
Abstract
Keywords
Introduction
Lung cancer is the most rapidly growing malignancy, among leading causes of morbidity and mortality worldwide, and poses the greatest threat to human health and life activities. According to the latest data of the World Cancer Report released by the World Health Organization (WHO), lung cancer is the leading cause of cancer-related deaths, accounting for 18% of all cancer-related deaths. Non-small cell lung cancer (NSCLC) is the most important type of lung cancer, accounting for approximately 85% of all cases, and the most common subtype of NSCLC is adenocarcinoma, which accounts for 40% of all NSCLC cases. 1 Although we have developed a wide variety of treatment modalities and found many oncogenes that lead to the development of lung adenocarcinoma, the relapse rate of lung adenocarcinoma is still high; therefore, finding new prognostic markers and therapeutic targets is key for effective treatment of lung adenocarcinoma (LUAD). Owing to the complexity of the mechanism of tumor formation, we need to gain a deeper understanding of the genetic basis of cancer; our current knowledge being far from sufficient. Expression of oncogenes depends not only on the genes, but also on epigenetic alteration without changing the gene sequence. 2
Epigenetics is a research hotspot in recent years; previously, the epigenetic researchers were primarily focused on histone modification and DNA methylation. 3 However, with the development of technologies such as high-throughput sequencing, it has been gradually discovered that mRNA also undergo various modifications such as N6-methyladenosine (m6A), 4 which is dynamically and reversibly regulated through methyl-transferases (“writers”), binding proteins (“readers”), and demethylases (“erasers”). 5 RNA modification is also involved in mRNA export from nuclear, translational, alternative splicing, and stability, and after m6A modification, RNA shows changes in charge, base pairing, secondary structure, and protein-RNA interaction, which in turn affect the transport, localization, translation, and degradation of RNA, and subsequently regulates gene expression. 6 Evidence demonstrates that m6A modification relates to tumor proliferation, differentiation, occurrence, invasion, and metastasis, and plays the role of anti-oncogenes or oncogenes in malignant tumors in recent years.7–9 For example, in acute myeloid leukemia (AML), the fat mass and obesity-associated protein (FTO) inhibitor FB23-2 can significantly inhibit the proliferation of human AML cell lines and primary cells in vitro and promote cell differentiation/apoptosis. 10 Wang et al. found that METTL3 was highly expressed in breast carcinoma, and knockdown of METTL3 decreased the m6A content of B-cell lymphoma-2 (Bcl-2) and inhibited the development of breast cancer. 11 The study explored the role of Wilms tumor 1 associated protein (WTAP) in hepatocellular carcinoma (HCC) cells for the first time and found WTAP expression was significantly upregulated in HCC cells, and guided m6A modification to promote the development of HCC through the Hur-EtS1-P21/P27 axis, providing a potential new target for the treatment and prognosis of HCC. 12 However, in lung adenocarcinoma, the expression, clinical disease correlation, and prognostic values of RNA m6A methylation regulators have not been comprehensively studied. Therefore, it is important to study the function of m6A modification in lung adenocarcinoma and identify the key factor changes of m6A modification to elucidate the pathogenesis of lung adenocarcinoma.
This study used public databases to explore the multi-omics characteristics,clinicopathological features, prognostic predictive value of widely reported m6A RNA regulators in lung adenocarcinoma to identify four cardinal regulatory molecules and established a prognostic prediction model. We also performed immunohistochemical studies on patient tissue samples and validated the expression of four key regulators. Finally, we explored the possible mechanism and potential small molecule drugs that could be developed from these m6A key molecules.
