADGRD1 in LUAD: os and DFS (GEPIA).
We found high expression of ADGRD1 is significantly associated with shortened overall survival (OS) in patients and serves as a risk factor for OS; however, it shows no statistically significant association with disease-free survival (DFS) by the comparison of LUSC(Figure 6). This contrasts with the previous conclusion that ADGRD1 acts as a protective factor in lung adenocarcinoma (LUAD), demonstrating the heterogeneity of the prognostic role of this gene across different lung cancer subtypes.
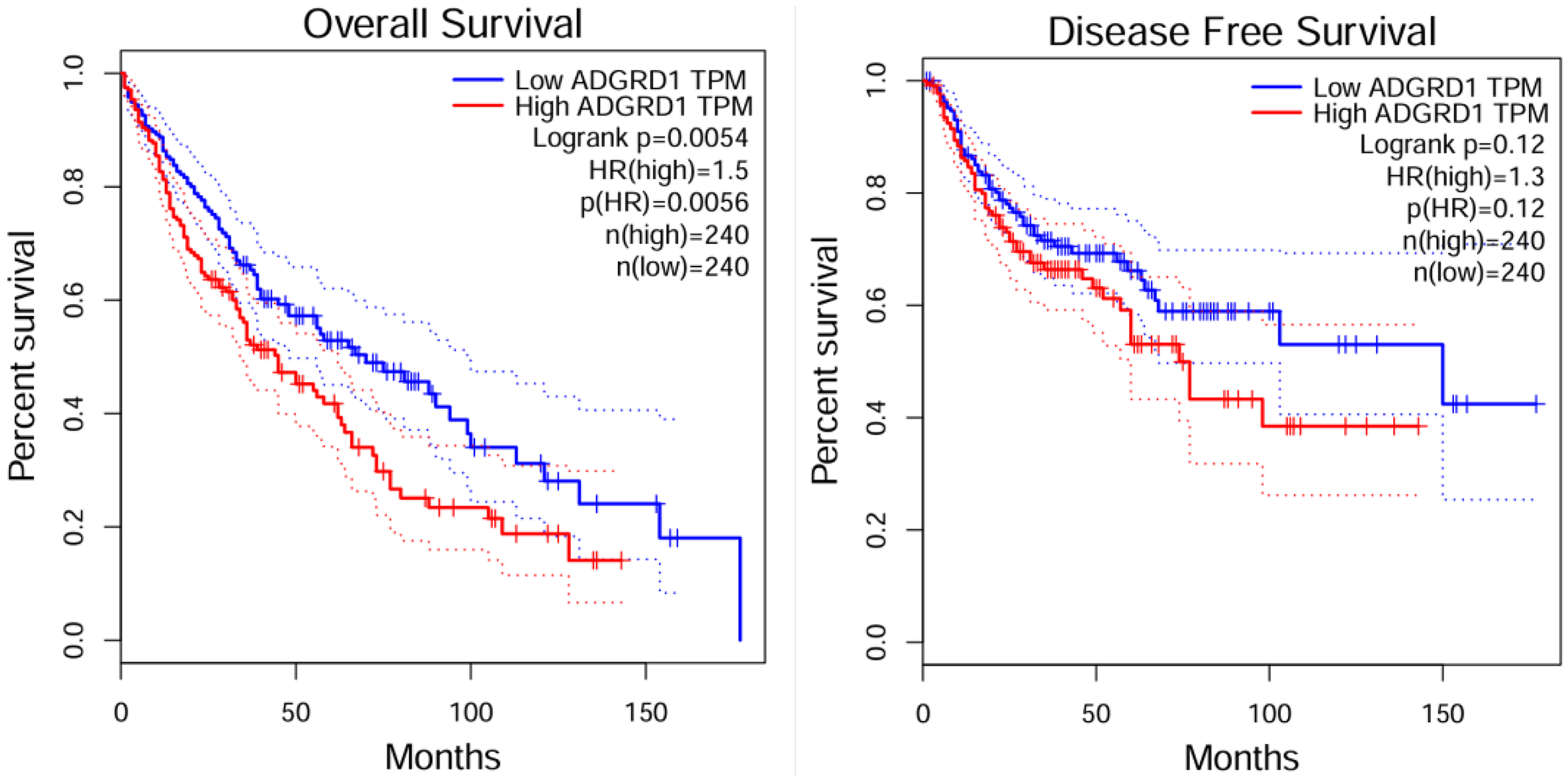
Prognostic relevance of ADGRD1 in LUSC: os and DFS (GEPIA).
