
Abstract
The development of 1,8-naphthalimide derivatives as cell probes, DNA targeting agents, and anti-tumor drugs is one of the research hotspots in the field of medicine. Naphthalimide compounds are a kind of DNA embedder, which can change the topological structure of DNA by embedding in the middle of DNA base pairs, and then affect the recognition and action of topoisomerase on DNA. Aminofide and mitonafide are the first 2 drugs to undergo clinical trials. They have good DNA insertion ability, can embed DNA double-stranded structure, and induce topoisomerase II to cut part of pBR322DNA, but not yet entered the market due to their toxicity. In this paper, the design and structure-activity relationship of mononaphthalimide and bisaphthalimide compounds were studied, and the relationship between the structure of naphthalimide and anti-tumor activity was analyzed and discussed. It was found that a variety of structural modifications were significant in improving anti-tumor activity and reducing toxicity.
Introduction
1,8-Naphthalimide is a multifunctional fluorescent construct, which is widely used in supramolecular and pharmaceutical chemistry. Naphthalimides have been developed into biological dyes, luminescent sensors, cell probes, DNA targeting agents, and intercalators due to their good chemical properties and biological activities.1–3 With the development of modern life science, it is an ideal research direction to design and synthesize new DNA intercalators and cutting agents by using the DNA variability between tumor cells and normal cells. There are 3 common modes of action of DNA intercalators: First is interaction with anion skeleton; Second, it interacts with the edge of base pair in DNA groove; Third, insert into base pairs. As a good intercalation agent, naphthalimide molecule shows stronger binding force, higher selectivity, and stronger specificity. 4
In 1980, Braña of Spain first reported the synthesis of 3-nitro-1,8-naphthalimide derivatives. 5 Later, it was confirmed that the compound can inhibit HeLa and KB cancer cells, and can be embedded into the base pairs of DNA double strands, resulting in the opening of double strands and affecting DNA synthesis, creating a precedent for these substances as DNA intercalators. Amonafide (compound 1) and mitonafide (compound 2) are the most famous compounds (Figure 1). With the deepening of the research on naphthalene imide and its derivatives, it has been proved that the amine on the side chain of N-imide plays a key role in its DNA chimerism. The 3 sites of naphthalene ring have the highest substitution activity, the substituents include nitro, amino, methoxy, etc. The modification of this basic structure is what most subsequent studies have done. A series of related compounds such as bis-naphthalimides, heterocyclic naphthalimides, and amino acid naphthalimides show broad application prospects in DNA interaction.6–8

Molecular structures of amonafide(compound 1) and mitonafide (compound 2).
Design and Structure-Activity Relationship of Mononaphthalimides and Their Derivatives