Materials and Methods
Data Collection and Processing
TCGA (http://cancergenome.nih.gov/) is a data-sharing platform for large-scale cancer patient information, including RNA expression, protein expression, mutation, RNA splicing, and corresponding clinical data. It is one of the most comprehensive public databases to date and has been widely used in various fields. 13 Gene expression of lung adenocarcinoma and clinical information of patients were obtained using the TCGA website tool GDC (http://portal.gdc.cancer.gov/), and a total of 535 lung adenocarcinoma samples at various stages and 59 normal control samples were obtained from the TCGA website. Gene expression and corresponding clinical information were collated using Perl software (https://www.perl.org/). The expression levels of genes including writers (WTAP, methyltransferase like 14[METTL14], METTL3, zinc finger CCCH domain containing protein 13 [ZC3H13], RNA binding motif protein [RBM15], and KIAA1429), erasers (FTO and α-ketoglutarate-dependent dioxygenase AlkB homolog 5 [ALKBH5]), readers (YTH N6-methyladenosine RNA binding protein1 [YTHDF1], YTH N6-methyladenosine RNA binding protein 2 [YTHDF2], YTH N6-methyladenosine RNA binding protein 3 [YTHDF3], insulin like growth factor 2 mRNA binding protein 1 [IGF2BP1], IGF2BP2, insulin like growth factor 2 mRNA binding protein 3 [IGF2BP3], HNRNPC, YTH domain containing 1 [YTHDC1], YTH domain containing 2 [YTHDC2], and HNRNPA2B1) were extracted. The discovery of these genes and their relationship with the m6A process has been reported previously.14–17
Mutation Pattern Analysis
CBioPortal (http://www.cbioportal.org/), a comprehensive website integrating various kinds of data such as somatic mutations, DNA copy-number alterations (CNAs), mRNA and microRNA (miRNA) expression, DNA methylation, protein abundance, and phosphorylated protein enrichment, which can facilitate the exploration of multidimensional cancer gene set data and can allow visual analysis across genes, samples, and data types. 18 Users can visualize the patterns of genetic alterations among multiple samples and compare the frequency of genetic alterations in a cancer study or sum up and generalize all related genomic alterations in an individual tumor sample. This website also supports biological pathway exploration, survival analysis, mutual uniqueness analysis among genetic alterations, and selective data download. 19 In this study, we performed mutation pattern analysis using copy number data of 656 patients in six lung adenocarcinoma datasets from the cBioPortal website and downloaded mutation pattern maps for 18m6A regulators.
Construction of Unsupervised Consensus Clusters
Unsupervised consensus clustering was performed to cluster tumor samples into subgroups that were based on the expression matrix of m6A regulators using the ConsensusClusterPlus R package. 20 The cumulative distribution function curve was plotted to obtain the optimal K value (K is the number of subgroups divided).The following parameters were applied for clustering: number of repetitions = 1000 bootstraps; pItem = 0.8 (resampling 80% of any sample); pFeature = 1(resampling 100% of any protein) and clustering algorithm = k means method. Clustering that showed the most significant survival difference was considered.
Construction of Risk Score Model
Lasso regression is a regression analysis method performing both regularization and variable selection to improve the interpretability and prediction accuracy of the statistical models it manufactures. The most excellent subset selection and the connections between lasso coefficient estimates can be identified to form the prognostic model.
21
We evaluated the prognostic performance of m6A regulators by a univariate Cox regression analysis in the training cohort. Followed by Lasso regression, that was based on 15m6A regulators to identify the m6A regulator gene signature associated with high lung adenocarcinoma. This was performed using the glmnet R package. Ten-fold cross-validation was used to determine the values of λ and we selected the λ with the smallest partial likelihood deviance. According to the λ value, each remaining gene was assigned with a LASSO coefficient, and the m6Sig score was generated using the following formula:

The risk score of each sample was obtained using the risk score formula and correlated with the clinicopathological parameters of the patient.