In addition, existing literature has shown that both in LUSC and LUAD, the relative mRNA expression level of ADGRD1 is significantly lower than that in their corresponding adjacent normal tissues (P < .05); Kaplan-Meier survival analysis results indicated that the 10-year survival rate of lung cancer patients with high ADGRD1 expression is significantly higher than that of patients with low ADGRD1 expression (P < .001), suggesting that ADGRD1 may function as a tumor suppressor gene in lung cancer tissues, and its high expression is beneficial to patients’ prognosis. 43
A further comparison of analysis results from two independent human tissue expression resources—the Human Protein Atlas (HPA) and the Genotype-Tissue Expression (GTEx) Project—revealed that ADGRD1 exhibits lung tissue-enriched expression. As shown in Figure 7, ADGRD1 expression levels ranked among the highest in both resources and were significantly higher than those in most non-lung tissues. This lung tissue-specific enrichment, combined with the expression patterns observed in tumor datasets, indicates that ADGRD1 is closely associated with lung physiological functions, may play an important role in the initiation and progression of lung cancer, and highlights its potential as a NSCLC biomarker.
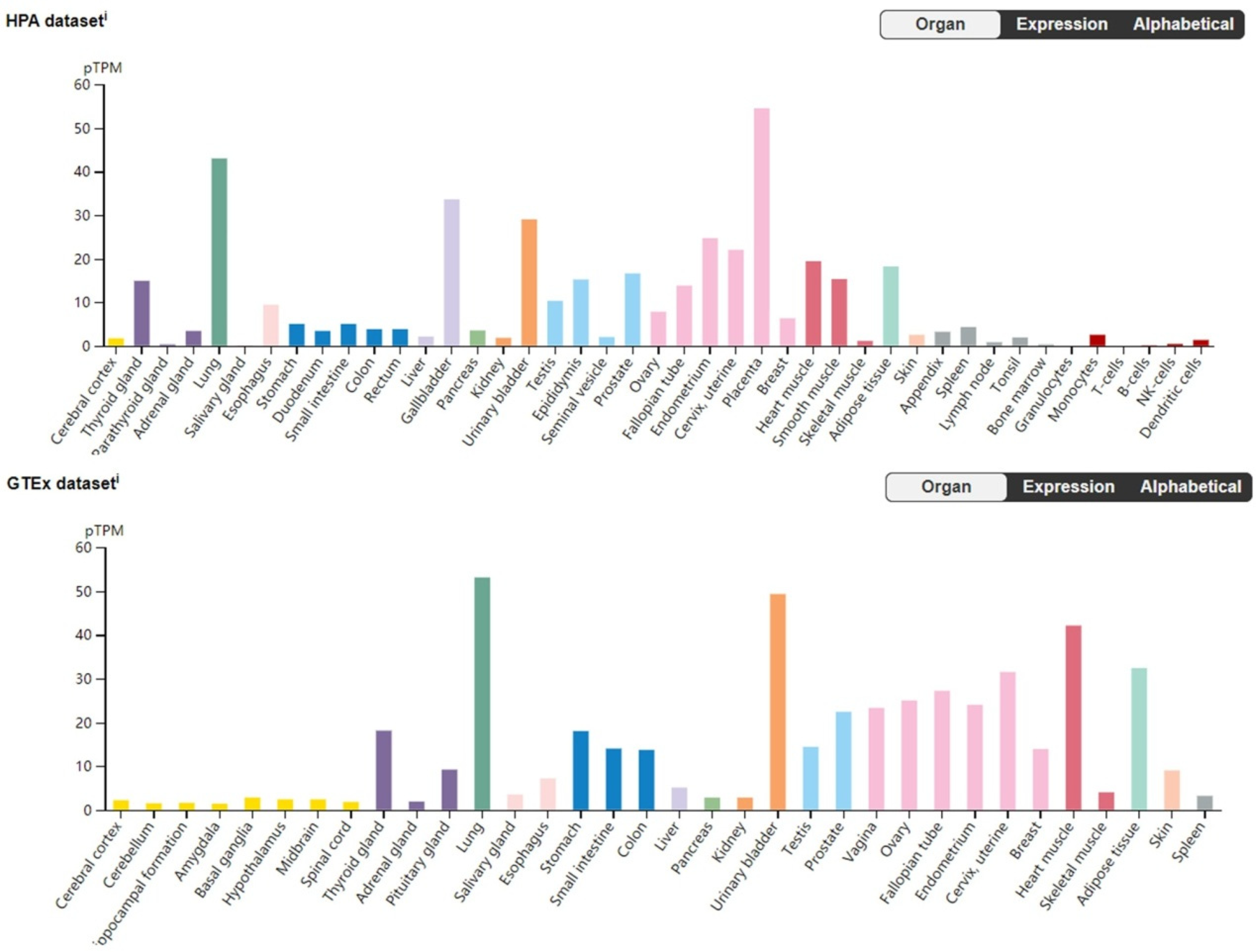
Comparative results across HPA and GTEx.
Discussion
This study proposes an AI-driven strategy for NSCLC biomarker discovery, which proceeds as follows:First, the overall tissue-specific molecular structure was identified using the Principal Component Analysis-K-means Clustering (PCA-K-means Clustering) method; second, a bilinear model combined with anchor-aware weighting was employed to fit known lung cancer-related genes, and a near-line filtering mechanism based on perpendicular distance was constructed; finally, cross-cohort data on differential expression and survival outcomes from GEPIA were integrated into an interpretable scoring system.
