
Abstract
Triple-negative breast cancer (TNBC) comprises a group of aggressive and heterogeneous breast carcinoma. Chemotherapy is the mainstay for the treatment of triple-negative tumors. Nevertheless, the success of chemotherapeutic treatments is limited by their toxicity and development of acquired resistance leading to therapeutic failure and tumor relapse. Hence, there is an urgent need to explore novel targeted therapies for TNBC. Receptor tyrosine kinases (RTKs) are a family of transmembrane receptors that are key regulators of intracellular signaling pathways controlling cell proliferation, differentiation, survival, and motility. Aberrant activity and/or expression of several types of RTKs have been strongly connected to tumorigenesis. RTKs are frequently overexpressed and/or deregulated in triple-negative breast tumors and are further associated with tumor progression and reduced survival in patients. Therefore, targeting RTKs could be an appealing therapeutic strategy for the treatment of TNBC. This review summarizes the current evidence regarding the antitumor activity of RTK inhibitors in preclinical models of TNBC. The review also provides insights into the clinical trials evaluating the use of RTK inhibitors for the treatment of patients with TNBC.
Introduction
According to the GLOBOCAN 2020 estimates of cancer incidence and mortality, breast cancer is the most common cancer worldwide, excluding basal cell carcinoma. 1 It accounted for 23 million new cancer cases and 685 000 deaths in 2020 globally. 2 The burden of breast cancer is anticipated to increase to more than 3 million new cases and 1 million deaths in 2040. 3 Despite a common tissue of origin, breast cancer is highly heterogeneous comprising a spectrum of tumors with complex variations in biological behavior, pathological and clinical features, and response to therapy. Early in the 2000’s, Perou and colleagues revolutionized the understanding of breast cancer biology by introducing the molecular classification of the disease into distinct subtypes based on the gene expression profiles of tumors. 4 Accordingly, the main molecular subtypes identified of breast cancer included the luminal-like, the human epidermal growth factor receptor 2 (HER2)-positive, the basal-like, and the normal-like cancers. 4 Luminal breast cancers are characterized by the expression of the estrogen receptor (ERα) gene and luminal epithelial cell keratins 8 and 18. 4 This subtype is further divided into two distinct subgroups based on gene expression profile. 5 The luminal subtype A tumors revealed the highest expression of the ERα gene while luminal subtype B tumors displayed low to moderate expression of the luminal-specific genes including the ER cluster. 5 The HER2-positive subtype is characterized by the amplification of the ErbB2 gene. 4 The basal-like subtype is known for the high expression of laminin and the basal cell keratins 5 and 17. 4 Besides, the basal-like subtype is characterized by a high frequency of TP53 mutations.5,6 Both HER2-overexpressing and basal-like breast cancers do not express ER and most of its related genes. 4 Normal breast-like tumors demonstrate high expression of genes expressed by adipose and basal epithelial cells. These tumors also showed low expression of luminal epithelial genes.4,5 The different molecular subtypes are associated with remarkable differences in patient outcomes. The HER2-positive and the basal-like subtypes are correlated with poor prognosis and the shortest survival times compared to other molecular subtypes.5,6
Triple-Negative Breast Cancer
Triple-negative breast cancer (TNBC) is a distinct subtype of invasive breast cancer characterized by poor prognosis and lacks the expression of ER, progesterone receptor (PR), and HER2 overexpression.7,8 It accounts for 15%-20% of all breast cancer cases diagnosed. TNBCs represent an aggressive phenotype with a high level of invasiveness and biological heterogeneity. 9 TNBC is likely to metastasize to the central nervous system and the visceral organs such as the liver, lungs, and bones. 10 This subtype is frequently diagnosed in women under 40 years of age and has a mortality rate of 40% within the first 5 years of diagnosis. 11 Besides, TNBC is associated with an early recurrence rate in 25% of cases following surgery and a short median survival time after metastasis.12,13 Although the majority of TNBCs share huge similarities with the genetically defined basal-like molecular subtype, the two terms, however, are not biologically synonymous.8,14 In this regard, not all TNBCs show a basal-like phenotype indicated by a strong expression of basal markers such as cytokeratins 5 and 17. 8 Claudin-low breast cancer represents a subgroup of triple-negative invasive ductal tumors that are characterized by the low expression of luminal differentiation markers and enrichment of epithelial-to-mesenchymal transition (EMT) markers and stem cell-like features.15,16 The claudin-low carcinomas are distinctive by the low expression level of critical cell-to-cell adhesion molecules such as claudins 3, 4, and 7, occludin, and E-cadherin. 16
Classification of TNBC
In 2011, the gene expression pattern of tumor samples from 587 cases of TNBC was analyzed, revealing six main subtypes of TNBC with unique gene expression profiles. 17 These subtypes included the basal-like 1, basal-like 2, mesenchymal-like (MES), immunomodulatory (IM), mesenchymal stem-like, and luminal androgen receptor (LAR) subtypes. 17 Subsequently, transcriptome profiling analysis of 198 TNBC tissues was conducted proposing four distinct molecular subtypes of TNBC: basal-like immunosuppressed (BLIS), basal-like immune-activated, MES, and LAR subtypes. 18 Liu et al identified four new subtypes by using transcriptome microarrays integration of both long non-coding and messenger RNAs in 165 samples of TNBC as follows: BLIS, MES, IM, and LAR subtypes. 19
The BLIS Subtype
The BLIS subtype is the most prevalent subtype of TNBC. It represents highly proliferative cancer cells due to their enrichment with genes associated with cell division and cell cycle pathways. 19 Additionally, the BLIS subtype is characterized by the downregulation of genes regulating innate immune response pathways.18,19 Patients harboring BLIS tumors are at high risk of recurrence and reduced overall survival (OS).18–20
The MES Subtype
The mesenchymal-like (MES) subtype is characterized by the downregulation of genes related to cell proliferation. 19 It demonstrated a unique cluster of genes regulating interaction with the extracellular matrix, EMT, and growth factor signaling pathways.17,19 Compared to the other subtypes of TNBC, the MES tumors overexpress the ‘Inducing Angiogenesis’ hallmark of cancer cells rendering them of a higher angiogenic capability. 21
The IM Subtype