Patients
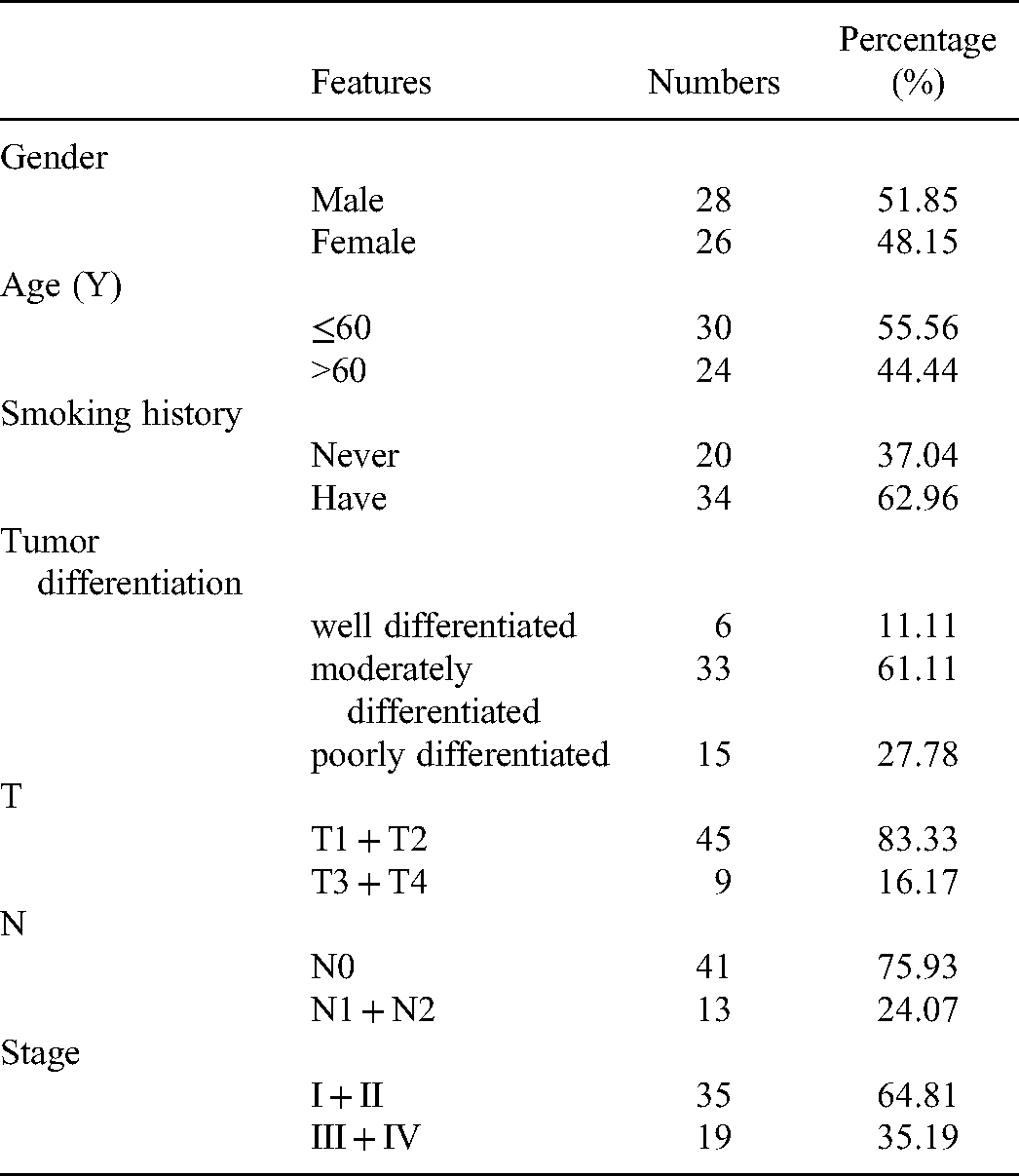
As a retrospective study, we collected historical case data of patients from the First Affiliated Hospital of Xi’an Jiaotong University (our study was approved by the Ethics Committee of the First Affiliated Hospital of Xi’an Jiaotong University [ Approval number: XJTU1AF2020LSK-169 ]), who were enrolled in the hospital between June 2015 and May 2020 and finally selected 54 patients who were eligible for inclusion in our study according to the following criteria: 1. Patients with a confirmed diagnosis of lung adenocarcinoma based on histopathological and radiological features; 2. Patients who had complete clinical, pathological, laboratory, and follow-up data; 3. Patients who were fully informed of the aims and scope of the study and signed an informed consent form; and 4. Patients who were able to participate in the entire follow-up process. The end of the follow-up period was May 2021 and the endpoints were overall survival (OS) and Progression-Free-Survival (PFS). Verbal informed consent was obtained from the patient(s) for their anonymized clinical information to be published in this article.The reporting of this study conforms to STROBE guidelines 22 and REMARK guidelines. 23
Immunohistochemistry
We performed immunohistochemical staining analysis using tissues from these 54 patients with lung adenocarcinoma and corresponding normal lung tissues to explore the relationship between the protein expression level of m6A regulators and the prognosis of patients. According to instructions of the reagent manufacturer, m6A regulators were analyzed by IHC applying formalin-fixed, paraffin-embedded tissue blocks. Briefly, paraffin parts were dewaxed, and endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide for 10 min at 37corresponding primary antibodies (Proteintech Group, China) overnight at 4℃ overnight and then with a secondary antibody at 25 °C overnight. The sections were counterstained with hematoxylin. The whole segments were evaluated by two pathologists who were blinded to the patients’clinical information. The samples were scored based on the intensity and proportion of the cells stained.