When extending this method to biomarker screening for other tumors in the future, several limitations should be considered. First, reliance on public datasets may introduce heterogeneity due to batch effects and incomplete clinical annotations; despite data standardization and harmonization, residual variability may still affect the generalizability of results. Second, the near-line criterion assumes the existence of an approximate linear manifold in the PCA space; although robust thresholds and anchor weighting reduce sensitivity to this assumption, results may still vary with the number of clusters and distance thresholds.
Conclusion
Through an AI-based dataset discovery and validation approach, this study found that multiple previously confirmed lung cancer-associated genes achieved high scores, demonstrating the reliability and accuracy of the proposed method. Additionally, during gene screening, it was discovered that ADGRD1 expression and its regulatory mechanisms may serve as prognostic indicators for NSCLC. The method proposed in this study provides a transparent and reproducible research pathway for prioritizing the screening of prognostic biomarkers and potential therapeutic targets for NSCLC.44,45
Footnotes
Abbreviations
Acknowledgements
The authors sincerely thank all public databases used in this study. They also express gratitude to the anonymous reviewers and associate editor for their valuable comments and suggestions.
Ethical Approval
This article does not involve any studies with human or animal subjects conducted by the authors.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Authors’ Contributions Statement
Xiaoyue Wang designed the study and drafted the manuscript; Na Liu and Shu Xu collected and analyzed the data; Ting Xu revised the entire manuscript. All authors have read and approved the final version of the manuscript and confirm that this manuscript has not been published previously and is not under consideration by another journal.
Data Availability
The original data used in this research are publicly available from online databases. All data supporting the findings of this study are included in the article. GSE databases: http://www.broadinstitute.org/gsea; GEPIA: (http://gepia2.cancer.pku.cn/); TCGA: ![]()