The IM subtype is saturated with genes primarily connected with immune cell signaling pathways. The upregulation of such genes is tightly linked to immune responses such as innate immune function and the stimulation of T lymphocytes.17,19 Notably, genes associated with immune checkpoints are highly expressed in IM tumors. These include the programed cell death 1 (PD-1) protein and programed cell death ligand 1 (PD-L1) which together are considered poor prognostic predictors.21,22
The LAR Subtype
The LAR subtype accounts for 10%-22% of TNBC. 23 Interestingly, the estrogen signaling pathway was found to be upregulated in this subtype. LAR is highly enriched with genes involved in hormone signaling pathways regulating biosynthesis and metabolism of steroid hormones. It is also characterized by the upregulation of androgen receptor (AR) and its related signaling pathways with a 10-fold greater expression of AR compared to other TNBC subtypes.17,19,24
Treatment of TNBC
The treatment of TNBC has been a constant challenge to oncologists. Due to the lack of hormone receptors and HER2 overexpression, TNBC is unresponsive to endocrine and HER2-targeted therapies. 25 The treatment of TNBC is further complicated by the heterogeneous nature of the tumor and the risk of disease metastasis and recurrence. Therefore, chemotherapy remained the gold-standard systemic modality for the treatment of TNBC. 25 The National Comprehensive Cancer Network guidelines recommend the use of anthracyclines, taxanes, cisplatin, cyclophosphamide, and fluorouracil as the basis for the combination treatment regimens.20,26 Neoadjuvant chemotherapy is the standard of care and the preferred option for the treatment of early TNBC. 27 A substantial number of patients with TNBC achieved pathological complete response (pCR) using neoadjuvant chemotherapy. 28 Regularly, adjuvant chemotherapy is composed of anthracyclines, taxanes, and alkylating agents. Anthracycline or taxane-based adjuvant chemotherapy alone has limited benefits in TNBC treatment.29,30 A recent meta-analysis indicated that the addition of capecitabine to anthracyclines, with or without taxanes, improves response in adjuvant treatment in patients with TNBC. 31 Besides, the pCR is remarkably improved upon incorporating the platinum derivative carboplatin to paclitaxel-based regimens in adjuvant and neoadjuvant settings in early TNBC. 32
TNBC is characterized by lymphocyte infiltration into the tumor microenvironment and a high expression of PD-L1 in cancer cells. 33 The immune checkpoint inhibitors atezolizumab and pembrolizumab are currently approved for the treatment of TNBC in combination with chemotherapy.33,34 BRCA1 mutations are found in approximately 11%-20% of patients with TNBC. 35 Thus, poly-ADP-ribose polymerase (PARP) inhibitors such as olaparib and talazoparib have been approved for the treatment of HER2-negative patients with germ-line BRCA1/2 mutations. 36 Despite the availability of a few targeted therapies, chemotherapy remains the cornerstone treatment modality for patients with triple-negative carcinoma. However, the survival advantage reported with chemotherapy is mainly observed in patients with early-stage disease with limited effectiveness in advanced disease. 37 Additionally, chemotherapeutic drugs are associated with significant toxicities and the emergence of acquired resistance which together limit the usefulness of these drugs. 38 Hence exploring novel targeted therapy for the treatment of TNBC is urgently needed to expand treatment options and improve outcomes in patients. 39
Receptor Tyrosine Kinases
Tyrosine kinases are a family of enzymes that selectively catalyze the phosphorylation of tyrosine residues in different targeted substrates. 40 The phosphorylation by tyrosine kinases is an important regulatory mechanism of cellular activity and for signal transduction in response to intracellular and extracellular stimuli. Therefore, tyrosine kinases are key signaling molecules for the regulation of cell proliferation, differentiation, metabolism, and survival.41,42 The tyrosine kinase family comprises 90 enzymes that are classified into non-receptor and receptor tyrosine kinases (RTKs). 40 The RTKs are a conserved superfamily of cell surface receptors that transduce transmembrane signaling. 43 RTKs are expressed throughout the body tissues regulating several cellular processes during intrauterine development and throughout adulthood. In humans, there are 20 known RTK families comprised of 58 unique receptors. 44 RTKs are regularly classified based on the molecular basis of their natural ligands and their structural properties. 45 Table 1 lists the families of RTKs and their biological activities.
Classification of RTK Families with Their Members, Prototype Receptors, and Main Biological Functionalities

Abbreviations: AATYK, apoptosis associated tyrosine kinase; ALK, anaplastic lymphoma kinase; AXL, anex-elekto (uncontrolled), CCK; colon carcinoma kinase; DDR, discoidin domain receptor; EGFR, epidermal growth factor receptor; EPHR, ephrin receptor; ERBB, erythroblastic oncogene B; FGFR, fibroblast growth factor receptor; FLT3L, FMS-like tyrosine kinase 3 ligand; IGFR, insulin-like growth factor receptor; IR, insulin receptor; KIT, stem cell factor receptor; LMR, Lemur; LTK, leukocyte tyrosine kinase; M-CSFR, macrophage colony-stimulating factor receptor; MET, hepatocyte growth factor receptor; MUSK, muscle specific kinase; NGFR, nerve growth factor receptor; PDGFR, platelet-derived growth factor receptor; RET, rearranged during transfection; RON, recepteur d'origine nantais; ROR, receptor tyrosine kinase-like orphan receptor; RTK106, receptor tyrosine kinase 106; RYK, receptor related to tyrosine kinases; TEK, angiopoietin-1 receptor; TIE, tyrosine kinase receptor in endothelial cells; TRKA-C, tropomyosin receptor kinase A-C; TYRO3, protein tyrosine kinase 3; VEGFR, vascular endothelial growth factor receptor.
Structure and Activation of RTKs
Each RTK shares similar prototype structural protein domains along with receptor-specific characteristics. The prototype RTK consists of a ligand-binding extracellular domain and an intracellular cytoplasmic domain, separated by an alpha-helical single-pass hydrophobic transmembrane domain.44,46 The cytoplasmic domain has protein kinase activity containing regulatory juxta-membrane region, tyrosine kinase domain, and carboxyl-terminal tail region.44,46 The extracellular domains of RTKs are substantially variable in the different subfamilies. The subfamilies of RTKs are further characterized by structural diversity of the native conserved elements composing the extracellular region, including immunoglobulin-like or epidermal growth factor (EGF)-like domains, fibronectin type III repeats, or cysteine-rich regions. 45 Alternatively, the catalytic kinase domains display the highest level of conservation among the different RTK families. 41 Figure 1 is a general overview of the structure of an RTK and the subfamilies.