Exploration of Target Genes of m6A Regulators
To further explore the possible mechanisms, we took advantage of the m6A2Target to explore the downstream genes that these four key m6A molecules may act on. m6A2Target (http://m6A2target.canceromics.org/) is a comprehensive database for the target genes of writers, erasers, and readers (WERs) of m6A modification. It integrates high confidential targets ratified by low-throughput experimentations and potential targets with binding evidence indicated by high-throughput sequencing such as RIP-seq, ChIP-seq, and CLIP-Seq, or as inferred from m6A WER perturbation followed by high-throughput sequencing such as m6A-Seq, Ribo-Seq, and RNA-Seq. 24 Our results were presented in three parts: validated targets, binding, and perturbation, among which validated targets were not only predicted based on sequencing results, but also validated by a series of subsequent experiments; binding was limited to some omics data, which was predicted by the target genes of the interactions between proteins and DNA, proteins and RNA, and proteins and proteins. Regarding the perturbation column, after knockdown and overexpression of these three types of methylases, the RNA level, protein level, translation level, methylation level, and other changes in this gene can be shown in this column after confirmation from the m6A-seq, transcriptome, proteome,translation products, and other data.
Prospective Drug Prediction
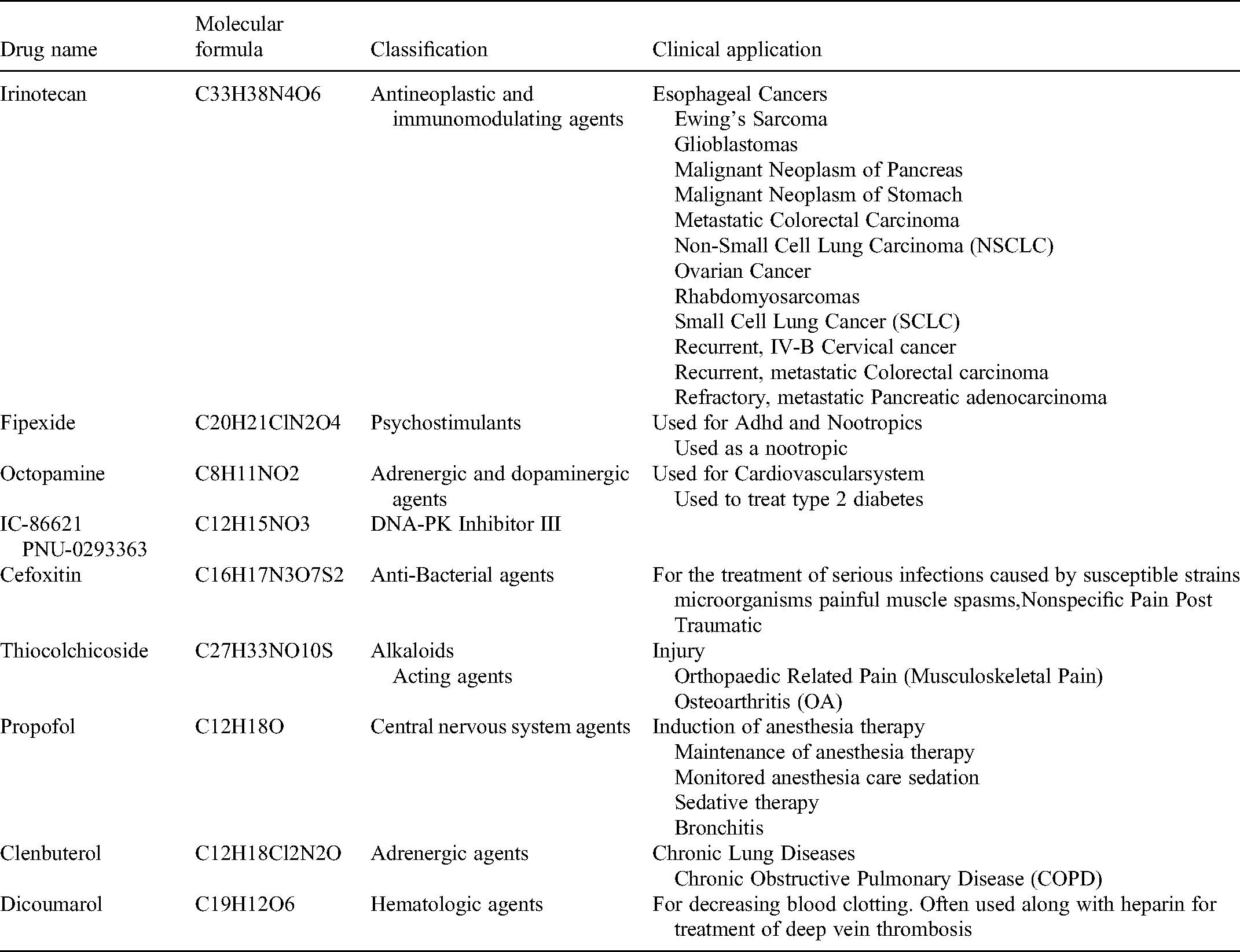
Connectivity map (CMap) is a biological application database established by researchers to measure the interconnection between small molecule drugs, gene expression, and diseases using the genetic differences shown by small-molecule drugs after treating human cells.25,26 We used this database to explore prospective agents for lung adenocarcinoma based on the resulting key genes. We fixed four key genes as UP (upregulated) genes and DOWN (downregulated) genes according to their expression trends in lung adenocarcinoma, and the screening conditions were: non-percent null≥50, p value < .05, and −1 < meanscore < 0 compounds, respectively, and obtained 31 small molecule compounds that may become prospective drugs for lung adenocarcinoma. The structure of these drugs was obtained from the DrugBank database (https://www.drugbank.ca/).
Statistical Analysis