Naphthalimides are synthesized by the reaction of 1,8-naphthalene dicarboxylic anhydride with amine, and the amino functionalization at position 3, 4, 5, or 6 of the ring produces compounds with internal charge transfer transition. Migration and solvent effects can be seen in the synthetic band of the absorption spectrum, and it has strong fluorescence; compared with the absorption spectrum, the fluorescence spectrum is batochromically shifted. 9 According to the CPK accurate molecular model, the nitro substitution at position 3 is closer to the plane of naphthalene ring, while the hindered nitro at position 4 rotates out of the plane. 10
Among the mononaphthalimide compounds, the most representative ones are amonafide and mitonafide. Amonafide and its structural analogues can destroy the cleavage release balance of DNA topoisomerase II covalent adducts and affect the normal metabolism of topoisomerase II. Mitonaphthylamine mainly inhibits DNA synthesis, both of which show high DNA binding and selectivity in clinic. Although amonafide has good biological activity, its main metabolites are acetylation of N at position 5 of naphthalimide ring and oxidation of side chain; the main metabolite is N-3 acetylated derivative and oxidation of side chain. The metabolite N-acetyl amonafide can cause severe cytotoxicity (Figure 2). Interestingly, the general slow acetylated population is more likely to cause cell damage and adverse drug reactions due to the accumulation of drugs in cells. 11 On the contrary, Asians with relatively large amount of N-acetyltransferase are more likely to have adverse reactions of bone marrow suppression. 11
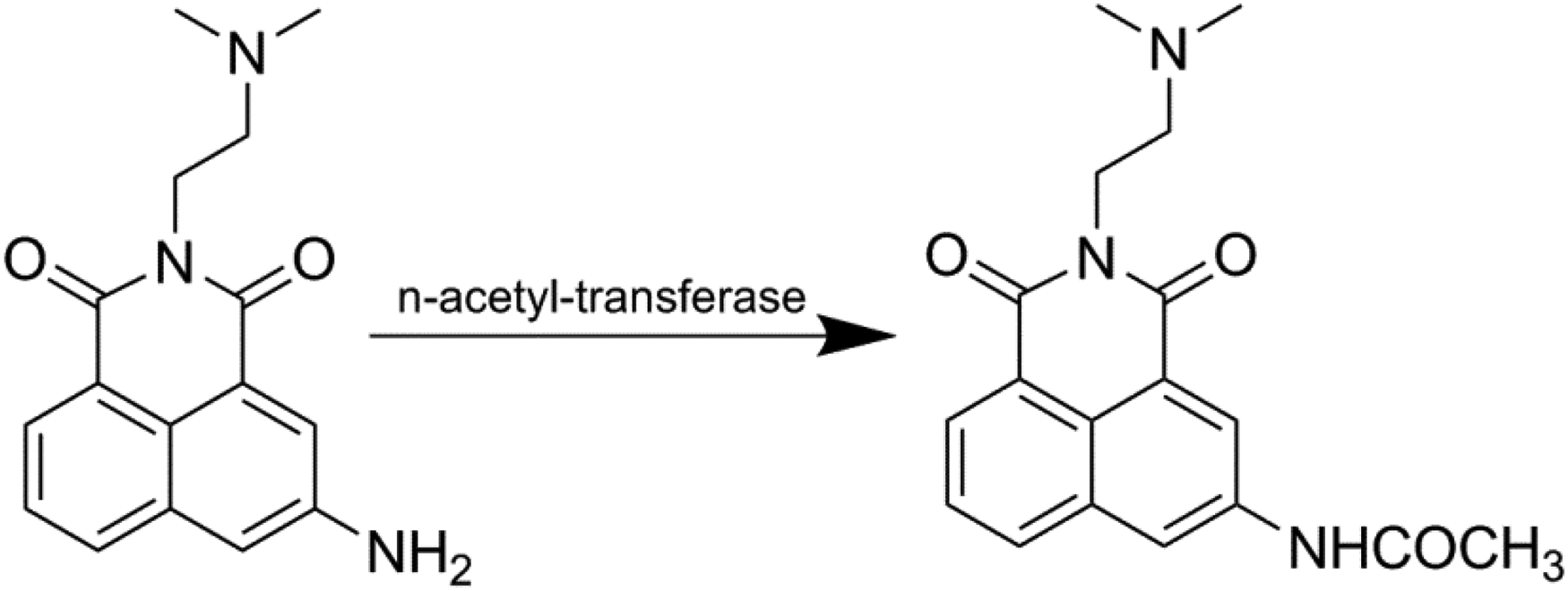
Acetylation pathway of amonafide.
Amonafide only opened the prelude of polycyclic aromatic amides as DNA intercalators, and the research on related derivatives continued for decades. In order to improve the antitumor activity and reduce the side effects, the researchers modified the branched chain structure of naphthalimide containing different elements and functional groups.
In 2007, Quaquebeke et al 12 designed and synthesized 5 classes of 40 naphthalimide analogues (Figure 3). Some compounds can be directly hydrolyzed to unsubstituted urea to avoid the side effects caused by acetylation. They include 11 kinds of amide (3a), 11 kinds of urea (3b), 7 kinds of imines (3c), 7 kinds of amine (3d) and 4 thiourea (3e). In addition, the cell lines of glioblastoma, colon cancer, breast cancer, and non-small cell lung cancer were tested in laboratory, showing good cell use. Comparing MTD (maximum tolerated dose) data, it was found that when urea imines were reduced to amines, the T/C value of the latter (T/C index is the ratio of median survival time of treatment group (T) to control group (C)) usually decreased. Moreover, once the oxygen atom on urea is replaced by sulfur atom to produce thiourea, the antitumor activity in-vitro is significantly lower than that of the corresponding urea, and the IC50 value is more than 2 times higher. When compounds (3b class) are replaced by trichloromethyl, it shows obvious mild hematotoxicity at the “therapeutic” dose. Because of its different mechanism, it can be further studied as a high-efficiency DNA intercalation agent.
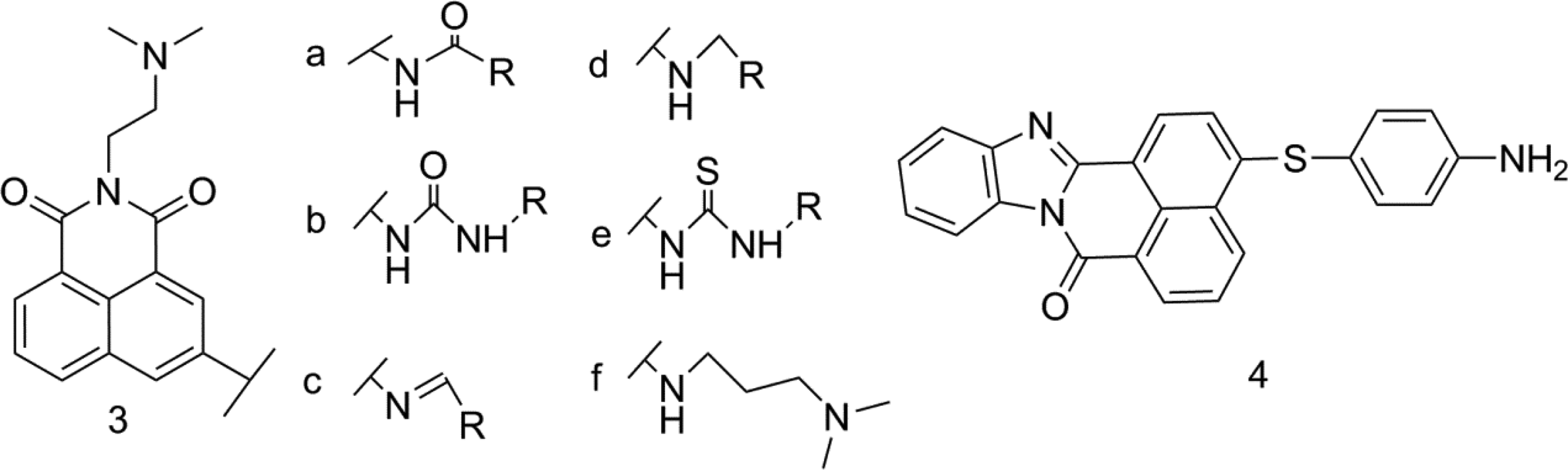
Amine substituted naphthalimide derivatives (compound 3(a-f) and compound 4).