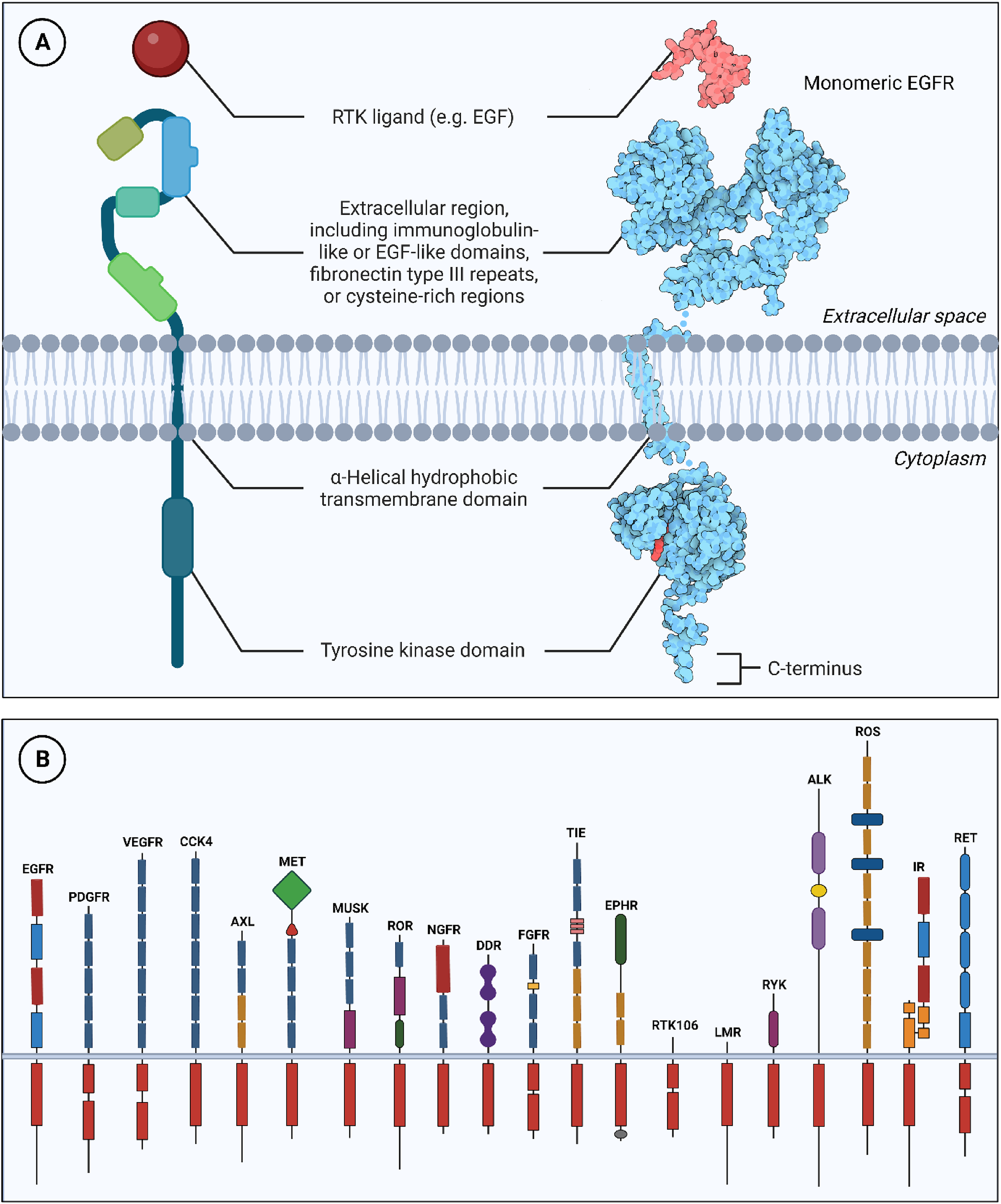
Prototype structure of an RTK illustrating extracellular, transmembrane, and intracellular domains. (a) The structure of the prototype receptor (EGFR) and its ligand (EGF) were obtained from the Protein Data Bank (PDB) under the accession entry of 2GS6 and 1EGF, respectively. The rendered three-dimensional structure of EGFR was obtained from Wikimedia Commons (File:126-Epidermal Growth Factor EGFR by David Goodsell under the Creative Commons Attribution 3.0 license). (b) The 20 RTK families and their corresponding structures. EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; RTK, receptor tyrosine kinase.
The activation of RTKs is mediated by ligand binding to the extracellular domain leading to the dimerization or oligomerization of receptors. 46 The dimerization results in trans-autophosphorylation of the tyrosine kinase domains, in which one subunit of the dimer phosphorylates the opposing subunit. 41 Upon tyrosine phosphorylation, the activation loop adopts an open conformation that gives access to ATP and substrates to catalyze the phosphorylation of specific tyrosine residues. 41 Autophosphorylation involves two different classes of tyrosine residues. One phosphorylation occurs on conserved tyrosine residues within the kinase domain of the RTK itself while the secondary autophosphorylation sites are regularly located outside the kinase domains and serve the fundamental function of creating docking sites for downstream transduction molecules containing Src homology-2 (SH2) and protein tyrosine binding (PTB) domains. 41 In turn, these proteins recruit other effector molecules having SH2, SH3, PTB, and Pleckstrin homology domains. 41 Multiple intracellular signaling pathways are activated by RTKs such as the phosphatidylinositol 3-kinase (PI3K), the mitogen-activated protein kinase (MAPK), the phospholipase C-γ (PLC-γ), and the signal transducers and activators of transcription (STATs).46,47 These pathways control cell growth, differentiation, neovascularization, and tissue repair. The activation of a prototype RTK is shown in Figure 2.

Mechanism of RTK activation and signaling. The binding of a ligand to the extracellular domain of an RTK monomer induces receptor dimerization followed by the autophosphorylation of the intracellular kinase domain and the subsequent phosphorylation of specific tyrosine residues near the carboxy-terminus. An active RTK recruits and activates several downstream substrates and signaling pathways. RTK, receptor tyrosine kinase.
Oncogenic Activity of RTKs
RTKs represent a prototypical class of oncogenes involved in most types of human malignancies. Aberrant RTK signaling drives tumorigenicity by increasing proliferation, survival, migration, invasion, metastasis, and decreasing apoptosis of cancer cells. 48 Constitutive activation is a principal oncogenic mechanism of RTKs. It results from gain-of-function mutations, genomic amplification, chromosomal rearrangements, and/or autocrine activation. 49 Several RTK mutations are ‘Driver Mutations’ leading to growth advantage for cancer cells promoting the initiation and progression of cancers.50,51 RTK gene amplification leads to greater levels of cell surface receptors available for signaling and increases the likelihood for clustering and dimeric partnering of RTKs. 52 Numerous chromosomal rearrangements have led to the formation of novel tyrosine kinase fusion oncoproteins resulting in constitutive ligand-independent kinase activation. 53 Another important mechanism for the constitutive activation of RTKs is the autocrine-paracrine loops. 41
RTK Inhibitors
Because of their key roles in oncogenesis, RTKs are substantial targets in cancer therapy. The inhibition of RTKs is clinically achieved by two classes of therapies: the monoclonal antibodies (mAbs) and the small-molecule tyrosine kinase inhibitors (TKIs). The mAbs (also known as the biologicals) bind to the natural ligands leading to their neutralization or to the extracellular domain of the RTK to inhibit ligand binding and/or receptor dimerization. 54 The TKIs are small molecules that specifically target the ATP-binding site of the intracellular tyrosine kinase domain of target RTKs.49,54 Table 2 lists RTK inhibitors currently approved for the treatment of human cancers.