All data and figures were obtained by R (version 3.6.3), a risk scoring system which was established via consensus clustering analysis and LASSO regression. Consensus clustering analysis was used to categorize LUAD patients into subtypes, and Kaplan–Meier plots, displayed as hazard ratios (HRs) with 95% confidence intervals (CIs), were performed to compare the OS of patients of different subclusters and risk groups. Univariate Cox regression was used to analyze the clinical features and the risk score for connection with overall survival (OS). The independent prognostic value was indicated by multivariate Cox regression analysis. The statistical difference between the two groups was calculated using the Wilcoxon rank sum-test. Statistical significance was set at p < .05.
Results
Differential Expression, Correlation, and Mutation Pattern of m6A RNA Methylation Regulatory Genes in Lung Adenocarcinoma
Using the TCGA platform, the mRNA expressions of 18m6A regulators were downloaded, including writers (WTAP, METTL14, METTL3, ZC3H13, RBM15, and KIAA1429), erasers (FTO, and ALKBH5), readers: (YTHDF1, YTHDF2, YTHDF3, IGF2BP1, IGF2BP2, IGF2BP3, HNRNPC, YTHDC1, YTHDC2, and HNRNPA2B1), and the data contained 535 cancer samples and 59 normal samples. First, we explored the interrelationship between these 18m6a-regulated genes. In the spearman correlation analysis (Figure 1A), YTHDC1, YTHDC2, FTO, and RBM15 and METTL14 were positively correlated with each other. In addition, ALKBH5 was not significantly associated with any of the genes in lung adenocarcinoma.

Multi-platform features of RNA m6A methylation regulators in LUAD. (A) Spearman correlation analysis of 18 RNA m6A methylation regulators,Red color represents a positive correlation and blue color represents a negative correlationn Genetic alteration of RNA m6A methylation regulators; (B) Mutation pattern maps of 18 RNA m6A methylation regulators; (C) Expression of m6A methylation genes in lung adenocarcinoma.*p < .05; **p < .01; ***p < .001.
Furthermore, FTO was negatively correlated with HNRNPA2B1, HNRNPC, and IGF2BP1. Copy number data from 656 patients in six lung adenocarcinoma datasets of the CBioPortal (http://www.cbioportal.org/) website were used for mutation pattern analysis, and we found that 32% of 656 patients had genetic alterations and obtained mutation pattern maps for each gene (Figure 1B).