In 2008, in experiment of Norton et al 13 4-amino group was alkylated to avoid N-acetylation while maintaining anticancer activity, significantly reduced cytotoxicity, and IC50 value was in the range of 10−6 to 10−7 M. In 2009, Xie et al 14 synthesized and evaluated 8 3-aminoalkyl substituted derivatives. The absence of primary amine at 3 position successfully avoided acetylation toxicity and maintained biological activity higher than that of amonafide. Compound 3f (Figure 3) had good cellular activity against HeLa and P388D1 (IC50 value was 0.71 and 0.23 μM, respectively).
In 2013, Verma et al 15 introduced N-containing groups at position 4 of naphthalimide and evaluated 60 human cancer cell lines. It was confirmed in in-vitro experiments that compound 4 has extensive anti-tumor activity. The MG-MID TGI (average total growth inhibition concentration) of compound 4 on 8 common cancer cells is 38.71 μM, including breast cancer, prostate cancer, and non-small cell lung cancer, it is 5 times more than the standard antitumour drug 5-fluorouracil.
In 2017, Wang H et al 16 reported that 3-amino 1,8-naphthalimide (3-A1, 8NI), an active inhibitor of Vibrio cholera, has the same inhibitory effect compared with the clinically applied cholera drug virstatin, and it can inhibit the production of its pathogenic factor cholera toxin, and hinder the swimming or cluster movement of Vibrio cholera and Vibrio parahaemolyticus. It is expected to be developed as a drug directly targeting the virulence factor of Vibrio cholera for the combined treatment of moderate and severe cholera.
In conclusion, the study of mononaphthalimide has been relatively sufficient. Some compounds of mononaphthalimide can induce G2/M phase cell cycle arrest by inhibiting PI3K/Akt signaling pathway, and have an effective effect on Topo II and lead to DNA double bond break. 17 One of the important modification is in the structure of naphthalene ring. Compared with 4-position substitution, 3-position nitro substitution on naphthalene ring has better antitumor activity. 18 Electron-pulling groups attached to naphthalene rings can enhance binding to DNA, but when 3-position substitution is not available, 4-position substitution can significantly enhance the activity. The structure-activity study shows that there is a basic terminal group in the side chain, and the terminal nitrogen of the side chain is separated from the naphthalene ring by 2 or 3 methylene units, which plays a key role in their anticancer activity. 19
Study on bis-Naphthalimide Derivatives
Bis-naphthalimide derivatives usually have higher activity than 1,8-naphthalimide monoderivatives and have good effects on a series of human cancer cell models. Attachment of 2 pharmacophores to bifunctional linker is often used to connect 2 pharmacophores. In all types of linkers, 20 polyamine spacers have been widely developed in the construction of double insertions. Double insertion can better induce the expansion and elongation of double helix, so as to cause structural changes, affect the condensation of chromatin and the interaction between DNA and related enzymes, and have better DNA binding ability and anticancer activity.
The Beginning of bis-Naphthalimides
The research on the bis-naphthalimide derivatives also began with the Braña research group. They used aminonaphthalimide as the lead compound to connected with 1,7-heptenyl linker to connect the 2 parent structures of naphthalimide through the “arm,” and to obtain the first symmetrical bis-naphthalimide derivative (Figure 4). 21

The first bis-naphthalimide derivative (compound 5).
Among the bis-naphthalimide derivatives, the first to enter the clinical trial are irinafide and bisnafide (Figure 5). Elinafide is a kind of bis-naphthalimide with the most basic structure. There is no substituent on the naphthalene ring, but it can act on the large groove of DNA and has good antitumor activity. Bisnafide DMP-840 was developed by Merck. 22 Condensation of 2 3-nitro-1,8-naphthalanhydride molecules with (2R,2R’)-N1, N1’-(ethane-1,2-diyl) dipropane-1,2-diamine leads to Bisnafide (DMP840) molecule in free form. The mechanism study found that number of appropriate compounds inhibited the biosynthesis of DNA and RNA by interfering with the normal binding of thymine and uracil to inducing DNA single strand break. 23 In addition, it can also be used as a eukaryotic topoisomerase II poison to stabilize the cleavage complex between topoisomerase II and DNA, resulting in cell death. 24 Compared with irinafide, it has no muscle toxicity. The substituents on the naphthalene ring change its planar aromatic ring structure and improve its toxicity and biological activity.