Summary of US FDA-Approved RTK Inhibitors Including mAbs and Small-Molecule TKIs Used in Cancer Therapy
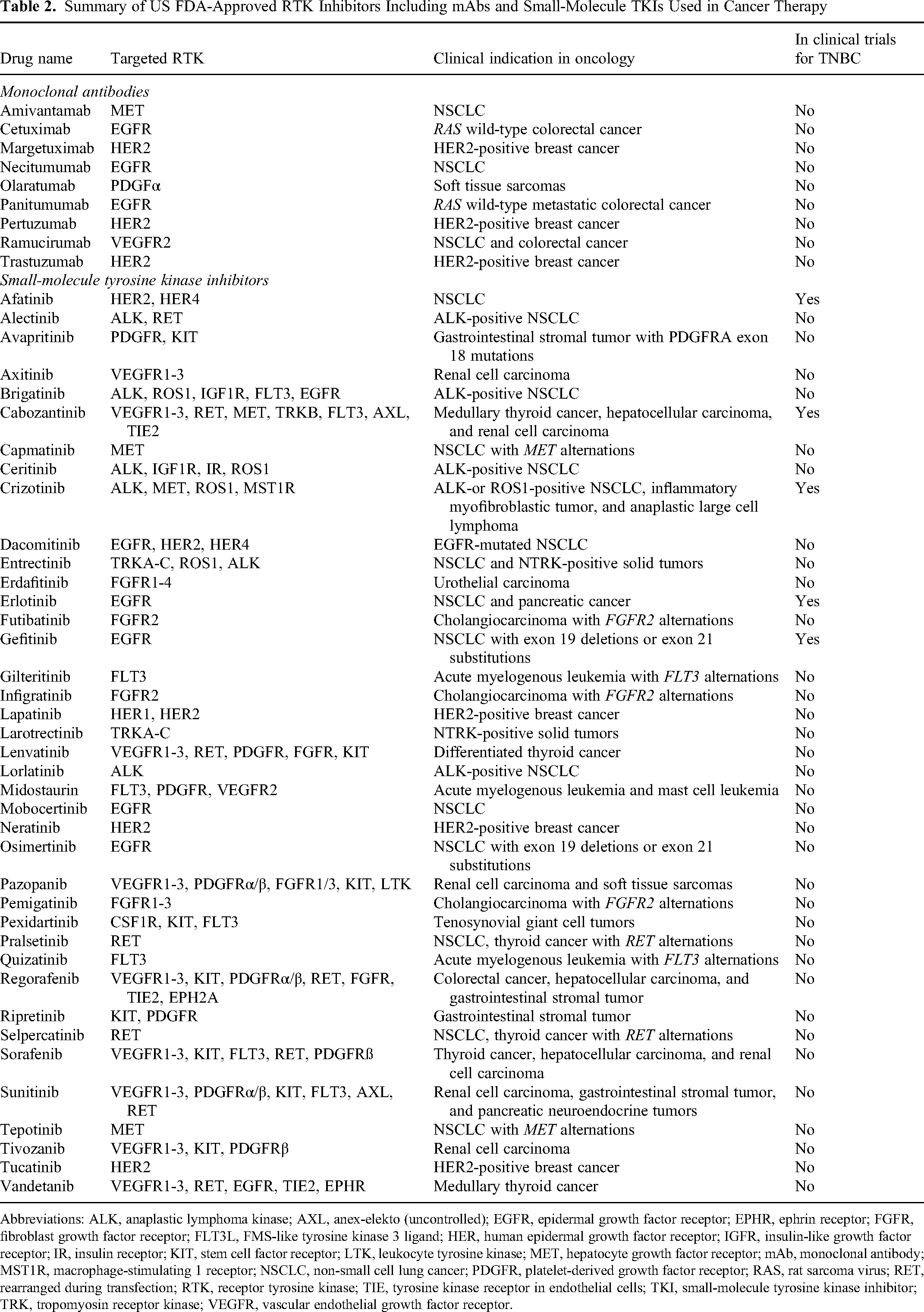
Abbreviations: ALK, anaplastic lymphoma kinase; AXL, anex-elekto (uncontrolled); EGFR, epidermal growth factor receptor; EPHR, ephrin receptor; FGFR, fibroblast growth factor receptor; FLT3L, FMS-like tyrosine kinase 3 ligand; HER, human epidermal growth factor receptor; IGFR, insulin-like growth factor receptor; IR, insulin receptor; KIT, stem cell factor receptor; LTK, leukocyte tyrosine kinase; MET, hepatocyte growth factor receptor; mAb, monoclonal antibody; MST1R, macrophage-stimulating 1 receptor; NSCLC, non-small cell lung cancer; PDGFR, platelet-derived growth factor receptor; RAS, rat sarcoma virus; RET, rearranged during transfection; RTK, receptor tyrosine kinase; TIE, tyrosine kinase receptor in endothelial cells; TKI, small-molecule tyrosine kinase inhibitor; TRK, tropomyosin receptor kinase; VEGFR, vascular endothelial growth factor receptor.
Targeting RTKs in TNBC: Evidence from Preclinical Studies
Taking into consideration the limited number of therapeutic options for the treatment of TNBC and the scarcity of targeted therapies, exploring new targets is mandatory. Because of the key role of RTKs in driving oncogenesis in several types of epithelial tumors and the success of several inhibitors in clinical use, it is of paramount importance to shed light on the utilization of RTK inhibitors for the treatment of TNBC. The following part provides evidence on the role of specific RTKs in the tumorigenicity of TNBC and the outcomes of targeting them with inhibitors in preclinical models.
The Epidermal Growth Factor Receptor: EGFR
Epidermal growth factor receptor (EGFR) (HER1/ErbB1) is an RTK that belongs to the EGFR family.55,56 Human EGFR is mapped to the short arm of chromosome 7. 57 EGFR is expressed in the majority of human cells and is activated by multiple ligands including EGF, transforming growth factor-alpha, amphiregulin, and epiregulin. 57 When activated, EGFR drives a series of intracellular signaling pathways such as the RAS/MAPK, PI3K/AKT, STATs, and protein kinase C (PKC) to regulate cell proliferation, survival, and angiogenesis.57,58 EGFR deregulations in cancer cells include gene amplification, activating mutations, autocrine and paracrine activating loops. 59 The expression of EGFR is elevated in TNBC compared to luminal tumors.60,61 The expression of EGFR varied between 13% and 78% in TNBC. 62 Besides, activating EGFR mutations, exon 19 deletions and exon 21 missense (L858R), were found in 11.4% of patients with triple-negative tumor samples. 63 These mutations cluster around the ATP-binding pocket of the kinase domain promoting ligand-independent activation compared to wild-type receptors. 63 Other studies, however, rarely reported EGFR mutations in TNBC.62,64 High EGFR expression is also associated with resistance to breast cancer treatments. 65
EGFR inhibitors were evaluated in preclinical models of TNBC. Afatinib is a competitive TKI of EGFR, HER2, and HER4. 66 Afatinib suppressed growth of TNBC cells and synergized with dasatinib (a TKI of ABL and KIT) through G1 cell cycle arrest and reduced tumor growth in a xenograft model of HCC1806 cells. 67 Triple combination of afatinib, dasatinib, and trametinib (a MEK inhibitor) induced synergistic antiproliferative and cytotoxic activity in TNBC cells by inhibiting ERK and PI3K/AKT pathways. 68 In another study, afatinib combined with an inhibitor of PIK3CA induced synergistic growth inhibition in PI3K-overactivated TNBC patient-derived xenografts. 69 The combination of the EGFR inhibitor gefitinib with chemotherapy produced synergistic anticancer activity mediated by cell cycle arrest in TNBC. 70 Besides, the combination of gefitinib with PI3K/AKT inhibitors 71 or autophagy inhibitors 72 induced cytotoxicity in TNBC cell lines and xenograft animal models, respectively.71,72 Combined gefitinib and mammalian target of rapamycin (mTOR) inhibitors, temsirolimus or everolimus, remarkably suppressed growth and induced cytotoxicity in TNBC cells. 73 Erlotinib is an ATP-competitive TKI of EGFR. 66 Erlotinib inhibited tumor growth and metastasis in preclinical TNBC models.74,75 The combination of erlotinib with gemcitabine synergistically reduced the viability of MDA-MB-468 and BT549 TNBC cells by inhibiting AKT activation. 76 In another combination, erlotinib and palbociclib, an inhibitor of cyclin-dependent kinase (CDK) 4/6, induced an additive antiproliferative effect in culture and delayed tumor growth of triple-negative xenografts. 77 Myeloid cell leukemia 1 (MCL1) is an anti-apoptotic protein related to the BCL-2 family that mediates resistance to EGFR inhibitors. The addition of the MCL1 inhibitor, S63845, to erlotinib enhanced its sensitivity in multiple TNBC cell lines. 78
Cetuximab and panitumumab are anti-EGFR mAbs. 79 Menendez et al reported that the triple-negative subtype was highly responsive to cetuximab compared to other breast cancer subtypes. 80 Low doses of cetuximab and cisplatin synergistically inhibited proliferation and induced apoptosis in MDA-MB-468 cells. 80 In another combination, cetuximab and dasatinib significantly reduced tumor volume in MDA-MB-468 tumor-bearing mice compared to the control group. 81 In the study by Ferraro et al, the anti-EGFR mAb 111 was combined with cetuximab or panitumumab, as it binds EGFR independently from the binding sites of these mAbs. 82 The combination of EGFR mAbs induced robust EGFR degradation, reduced colony formation, cell invasion, and in vivo tumor growth of TNBC. 82 Besides, chemotherapy and anti-EGFR mAb treatment induced a synergistic effect in BRCA1-mutated TNBC cells mediated by G1 arrest and inhibition of EGFR and ERK1/2 phosphorylation. 83 In an interesting study, dual targeting of EGFR with a mAb (cetuximab or panitumumab) and a TKI (gefitinib or erlotinib) induced a synergistic inhibition of cell proliferation and the RAS/MAPK pathway in MDA-MB-468 and SUM1315M02 cells. 61 Furthermore, in a panel of TNBC cell lines, a dual combination of several inhibitors of EGFR synergized with PHA-767491, an inhibitor of cycle 7 protein kinase and CDK9. 84 The combination of lapatinib, gefitinib, or erlotinib with PHA-767491 inhibited cell proliferation and induced G2/M cell cycle arrest and apoptosis via reduced expression of ERK/mTOR-related genes. 84 Despite promising findings in preclinical studies, EGFR inhibitors failed to demonstrate remarkable therapeutic activity in patients with TNBC in clinical trials as discussed below. In addition, claudin-low TNBC cells such as MDA-MB-436, BT-549, and MDA-MB-231 have limited sensitivity to EGFR inhibitors based on the Genomics of Drug Sensitivity in Cancer dataset (GDSC2). 85
The Hepatocyte Growth Factor Receptor: MET