A heatmap of the expression of these m6a-regulated genes was constructed using TCGA data (Figure 1C), and we found that the expression of eraser ALKBH5 (p = .235), readers: YTHDC1 (p = .419) and YTHDC2 (p = .339) in lung adenocarcinoma data was not statistically significant (Table 1). Thus, we focused on the remaining 15 genes for the subsequent observations. Heatmap indicated that the expression of WTAP, ALKBH5, FTO, and ZC3H13 and METTL14 was decreased in lung adenocarcinoma compared to the control tissues, and the expression of the remaining nine m6A genes was increased compared to control tissues, wherein HNRNPC expression differed most significantly (p < .001).The above analysis revealed the multi-omics characteristics of m6A regulators in LUAD, suggesting that these m6A regulators were different from each other in LUAD.
Gene expression analysis.

Consensus Clustering of LUAD Tissue Samples Based on the mRNA Expressions of m6A Regulators
To investigate the relationship between gene expression and population prognosis, we divided the samples into different groups with K = 2–9, using a class discovery tool “ConsensusClusterPlus” and the “K” was used to indicate the number of groups (Figure S1). We referred to the cumulative distribution function and the area under the curve to determine the optimum K value at which the sample distribution achieves the maximal stability and found that at K = 2, the lung adenocarcinoma samples could be divided into groups with high non-overlap and intra-group correlation (Figure 2A–C). We further examined whether there were significant differences in demographic and clinicopathological characteristics such as age, sex, tumor stage, T (T represents tumor size, including T1, T2, T3, T4), M (M represents tumor metastasis, including M0, M1, and MX), N (N represents tumor lymph node metastasis, including N0, N1, N2, and N3), and survival status between the two groups and constructed a heatmap. The sample clusters were significantly related to clinical characteristics, including sex (p < .05) and M (p < .01) and survival status (p < .001) (Figure 2D). We performed survival analysis of these two sample groups (n = 494), and observed that there was a statistically significant difference between the survival of group 1 and group 2 (p = .00017) (Figure 2E). This indicated that typing based on m6A-related genes was significantly associated with prognosis. These m6A-related genes can be used as potential prognostic factors in patients with LUAD.
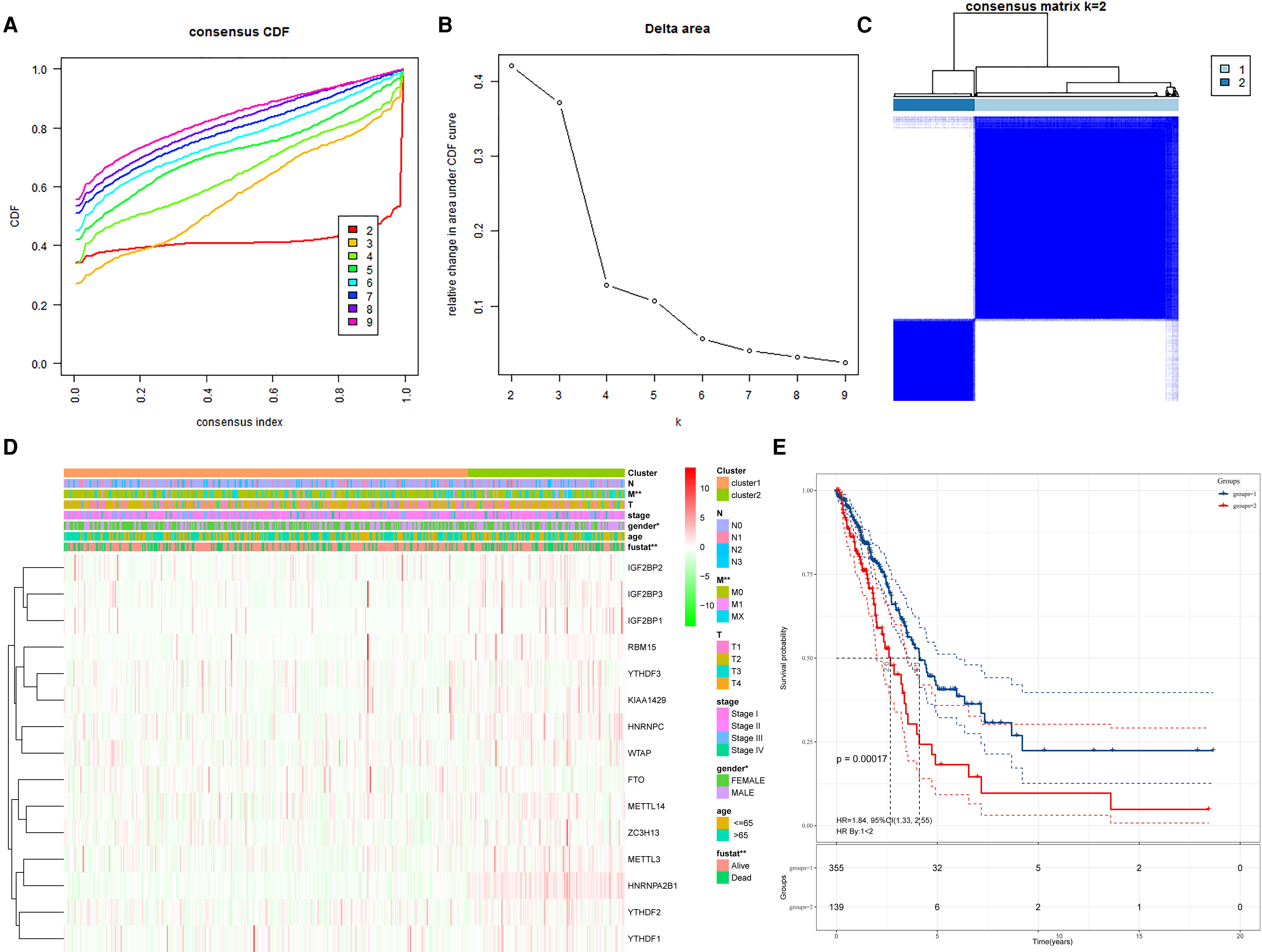