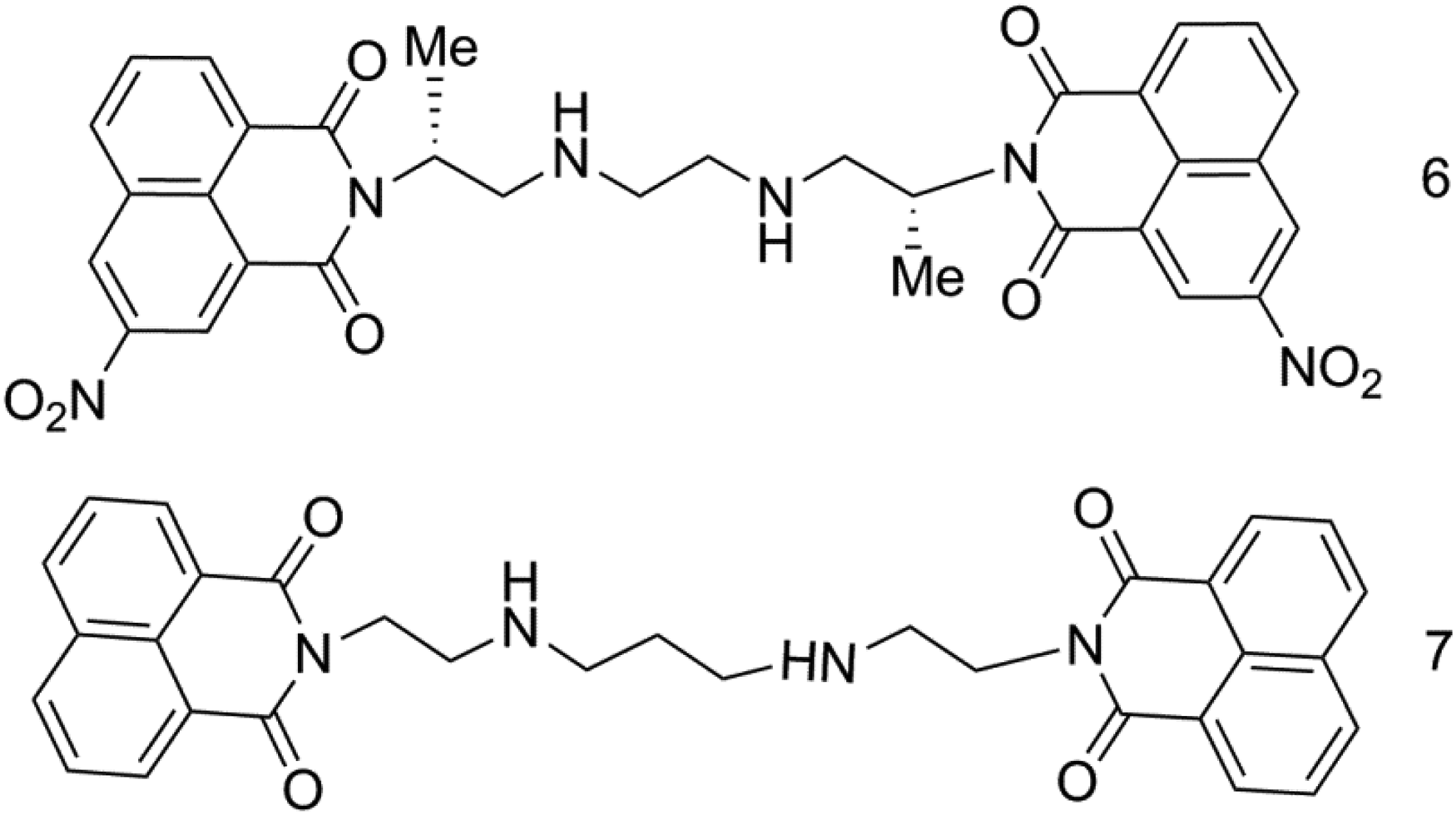
Molecular structure of bisnafide (compound 6) and irinafide (compound 7).
Novel Diamine bis-Naphthalimide Derivatives
In 2018, Song Yu et al 25 synthesized 5 naphthalimide derivatives (Figure 6) from 1,8-napthalic anhydride. Firstly, 1,8-napthalic anhydride and L-lysine were refluxed in ethanol to obtain the intermediate compound, then reacted with di-tert-butyl dicarbonate to protect the amino group, and then the carboxyl group was condensed with different diamines. After deprotection, new bis-naphthalimide derivatives (compound 8-12) were obtained modified with L-lysine and different diamine linkers. The results showed that all compounds had good DNA binding ability and showed a preference for double strands with high proportion of adenine and thymine. The DNA binding constants and binding modes of the compounds were measured by UV–Vis titration and viscosity experiments. It was found that the binding properties of the bis-naphthalimide derivatives were greatly affected by the length of diamine linker, and the short chain compounds had large binding constants and relatively classical binding modes. However, in the cytotoxicity experiment, compound 11 with rigid 1,4-bis(aminomethyl)benzene linker showed better cytotoxicity than other bis-naphthalimide derivatives.

Molecular structure of 5 diamine bis-naphthalimide derivatives (compound 8-12).
Platinum Complexes of bis-Naphthalimide
As one of the most successful series of drugs in the medical field, platinum drugs account for about 50% of the clinical chemotherapy regimen. They can effectively kill and prevent DNA replication, but they have serious nephrotoxicity and hematotoxicity. 26 Looking for new compounds that can overcome the inherent defect potential of platinum (II) drugs has been pursued by the majority of researchers.