MET (also known as c-MET) is an RTK that belongs to a subfamily that includes RON and SEA receptors. 86 MET proto-oncogene is found on the long arm of chromosome 7 in the human genome. The hepatocyte growth factor (HGF) is the natural ligand for MET. Under physiological conditions, MET is expressed by epithelial cells and activated by HGF through paracrine ligand binding. 87 The activation of MET drives a complex genetic program referred to as ‘invasive growth’ which consists of a series of physiological processes including cell proliferation, invasion, angiogenesis, morphogenesis, and branching tubulogenesis. 87 The HGF/MET axis is essential for organ development during embryogenesis and in tissue homeostasis during adulthood. 88 Deregulations of MET are reported in different types of tumors, including gene mutations, gene amplification, overexpression, and autocrine loop activation.87,88 Such deregulations induce aberrant activation of MET which in turn promotes aggressive and invasive tumor growth by driving cancer cell proliferation, scattering, EMT, migration, invasion, and metastasis. 87 Additionally, the high potential of crosstalk between MET with several other RTKs, most importantly members in the EGFR family, makes MET a key player in mediating drug resistance to targeted anticancer drugs.86,89 Besides, MET promotes resistance to other targeted therapies such as BRAF and MEK inhibitors and cytotoxic chemotherapy. 90
Several reports demonstrated MET amplification and receptor overexpression in invasive breast cancer, particularly triple-negative tumors.91–94 Veenstra et al showed that among patients with triple-negative tumors, MET amplification was linked to a higher relapse rate compared to patients without amplification. 94 The authors also revealed similar findings in patients with MET gain. Breast cancer patients with high-expression levels of MET had reduced survival rates compared with patients whose tumors showed low expression levels of MET.95,96 Further evidence from a meta-analysis indicated that MET overexpression was associated with an increased risk of recurrence and mortality in patients with TNBC. 97 MET expression was associated with adverse prognosticators such as increased Ki-67 expression, tumor size, tumor stage, high grade, and high metastatic burden in TNBC patients.92,96,98 Interestingly, Xing et al revealed that MET was overexpressed in the brain metastatic lesions compared to other sites of metastasis in patients with TNBC. 99 The co-expression of MET and EGFR was associated with worse disease-free survival than that in TNBC patients expressing EGFR alone. 93
The effects of MET inhibitors in preclinical models of TNBC have been examined in several studies. Crizotinib is a potent and specific small-molecule TKI of both MET and the anaplastic lymphoma kinase (ALK). 100 It inhibited the proliferation, migration, and invasion of MDA-MB-231 triple-negative cells in a dose-dependent fashion in vitro. 101 Besides, the combination of crizotinib with chemotherapy resulted in synergistic growth inhibition of MDA-MB-231 cells. 101 Synergism was also reported for the combination of crizotinib with either a PKC inhibitor or a CDK4/6 inhibitor in preclinical models of TNBC. 102 Hyperactivation of MET has been shown to contribute to acquired resistance to PARP inhibitors in TNBC. 103 The combination of talazoparib, a PARP inhibitor, and crizotinib synergistically inhibited the proliferation and colony formation of TNBC-resistant cells. 103 Cabozantinib is a potent small-molecule inhibitor of MET, vascular growth factor receptor 2, RET, KIT, and AXL. 104 Cabozantinib effectively inhibited the growth and invasion of TNBC cells in both monocultures and cocultures with HGF-overexpressing or cancer-associated fibroblasts. 105 Cabozantinib effectively suppressed the growth and metastasis of TNBC in a novel xenograft model that expressed human HGF and supported paracrine MET signaling. 105 Yi et al identified various MET inhibitors as inducers of synthetic lethality in combination with EGFR inhibitors in MDA-MB-231 cells. 106 The combination of the MET small-molecule TKI SU11274 with various EGFR inhibitors produced synergistic growth inhibition of TNBC cells by induction of G2 arrest and decreased ribosomal protein S6 levels in treated cells. 106 Furthermore, the combination of SU11274 with a γ-secretase inhibitor suppressed TNBC cell growth suggesting a potential therapeutic benefit by dual targeting of MET and NOTCH for TNBC patients. 107 In MDA-MB-231 cells, the knockdown of MET induced type II topoisomerase expression and increased cancer cell sensitivity to the chemotherapeutic drug daunorubicin. 96 The silencing of MET via small interfering RNA in the high-MET expressing TNBC cell lines BT549 and HCC1954 significantly reduced cell proliferation and migration, but not invasion. 93
MET and EGFR are highly expressed in TNBC. 108 MET has been also shown to cross-react with EGFRs providing alternate pathways to substitute for their activity, thus promoting resistance to EGFR inhibitors. 109 Hence, targeting MET and EGFR is a promising strategy that could restore the sensitivity of targeted therapy in TNBC. The combined treatment of the MET inhibitor foretinib with the dual EGFR and HER2 inhibitor lapatinib reduced the viability of MDA-MB-231 and BT549 cells synergistically. 110 These effects were associated with G2/M arrest and reduction of phosphorylated AKT levels. Further, cancer cell migration and invasion were more effectively inhibited with the combination treatment than with monotherapy. 110 The silencing of EGFR increased sensitivity to PHA-665752 treatment in TNBCs and the combination with erlotinib synergized to decrease the viability of MDA-MB-468 cells. 93 In another study, Linklater et al demonstrated that the combined treatment of MGCD265/erlotinib was the most effective in suppressing the growth of the TNBC tumorgrafts compared to the crizotinib/erlotinib combination treatment, individual drugs, and vehicle-control groups. 108 The combination of MGCD265 and erlotinib decreased phosphorylated levels of ERK and S6 ribosomal protein levels, proliferation, and increased necrosis in the treated tumors. The combined treatment of the MET inhibitor capmatinib with the EGFR inhibitor AZD-8931 inhibited the proliferation of chemoresistant MDA-MB-468 cells in vitro and reduced tumor growth in vivo. 111
Recepteur D'origine Nantais: RON
RON, also known as macrophage-stimulating 1 receptor, is an RTK that belongs to the MET family.112–114 The tyrosine kinase signaling domain of RON and MET share 63% of their sequence identity, while 25% of the extracellular domain of RON is identical to MET. 112 RON is mainly expressed in epithelial cells and is essential for embryonic development.113,114 The macrophage-stimulating protein (MSP) is the natural ligand of RON. 112 The binding of MSP to RON activates the PI3K/AKT, RAS/MAPK, focal adhesion kinase (FAK), Src, Jun kinase, STAT3, and β-catenin signaling pathways.112,114 RON regulates epithelial cell motility by mediating cell dissociation, migration, and matrix invasion.113,114 Hence, the oncogenic activity of RON is highly associated with the development of invasive and malignant tumor phenotypes. 114
The activation of RON induced proliferation, migration, and invasion, and prevented apoptosis of breast cancer cells in vitro.115–117 MSP induced the invasion of the triple-negative MDA-MB-231 and MDA-MB-468 cells but not the luminal MCF7 breast cancer cells. 118 Zinser et al demonstrated that RON overexpression effectively induced mammary transformation and metastasis in transgenic animals mediated by the up-regulation of genes controlling cell cycle and proliferation such as cyclin D1 and c-Myc. 119 The PI3K/mTORC1 pathway is fundamental for spontaneous metastasis induced by RON in vivo. 120 In this regard, the inhibition of mTORC1 with everolimus reduced RON-dependent metastatic lesions, and the combined blockade of mTORC1 and RON delayed tumor progression in vivo. 120 Recently, Bourn et al showed that RON signaling promoted tumor cell survival and proliferation along with an immune-tolerant microenvironment mediated by decreased recruitment of M1 macrophages, natural killer cells, and CD8-positive T cells. 121 Accordingly, the activation of RON signaling could promote the growth and progression of breast cancer, at least in part, through immune evasion. Besides, RON has been associated with resistance to targeted therapy in breast cancer.122,123