Identification of subgroups with distinct prognosis using consensus clustering. (A) Consensus clustering distribution function (CDF) for LUAD; (B) Relative changes in the area under the CDF curve for LUAD; (C) Consensus clustering matrix for k = 2; (D) The heatmap of clusters and lung cancer-related clinical characteristics; (E) Kaplan-Meier curves of overall survival (OS) in LUAD patients between group 1 and group 2. *p < .05; **p < .01; ***p < .001.
Construction of Risk Score Model
To considerably predict the outcome of patients with LUAD, we randomly divided the samples with complete survival information (n = 494) into training cohort (n = 248) and test cohort (n = 246) and evaluated the prognostic performance through a univariate Cox regression analysis in the training cohort. Among these 15 genes, IGF2BP2, IGF2BP1, HNRNPC, KIAA1429, IGF2BP3, METTL3, WTAP, and HNRNPA2B1 were significantly correlated with patient prognosis (Table 2). To accurately predict survival of lung adenocarcinoma with m6A RNA methylation genes, we conducted LASSO Cox regression on 15m6A-related genes. Finally, according to the combined algorithm of lasso regression and multivariate Cox regression, and considering the functional roles of these genes in biological processes, four genes were selected (writer: METTL3 and three readers: IGF2BP2, HNRNPC, and HNRNPA2B1) to construct a survival risk prediction model in the training cohort, and we used a flowchart to show this process (Figure S3).The formula was generated as follows:
Prognostic performance by a univariate Cox regression analysis.

This methodology allowed us to calculate the risk scores for all samples by combining the expression levels and risk coefficients of genes. According to this formula, the patient was categorized by us into the high-risk group with all patients’ risk score ≥ median value, and patients in the low-risk group with a risk score < median value (Figure 3A and B). A heatmap between the risk group and clinical features was constructed (Figure 3C and D). In the training cohort (Figure 3C), the risk group was correlated with T (p < .05) and survival status (p < .01), whereas in the test cohort (Figure 3D), we also found that the risk was significantly correlated with T (p < .05), M (p < .05), stage (p < .01), and fustat (p < .01). The survival curve was obtained via the Kaplan–Meier analysis, which demonstrated that patients with higher risk scores tended to have poorer prognosis either in the training set (p < .01) (Figure 3E) or in the test set (p < .01) (Figure 3F).