In 2018, Tan Xiaoxiao et al 27 designed and synthesized a series of bis-naphthalimide platinum (IV) complexes (Figure 7) with oxaliplatin as the core as antitumor drugs. In-vitro biological activity showed that all compounds had moderate effective antitumor activity against all tested tumor cell lines, especially compound 14, whose IC50 value for SKOV-3 and HeLa was 3.1 and 12 μM. Its activity is comparable or even better than cisplatin and oxaliplatin. Compound 14 shows low toxicity and strong safety in vivo, and its LD50 and MTD values are twice higher than oxaliplatin. Naphthalimide platinum(IV) complex has a dual DNA damage mechanism, it can interact with tetravalent dsDNA to cause the first DNA damage. The further reduction of naphthalimide platinum(IV) would release platinum(II) complexes and naphthalimide acids which would induce remarkable secondary damage to DNA. It is a good scheme for the structural modification of bis-naphthalimide.

Molecular structure of the bis-naphthalimide platinum (IV) complexes (compound 13-17).
In addition, 2 ruthenium(II) polypyridyl complexes containing 1,8-naphthalimide group as inhibitors of topoisomerases were reported.
Bis-naphthalimide derivatives have a double insertion layer planar rigid structure. Researchers have modified the parent ring and adjusted different connecting arms, and achieved good results. Mekapati et al 28 studied the quantitative structure-activity relationship of naphthalimide and found that the substitution of amino group at position 3 of naphthalimide mother nucleus is conducive to improving its activity. Yang Qing et al 29 confirmed that the chiral structure is also of research significance through the study of naphthalimide derivatives connected with aminopyrrolidine fragments with chiral carbon.
Studies have shown that there are two types of binding of dinaphthimide derivatives to DNA: embedding binding and electrostatic binding. The strength of the embedding bond is related to the length of the linking groups and substituents. The longer the linking group or the smaller the spatial volume of the substituent, the stronger the flexibility and the more flexible the spatial structure of the compound. Such a compound structure transforms into an optimal spatial structure when embedded between DNA base pairs, resulting in base accumulation and local unspinning of the DNA double helix. However, electrostatic binding is not directly related to the length of the linking group and the substituent.
Studies on Other Naphthalimide Derivatives
The study of naphthalimide derivatives confirmed that the good insertion effect is characterized by the presence of tricyclic or tetracyclic ring planes and aromatic rings, which can intercalate in between 2 base pairs. The binding of intercalators can induce the expansion, elongation, and hardening of double helix, resulting in structural changes and affect the interaction between DNA and related enzymes. These changes of special structure-activity relationship can also give DNA sequence selectivity, make aromatic heterocycles in appropriate positions and interact with topoisomerase, so as to interfere with DNA replication and transcription. 30 These compounds include polyamine derivatives, benzimidazole and thiazole derivatives, and other heterocyclic conjugates.
Study on Polyamine Naphthalimide Derivatives
In 2015, Qian Xuhong et al 31 designed and synthesized a series of naphthalimide cyclic amine conjugates (Figure 8). These compounds have long lipophilic alkyl chains, and their activity on human tumor cell lines is equivalent to or stronger than that of aminonaphthalene, and they show anti-proliferative activity on human non-small cell lung cancer, cervical cancer, and colon cancer cells (IC50 value 10-30 μM). It was found that the introduction of long alkyl chain could improve the cytotoxic activity in a certain range. The representative compound 19 has strong inhibitory effect on topoisomerase I and II, and has moderate DNA insertion activity. The IC50 values of human cervical cancer and colon cancer cells are 8.4 and 8.1 μM respectively. Molecular model studies showed that compound 19c may interact with molecular targets by forming topoisomerase or DNA or drug ternary complexes.

Molecular structure of the naphthalimide cyclic amine conjugates (compound 18 and 19).
Tian Zhiyong et al 32 studied the interaction between 7 naphthalimide polyamine conjugates and herring sperm DNA. The analysis of fluorescence spectroscopy and molecular modeling technology shows that these compounds have a rigid planar structure of aromatic ring, which can intercalate into DNA to cause vertical separation of base pairs, resulting in distortion of phosphate skeleton, and polyamine sequences may be combined into small grooves through hydrogen bonds and electrostatic forces. Therefore, the complexes show excellent DNA conformational change ability. It has good anti-tumor research value. Among them, triamine compound 20 (Figure 9) has the strongest DNA binding ability.

Molecular structure of the naphthalimide triamine conjugate (compound 20).
And in 2020, Ufuk Yildiz et al 33 has reported 2 new quaternized 1,8-naphthaliimide derivatives have moderate antimicrobial activities against gram positive bacteria Staphylococcus aureus ATCC 29213, Enterococcus faecalis ATCC 29212 and inhibitory activities against Taqpolymerase and transcriptase. This study has important significance for naphthalimide derivatives to play a role in the field of bacteria.
Structure-activity relationship studies have shown that the N-alkylation of terminal nitrogen atom in polyamine linker is very important for inhibiting the proliferation of tumor cells, and the fragment with N-alkylation of no more than 2 carbon atoms has the best anti-tumor effect. In drug design, the polyamine structure is usually introduced into lead compound molecules to increase the selectivity and affinity of drugs to effective targets, 34 and to inhibit the proliferation of cancer cells by acting on multiple targets. Many compounds designed and synthesized by polyamine modification have achieved good results. Both phthaline-polyamine conjugates and anthrace-polyamine conjugates exhibit better proliferative inhibition than before modification, and the polyamine metabolic pathway has gradually become a promising target for cancer chemopprophylaxis and an effective strategy for chemotherapy.