RON overexpression and constitutive activation were reported in clinical breast cancer samples.115,119 Breast cancer patients with high tumoral expression of RON had poor relapse-free survival and OS. 124 Previous studies indicated the overexpression of RON in patients with primary TNBC.95,125 Tumors from patients with high expression of RON had a worse survival rate than patients with RON-low tumors. 95 Furthermore, poor prognosis and short survival were observed in patients with tumoral RON and MET co-overexpression. 95
Targeting RON and MET with the small-molecule TKIs BMS-777607 and tivantinib increased apoptosis of the TNBC cell lines HCC2185, MDA-MB-231, and SUM52PE in a dose-dependent manner. 95 Besides, BMS-777607 and tivantinib inhibited tumor growth in a xenograft model of TNBC. 95 Millar et al showed that treatment with the RON inhibitor, BMS-777607 effectively reduced MSP-induced phosphorylation of AKT and ERK1/2 in TNBC cells. 126 Further, BMS-777607 suppressed tumor progression in animal models. In other studies, the anti-RON mAb Zt/g4-MMAE reduced viability and induced death of TNBC stem-like cells.125,127
The Growth Arrest-Specific Protein 6 Receptor: AXL
AXL is an RTK that belongs to the TAM family that contains TYRO3 and MER receptors.128,129 AXL was first characterized as a transforming agent in human chronic myelogenous leukemia patients. The activation of AXL by its ligand the growth arrest-specific protein 6 stimulates AKT, mTOR, NFκB, and MAPK at which it also acts as a signaling hub regulating crosstalk between complex pathways.129,130 AXL plays key roles in cancer cell proliferation, survival, apoptosis, migration, angiogenesis, and metastasis.129,130 Overexpression of AXL is associated with aggressive tumor phenotypes and poor patient outcomes such as disease recurrence and reduced survival.129,130 Furthermore, increased expression of AXL is linked to the development of acquired resistance to chemotherapy and targeted anticancer drugs.130,131
Overexpression of AXL was demonstrated in TNBC cell lines and patient tumor samples.129,132–134 Meyer et al reported that the expression of AXL was exceptionally predictive of a lack of response to EGFR-targeted inhibitors. 135 Additionally, AXL can be transactivated by EGFR in a ligand-independent fashion and was remarkably associated with EGFR and MET-forming receptor dimers resulting in diversified signaling pathways in TNBC. 135 High AXL expression levels were strongly associated with reduced survival rates and poor prognosis in patients with triple-negative tumors.134,136,137 D'Alfonso et al showed that high AXL expression correlated with mesenchymal phenotype and lymphovascular invasion in triple-negative tumors. 132 AXL overexpression was also associated with therapeutic resistance in breast cancer.129,138
Several studies evaluated the effect of AXL inhibitors on TNBC. Zajac et al indicated that AXL depletion using siRNA or the AXL inhibitor bemcentinib (R428) reduced the directional migration of Hs578t and MDA-MB-231 cells. 133 In agreement, multikinase inhibition using a combination of non-RTK inhibitors and ALK inhibitors reduced the motility of AXL-overexpressing TNBC cell lines. 139 Wu et al showed that targeting AXL with the antibody hMAb173 inhibited the growth of TNBC cells in culture and tumor growth in animal models and reduced the phosphorylation level of MET. 134 In another study, the combination of the AXL inhibitor, ER-851 with antimitotic drugs induced antitumor activity and prolonged relapse-free survival in a mouse xenograft model of human TNBC. 131 Targeting AXL with mAbs blocked ligand-dependent receptor activation and TNBC cell migration and invasion.138,140 Leconet et al showed that 20G7-D9, an anti-AXL mAb, inhibited tumor growth and bone metastasis in AXL-positive TNBC xenografts. 140 In an interesting report, Wei et al engineered T cells with a CAR consisting of a novel variable fragment against AXL and indicated its antigen-specific cytotoxicity in vitro and in vivo. 129
Insulin-Like Growth Factor 1 Receptor: IGF1R
IGF1R belongs to the insulin receptor (IR) family of RTKs. 141 IGF1R and IR share 57% sequence identity and high structural similarity. IGF1R is activated by insulin-like growth factors (IGFs). 141 The IGF/IGF1R axis plays critical roles in regulating cell proliferation, differentiation, survival, migration, and invasion through the PI3K/AKT, MAPK, JAK/STAT, Src, and FAK signaling pathways. 142 The overexpression and/or activation of IGF1R is associated with a high risk of metastasis and poor prognosis in different types of human malignancies. 142
In breast cancer, IGF1R expression is elevated in about 50% of breast cancers, and more frequently in luminal A tumors compared to other subtypes. 143 However, Davison et al revealed that the expression level of IGF1R was similar in triple-negative and estrogen-responsive cell lines. 144 The impact of the IGF1R expression on clinicopathologic features and prognosis of breast cancer is inconsistent. In one study overexpression of IGF1R correlated with reduced survival rates and adverse prognosticators, 145 while the receptor expression correlated positively with good prognostic markers in another report. 146 The IGF/IGF1R axis is involved in resistance to targeted anti-HER2 drugs, in part, through the interaction with HER/ErbB receptors.147,148 Besides, increased expression and activity of IGF1R were linked to antiestrogen resistance in ER-positive breast cancer. 149
In triple-negative cancer cells, IGF1R is essential for maintaining mesenchymal morphologies and the expression of EMT-related markers, and its overexpression-induced cell migration and invasion mediated by FAK. 150 Nagle et al showed that loss of E-cadherin stimulates the IGF1R pathway and improves sensitivity to IGF1R/IR targeted therapy. 151 Serum levels of IGF/IGF1R were significantly higher in TNBC compared with the non-TNBC patients and were associated with reduced response rate and enhanced metastasis. 152
Targeting IGF1R has been proposed as a novel therapeutic approach in TNBC. Numerous studies indicated a synergistic growth inhibition for the combination of IGF1R inhibitors with metformin, 153 PI3K inhibitors, 154 MEK inhibitors, and mTOR inhibitors 155 in TNBC cells. The combined inhibitors induced cell cycle arrest in treated cells.153,155 In another study, a combination of IGF1R inhibitors remarkably suppressed the proliferation of TNBC cells to a greater extent than each inhibitor alone and reduced the expression levels of AR. 156 In a study by Litzenburger et al, the dual IGF1R/IR inhibitor BMS-754807, effectively reduced tumor growth in a triple-negative human tumorgraft mouse model characterized by high IGF1R expression and activity. 157 Besides, BMS-754807 and docetaxel showed superior tumor growth inhibition than either agent alone. Interestingly, complete tumor regression was achieved in some animals treated with the combination of BMS-754807 and docetaxel.