Glycosylated and Thioglycosylated Derivatives
In 2016, Moylan et al 35 designed the first glycosylated naphthalimide derivative (Figure 10) as a fluorescent probe for the triggered release of enzyme in-vitro. Glycosylated 4-amino-1,8-naphthalimide derivatives have natural glycosidic bonds. Glycosidases can selectively hydrolyze in situ and release naphthalimide as fluorescent imaging agent or therapeutic agent. In a tumor environment, β-Glucuronidase is secreted by inflammatory cells (monocytes/granulocytes) in extracellular necrotic areas, while its activity in healthy tissues is limited to lysosomes. In addition, high concentrations of glycosidase can be expressed in the colonic microflora and then release glycoside ligand probes (therapeutic drugs) in a specific manner in the colon. 36 In-vitro studies of various cancer cell lines have shown that naphthalimide can enter cells only after glycosylation is cleaved by enzymes, which can be used as a new method for probe (drug) targeted delivery.

The first glycosylated naphthalimide derivative (compound 21).
In 2018, Chen Wei et al 37 designed and synthesized a series of new thioglycosyl naphthalimide hybrid inhibitors (Figure 11) and compared their effects on 2 of the glycosyl hydrolase family β-N-acetylhexosaminidase (GH20, GH84). When m = 1 and n = 3, compound 22 had the strongest inhibitory effect (GH20ki = 3.46 μM); compound 23 (when k = 6, GH84ki = 24.81 μM). These compounds contain groups of different sizes and lengths in the connecting region between thioglycosyl and naphthalimide, showing different selectivity and superposition. GH84 β-N-acetylhexosaminidase, a nucleoplasmic enzyme, can catalyze the removal of GlcNAc (O-GlcNAc) attached to serine and threonine residues, and then affect the transcription and translation of proteins.
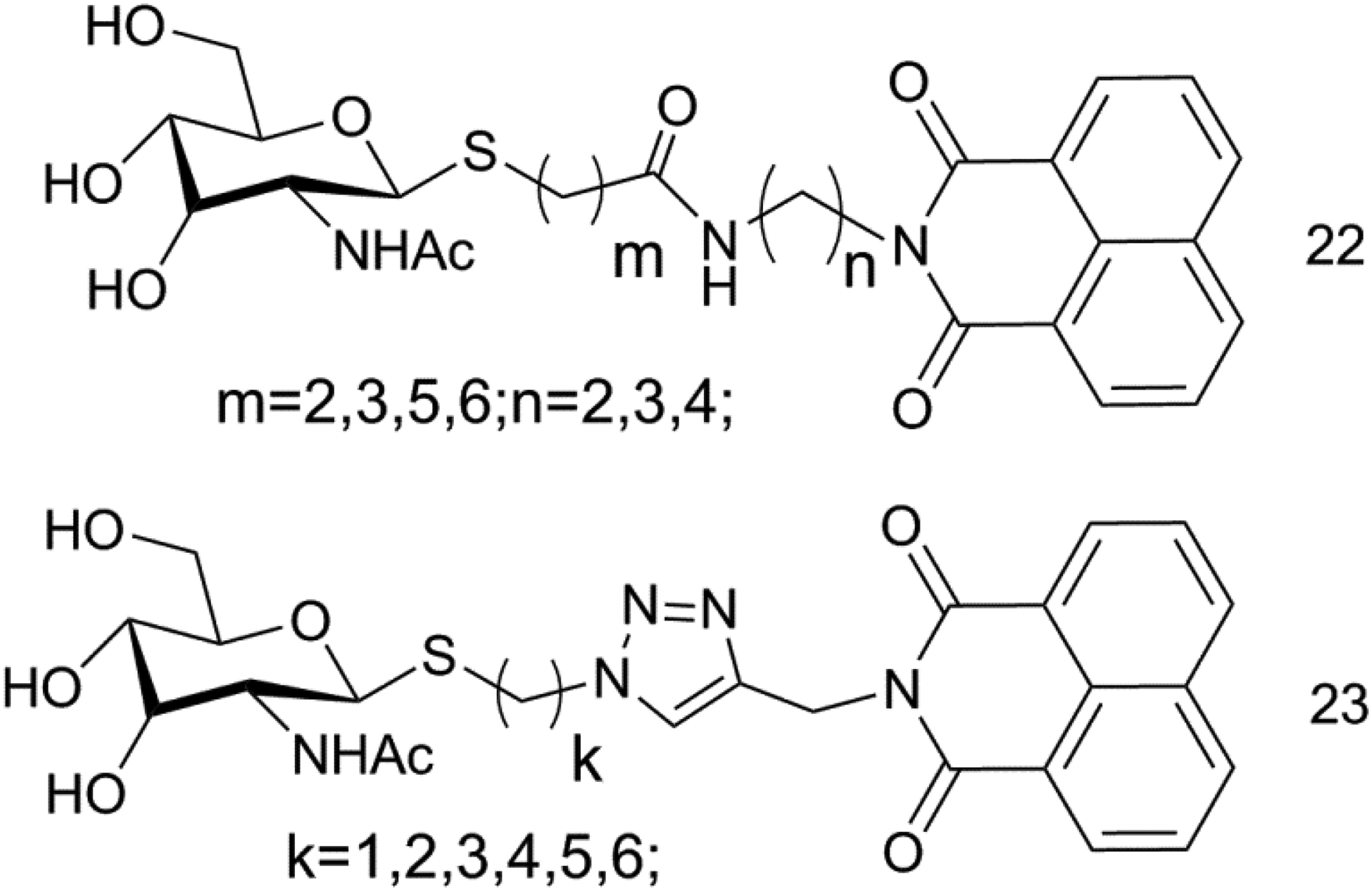
Thioglycosyl naphthalimide derivatives (compound 22 and 23).
Benzimidazole and Thiazole Derivatives
In 2019, Paul et al 38 synthesized 18 isomers of benzimidazole substituted naphthalimides (Figure 12) and studied their anti-proliferative activities on the 60 human cancer cell lines. Most compounds showed good activity, and the average anti-proliferative activity of compounds 24 and 25 was 1.4 and 1.83 μM. The results of UV-Vis spectrum, fluorescence spectrum, CD spectroscopy, and DNA melting experiment show that their fluorescence intensity is good, and DNA is obviously quenched, which may be a static quenching process. Cell cycle arrest experiment confirmed that compound 24 maintained its blocking effect on cells in G2/M phase and induced apoptosis.
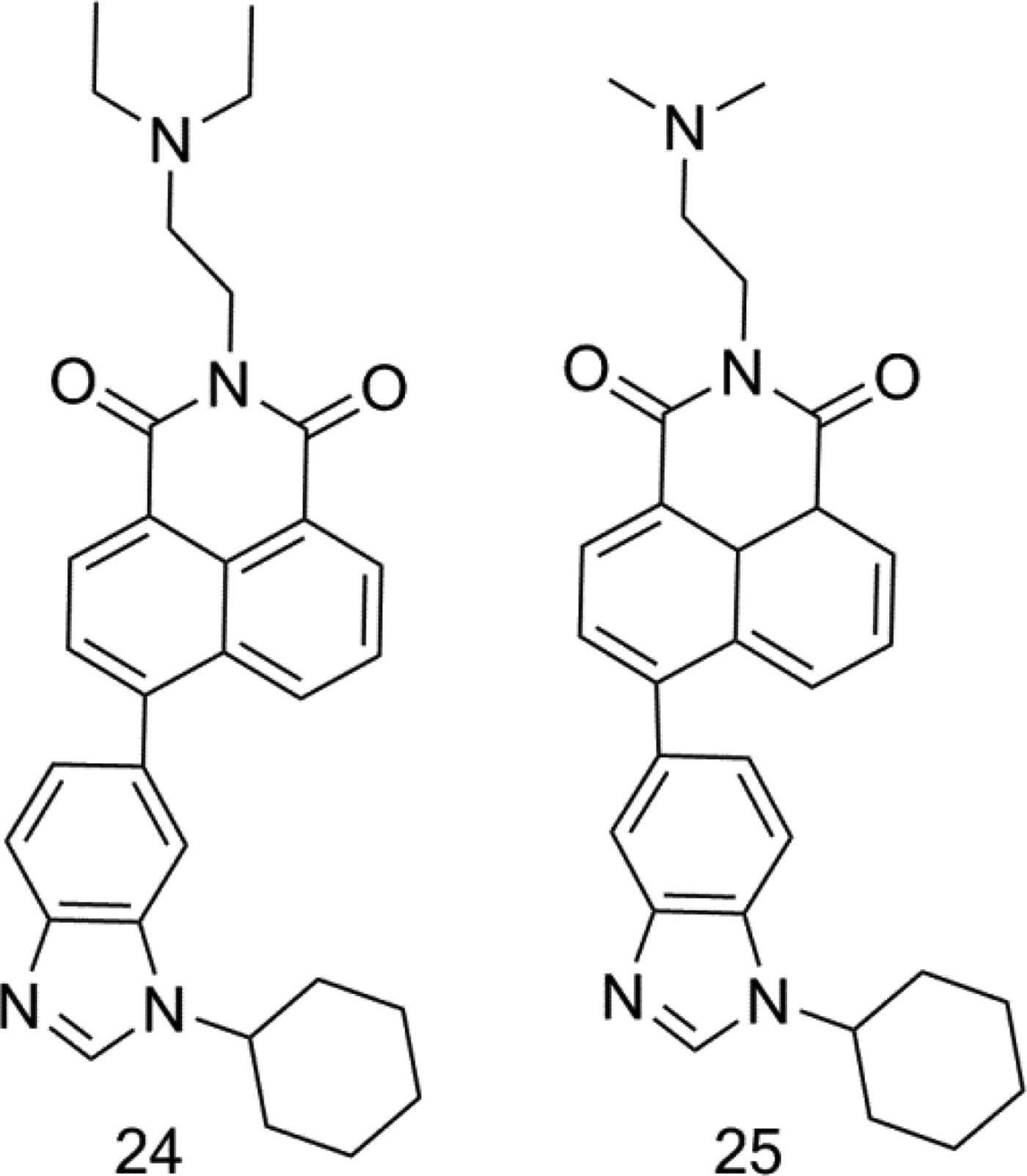
Benzimidazole substituted naphthalimide derivatives (compound 24 and 25).
In 2017, GE Chaochao et al 39 designed a class of compounds with fused thiazole rings (Figure 13), in which the IC50 values of 26a and 26c on human hepatoma cells (SMMC7721 and HepG2) were 1 to 10 μM range. The metastasis and diffusion of cancer cells is one of the main reasons why it is difficult to cure. Animal model studies have confirmed that compound 26a has strong inhibitory effects on 2 HCC models of primary tumor and lung metastasis. The study of cell mechanism and apoptosis showed that 26a blocked the deterioration of liver cancer mainly by inhibiting the growth of cancerous hepatocytes. The compound regulates cadherin and integrin α6 by up-regulating cyclin B1, CDK1, and p21 to inhibit cancer cell migration, and there is no obvious systemic toxicity within the therapeutic dose, which is worthy of further study.