The Stem Cell Factor Receptor: KIT
KIT (also known as c-KIT or CD117) is an RTK that binds stem cell factor (SCF) and is encoded by a proto-oncogene on the long arm of chromosome 4. 158 KIT regulates various biological functions such as homeostasis and fertility. 158 It activates RAS/MAPK, PI3K/AKT, and PLC-γ to regulate the proliferation, differentiation, and survival of KIT-expressing cells.159,160 KIT is known for its oncogenic activity and is expressed in solid and hematological tumors. Deregulations of KIT include gain of function mutations, point mutations, and overexpression. 158
KIT is expressed in triple-negative tumors. 161 The tumoral expression of KIT ranged from 30.0% 162 to 49.0% 163 in patients with TNBC. Jansson et al demonstrated a significantly higher expression of KIT in TNBC compared to non-TNBC patients (49% vs 10%, respectively). 163 Furthermore, the odds ratio of KIT-positivity, adjusted for tumor characteristics, was 6.8 times higher for TNBC than non-TNBC patients. 163 KIT gain and gene amplification were reported in breast cancer patients and were associated with ER-negativity, high grade, and reduced survival. 164 Mutations in KIT also correlated with poor prognosis in patients with TNBC.161,165
In a recent study by López-Mejía et al, the stimulation of KIT by SCF induced the migration of TNBC cells and the activation of STAT3, AKT, and ERK1/2. 166 Treating the cells with a panel of KIT inhibitors (imatinib, dasatinib, nilotinib, sorafenib, and sunitinib) produced variable antiproliferative activities; however, nilotinib was the most cytotoxic drug and induced a very homogenous response. 166
Other RTKs
Other RTKs were targeted in TNBC. Inhibiting the ephrin A10 receptor with mAbs induced significant tumor regression in a mouse model of TNBC accompanied by enhanced activation and infiltration of cytotoxic T lymphocytes. 167 Another inhibitor of the ephrin A2 receptor suppressed the growth of MDA-MB-231 cells in both culture and patient-derived xenografts in vivo through downregulating c-Myc and stabilization of p27Kip1. 168 In another study, the combination of the ALK inhibitor ceritinib with paclitaxel reduced the growth, induced apoptosis, and enhanced paclitaxel sensitivity in triple-negative tumors. 169 Figure 3 shows the RTKs mostly targeted in the treatment of TNBC and their downstream signaling pathways.

The major RTKs involved in the tumorigenesis of TNBC and their downstream signaling pathways. The activation of the EGFR, MET, RON, AXL, and IGF1R activates a wide range of downstream signaling hubs for pathways associated with cancer cell proliferation, survival, migration, and invasion. The RTKs can be targeted by mAbs and small-molecule TKIs. mAbs, monoclonal antibodies; TKIs, small-molecule tyrosine kinase inhibitors; TNBC, triple-negative breast cancer.
Clinical Trials Evaluating RTK Inhibitors for the Treatment of TNBC
None of the approved inhibitors of RTKs is currently indicated for the treatment of TNBC. EGFR inhibitors were evaluated in patients with triple-negative tumors; however, the results have been disappointing when these inhibitors were used alone or in combination with chemotherapy. 62 A multicenter phase I/II study (NCT00834678) that assessed erlotinib and bendamustine for patients with metastatic TNBC revealed a lack of complete response and disease progression in all patients within 7 months of the beginning of treatment. 170 The combination was also associated with serious toxicities such as prolonged lymphopenia and severe opportunistic infections. 170 Afatinib was evaluated in a neoadjuvant phase II trial including patients with stage II/III TNBC. 171 The results indicated modest effects for afatinib, however, certain molecular characteristics and genes were associated with the response to afatinib and paclitaxel combination therapy. 171 Despite the limited efficacy, clinical trials are still exploring EGFR inhibitors in patients with triple-negative tumors. The efficacy and safety of erlotinib are being evaluated in a phase II study in patients with TNBC who progressed on anthracyclines and taxanes, with time to progression being the primary endpoint (NCT00739063). In another phase II study (NCT00733408), maintenance treatment of erlotinib and bevacizumab after induction therapy of nab-paclitaxel and bevacizumab showed a median progression-free survival of 9.1 months among the 55 patients analyzed and <10% of the total incidence of serious adverse events, mainly infections and infestations (3.6%).
The efficacy and safety of cabozantinib are evaluated in a phase II study in patients with metastatic TNBC (NCT01738438). Published results based on a 60 mg dose of cabozantinib daily on a 3-week cycle for a median of three cycles failed to meet the primary endpoint in pretreated metastatic TNBC patients and did not achieve the target objective response rate. 172 However, favorable safety and encouraging efficacy signs were obtained. On top of that, this study supported MET to be a promising biomarker of response for further evaluation. 172 Similarly, a clinical trial for the combination of cabozantinib and nivolumab failed its primary endpoint of reaching an acceptable objective response rate in patients with TNBC (NCT03316586). Notably, no unexpected adverse events were detected, including elevated liver enzyme, hand-foot syndrome, fatigue, and hypothyroidism.
Sitravatinib is an oral, spectrum-selective TKI targeting the TAM family of RTKs. 173 A three-cohort, phase II trial is currently evaluating the safety and antitumor activity of sitravatinib in three different treatment combinations, in patients with locally recurrent or metastatic TNBC (NCT04734262). The overall response rate is the primary outcome of the three cohorts. Besides, a phase II, multicenter study was conducted to assess the activity and safety of bemcentinib, a selective AXL inhibitor, in combination with pembrolizumab in patients with locally advanced or metastatic TNBC (NCT03184558). However, the study was terminated because of a lack of response. Dasatinib is a multi-targeted TKI with potent activity against KIT. 174 A phase II study is evaluating the effect of dasatinib in patients with advanced TNBC (NCT00371254). The primary outcomes are the number and percentage of patients with complete or partial response. Additionally, an open-label, phase II clinical trial is evaluating dasatinib efficacy as a neoadjuvant treatment in locally advanced TNBC (NCT00817531). Table 3 summarizes clinical trials investigating RTK inhibitors for the treatment of TNBC.