Naphthalimide derivatives with fused thiazole rings (compound 26).
In 2022, Xiao Wang designed and synthesized a series of novel naphthalimide-benzotriazole conjugates (1a-3c) based on 1, 8-naphthalimide as a core skeleton, aiming at G-quadruplexes, that 3a can induce autophagy in A549 cells, which leads to autophagic cell death.
In 2019, Sankara 40 designed and synthesized a series of naphthalimide benzothiazole derivatives (Figure 14), and tested their cytotoxicity in-vitro. The compounds showed good selectivity for colon cancer and breast cancer, and the IC50 of the best derivative 28 was 0.3 to 0.8 μM. It is 10 times that of aminonaphthalene. Subsequently, DNA binding and molecular modeling of these new derivatives were carried out. It was found that the compounds had significant insertion between 2 strands of DNA, especially the naphthalimide ring of compound 28 obviously intercalates between G-C base pairs, and the benzothiazole fragment connected by piperazine extended to the small groove. These compounds are one of the most active derivatives in the current study.

Naphthalimide benzothiazole derivatives (compound 27-29).
The study of naphthalimide fusion heterocycles found that the larger the molecular plane structure, the stronger the ability to embed DNA. In terms of antitumor activity, sulfur heterocyclic fusion derivatives are generally more cytotoxic than their oxygen-containing analogs.
Flavonoids and Heterocyclic Conjugates
In 2017, Dai et al 41 reported a series of flavonoid naphthalimide derivatives (Figure 15). The IC50 value of most compounds on human hepatoma cells (HepG2, SMMC7721) was about 10 μM. In-vitro SAR study found that the small molecule compound 30c with high polypyrimidine motif can enter the target cells through polyamine transporters, locate in mitochondria, selectively generate reactive oxygen in liver cancer cells, and cause apoptosis and migration inhibition. Compared with other compounds, flavonoids derivatives have dual characteristics of biological imaging and anti-metastasis. They can effectively prevent lung metastasis and have fewer side effects. They are a kind of leading compounds of anti-metastasis drugs with research value.

Flavonoid naphthalimide derivatives (compound 30).
In 2019, Kremer et al 42 reported the design and synthesis of the first C-4 functionalized 1,8-naphthalimide heterocyclic hydrazine conjugate, and evaluated the inhibitory effect of Mycobacterium tuberculosis. However, none of the compounds inhibited the growth of Mycobacterium tuberculosis at the tested concentrations.
The introduction of polyamines into naphthalimide derivatives has a more significant inhibitory effect on the proliferation of most cells, which can inhibit topo II, induce LMP, and eventually lead to cell apoptosis and death. Further studies have shown that these derivatives can induce almost 100% apoptosis of HeLa cells through intrinsic mitochondrial pathways. Thus, it provides a new paradigm for the design of novel multi-target anticancer drugs. 43
Expectation
Naphthalimide anti-tumor drugs are promising components in the development of anticancer drugs. Naphthalimide is aromatic fluorophore, which has a good proliferation inhibition effect on a variety of common tumor cells. Naphthalimide compounds have been proved to be good inhibitors of DNA topoisomerase II. Aminofide and mitonafide are the first 2 drugs to be studied in clinical trials. They have good DNA insertion ability, but they have been found to have different degrees of myelosuppression, central nervous system toxicity, and other adverse reactions, which limits their clinical application. Therefore, it is of important research value to modify the structure of amonafide parent nucleus, optimize the synthesis method, reduce the cost, avoid or slow down the serious toxic and side effects of amonafide, and develop amonafide analogs with stronger affinity and better inhibition effect.
In recent years, researchers have modified naphthalimides as DNA intercalators with different structures and chromophores. Studies have shown that the strength of antitumor effect is usually related to the amount of DNA insertion. Therefore, we suspect that the combination of 2 anticancer molecules with different action modes should have better superposition and higher antitumor activity. Qian Xuhong's research group used naphthalimide as the basic framework to coupled amino acids, polyamines, indomethacin, and other structures. Some compounds showed good biological activity and reduced adverse reactions.
Although many compounds represented by amonafide have terminated clinical trials due to N-acetylation toxicity, selectivity, and other reasons, they have π conjugated polycyclic structure and have been confirmed to be good DNA inlays and anti-tumor candidate drugs. Amino acid conjugates have amino side chains and electron-deficient planar chromophores with flexible side chains, which promote DNA affinity through electrostatic or hydrophobic interaction, 7 and thus have better embedding ability and solubility. Amino acids can increase the transport of lipophilic compounds on the cell membrane. Coupling amino acids, aromatic amines, and aliphatic amines to the naphthalimide ring to obtain the target compound is a means to improve its biological activity.
In the future, based on abundant drug design means such as prodrugs, targeted drugs, and computer-aided drug design, and continuous in-depth research on action mechanism, more and more optimized design structures will appear through maintaining proliferation, apoptosis, and other action modes. Naphthalimide compounds have bright development prospects.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Statement
Our study did not require an ethical board approval because it did not contain human or animal trials.