Clinical Trials for Selected RTK Inhibitors for the Treatment of TNBC (Retrieved from: www.clinicaltrials.gov)

Abbreviations: EGFR, epidermal growth factor receptor; IGF1R, insulin-like growth factor 1 receptor; TNBC, triple-negative breast cancer; RTK, receptor tyrosine kinase.
Future Directions
The efficacy of RTK inhibitors in the treatment of TNBC is not yet definitive. A deeper understanding of the biology of triple-negative tumors is essential to identify RTKs critically linked to the development and progression of this aggressive subtype of breast cancer. In line with this, it is crucial to analyze the profile of deregulations of RTKs in TNBC. Based on the above, several RTKs were found to be upregulated and/or mutated in patients with triple-negative tumors. While growing evidence from preclinical studies supports the role of RTK inhibitors in the treatment of TNBC, the outcomes from clinical studies were disappointing in most cases. The limited clinical efficacy can be interpreted in terms of disease heterogeneity on both the molecular and cellular levels. While basal-like and claudin-low cancers share similarities, they are biologically distinguished, and they may respond differently to RTK inhibitors. Thus, there is an urgent need for a better classification of TNBC for treatment purposes. While the current treatment guidelines rely exclusively on the expression status for ER, PR, and HER2 to define the group of triple-negative patients, other tumor variables may be warranted to expand the treatment options. A large body of literature revealed that RTKs such as EGFR, MET, and AXL, could be very appealing targets for TNBC based on their overexpression and contribution to aggressive cancer cell behavior. However, the lack of clinical efficacy encountered with EGFR inhibitors brings to question the significance of the level of expression of an RTK as the sole determinant of response in patients. Despite the overexpression of EGFR in TNBC, limited clinical benefits have been demonstrated for the inhibitors in clinical trials. Thus, analyzing other factors that could influence the clinical response to RTK inhibitors is necessary. These include the presence of certain mutations and/or epigenetic alterations that could guide the selection of patients who could respond favorably to RTK inhibitors. Following the example of lung cancer, breast cancer patients with activating EGFR mutations might be more sensitive to TKIs. Another factor that could influence response to TKIs is changes in post-receptor signaling pathways. Such changes adversely impact the clinical usefulness of RTK inhibitors in the presence of constitutively active mitogenic and/or survival signaling pathways in triple-negative tumors further adding another layer of complexity to the treatment. To better identify TNBC patients who could be candidates for RTK inhibitors, an analysis of the genetic signature of key RTKs may help identify deregulated pathways and the likelihood of benefiting from currently available RTK inhibitors.
The crosstalk between RTKs is another factor that could lead to a lack of response in triple-negative tumors. As discussed above, the crosstalk between RTKs could diversify downstream signaling pathways and further promote resistance to inhibitors. This is particularly important as evidence indicated a superior activity for the combination of RTK inhibitors compared to individual drugs when used in TNBC. A panel of RTK inhibitors could be repurposed for the treatment of TNBC. While perceived as an advantage, it could be challenging to find the best compilation of RTK inhibitors for the treatment of triple-negative cancers.
Another challenge to the effective use of RTK inhibitors is the ability to assess response to therapy. Apart from the classical activating mutations known to enhance sensitivity to EGFR TKIs in lung cancer, few predictive biomarkers are available to assess response to RTK inhibitors. Li et al indicated that the activity of EGFR TKI was affected by the phosphorylated levels of HER3, ERK1/2, and p53 in urinary bladder cancer cells. 175 In resistant cells, phosphorylated p53 was highly expressed and the TKIs had no effect. In another study, Ki-67, STAT3, and p27 were predictive factors for the response to TKIs in patients with skin cancer. 176 A systematic review and meta-analysis revealed that EGFR gene copy number is a biomarker predictive for response to EGFR TKIs in patients with advanced non-small-cell lung cancer. 177 Another study indicated that mutations in PIK3CA and TP53 in circulating cancer cells may predict the efficacy of a pan-EGFR inhibitor in patients with HER2-positive metastatic breast cancer. 178 Collectively, several biomarkers have been shown to predict the response to RTK inhibitors in different types of cancer. The response to an RTK inhibitor could be determined by the expression levels, mutations, and/or alterations in receptors and their downstream signaling molecules. A combination of such biomarkers could improve the predictability of response to RTK inhibitors in TNBC.
Patients with advanced TNBC usually have poor survival and a higher risk for recurrence. An appealing aspect of utilizing RTK inhibitors is to enhance the sensitivity of chemotherapy in the treatment of TNBC. Introducing RTK inhibitors could reduce the emergence of acquired resistance by suppressing the upregulation of alternative signaling pathways associated with the survival and proliferation of cancer cells. Several ongoing clinical trials are currently evaluating the combined treatment of RTK inhibitors with chemotherapy in patients with TNBC (Table 3).
In a novel approach, RTKs have been targeted using antibody-drug conjugates (ADCs) in patients with TNBC. ADCs are typically composed of a mAb covalently attached to a cytotoxic drug (payload) with the support of a stable chemical linker.179,180 Trastuzumab deruxtecan is an ADC that targets HER2 and is conjugated with a topoisomerase I inhibitor, extecan derivative (Dxd). 181 Destiny-Breast04 was a phase III trial involving patients with HER2-low metastatic breast cancer who had received one or two previous lines of chemotherapy. 182 The study revealed that patients treated with trastuzumab-Dxd had significantly longer progression-free survival and OS than the patients receiving the physician's choice of chemotherapy. 182 Trastuzumab-Dxd is currently the first pharmaceutical with remarkable clinical efficacy in patients with HER2-low TNBC. 183 AVID100 is an anti-EGFR ADC directed against wild-type and mutant receptors and is conjugated with the microtubule inhibitor, DM1, a derivative of maytansine. 181 A clinical trial (NCT03094169) conducted to evaluate the efficacy and safety of AVID100 in patients with advanced epithelial carcinomas including metastatic TNBC was terminated because of a lack of efficacy. Together, ADCs provide a new horizon for the treatment of patients with TNBC and RTKs could be attractive targets for further development of this therapeutic strategy. 181
Conclusions
The lack of ER, PR, and HER2 expression makes TNBC a very challenging subtype to treat. Therefore, there is an urgent need for effective targeted therapies for patients with triple-negative tumors. RTKs are key regulators of cell proliferation, survival, and invasion. The overexpression and/or deregulations of RTKs in TNBC make them potential therapeutic targets. The combination of RTK inhibitors could be a novel approach for the treatment of TNBC since the coexpression of these receptors is associated with signaling crosstalk and further enhances survival and acquired resistance to targeted therapies. Despite the promising evidence from preclinical studies, RTK inhibitors showed limited clinical benefit in the treatment of TNBC. Further investigations are needed to evaluate the role of RTK inhibitors in clinical settings by better identification of patients who may respond to this therapy, in part, by a thorough analysis of candidate RTKs in the tumor tissue and the deregulations involved.
Footnotes
Abbreviations
Acknowledgments
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics Statement
Not applicable.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.