Construction of risk score model. (A) Risk scores for patients in the training cohort; (B) Risk scores for patients in the test cohort; (C) Heatmap between risk group and clinical features in the training cohort; (D) Heatmap between risk group and clinical features in the test cohort; (E) Kaplan–Meier cumulative curves of OS based on the risk scores calculated by the risk score model in the training cohort; (F) Kaplan–Meier cumulative curves of OS based on the risk scores calculated by the risk score model in the test cohort.*p < .05; **p < .01; ***p < .001.
Independent Prognostic Evaluation of the Risk Score Model
According to the risk score model, we evaluated the result of each cohort by plotting its receiver operating characteristics (ROC) and calculating the area under the ROC curve (AUC) (Figure 4A and B), AUC = 0.69 in the training cohort, while in the test set, AUC = 0.66, illustrating that our prognostic risk model has good validation power, and the risk score of each sample was based on the four m6A regulator gene signature (METTL3, IGF2BP2, HNRNPC, and HNRNPA2B1) were used as a risk factor in combination with clinical features to perform univariate and multivariate Cox regression analyses (Figure 4C and D). The univariate Cox regression analysis revealed that T (HR,1.623; 95% CI:1.310-2.011; p = 9.57 × 10−6), N (HR:1.793, 95% CI:1.465–2.194; p = 1.47 × 10−8), stage (HR:1.645; 95% CI:1.397–1.937; p = 2.42 × 10−9) and Risk score (HR:1.506, 95% CI:1.286–1.763; p = 3.61 × 10−7) were substantially related to patient prognosis. The multivariate Cox regression analysis revealed that only Risk score (HR:1.380, 95% CI:1.167 − 1.633; p < .001) had a strong correlation with patient prognosis. Thus, we can conclude that the risk score can be an independent prognostic factor for lung adenocarcinoma. Subsequently, a nomogram was constructed to facilitate clinicians in predicting the outcome from which we could acquire a score for each patient (Figure 5A). Age, gender, T, N, M stages, and risk score were given points according to their effects on the outcome, and by summing all the points up, we could obtain total points for each patient. To further validate our nomogram, we constructed calibration curves and calculated the 3-year and 5-year survival rates (Figure 5B and C).

Independent prognostic evaluation of the risk score model. (A) The ROC curve of risk score of training cohort; (B) The ROC curve of risk score of test cohort; (C) Univariate Cox regression analysis for evaluating the prognostic role of clinicopathological characters and risk score in LUAD; (D) Multivariate Cox regression analysis for evaluating the prognostic role of clinicopathological characters and risk score in LUAD. Hazard ratio (95% confidence intervals).

A nomogram built to facilitate clinicians to predict outcome of patients. (A) The nomogram plot to predict 3-year, 5-year overall survival. Summing up each point of clinicopathological characters and risk score to predict overall survival; (B) Alibration curves to compare the predicted 3-year survival by nomogram with the actual survival of the patient; (C) Alibration curves to compare the predicted 5-year survival by nomogram with the actual survival of the patient.
The Protein Expression of Four m6A Regulators in LUAD by IHC Analysis
To further validate whether the four key genes used to construct the risk model (writer: METTL3 and three readers: IGF2BP2, HNRNPC and HNRNPA2B1) are associated with patient prognosis, we performed immunohistochemical staining analysis using tissues from 54 patients with lung adenocarcinoma and corresponding normal lung tissues from the First Affiliated Hospital of Xi’an Jiaotong University to explore the relationship between the protein expression level of m6A regulators and the prognosis of patients. The clinical data of the patients are displayed in Table 3. As expected, we found that METTL3, IGF2BP2, HNRNPC, and HNRNPA2B1 were highly expressed in malignant LUAD tissues compared to normal lung tissues (Figure 6). An appraisal of whether the abnormal expression of m6A relevant genes was correlated with the outcomes and clinical progression of LUAD patients was done by analyzing the overall survival and progression-free survival data of our cohort. The results showed that these four genes were significantly related with overall and progression-free survival (Figure 7). Corroborating previous studies, our analysis revealed that the expression of the four m6A regulatory genes was mostly related to the prognosis of LUAD patients. Patients with high expression of METTL3 may have greater OS and PFS (Figure 7A and E). For the remaining three readers, IGF2BP2, HNRNPC, and HNRNPA2B1, their high expression was associated with an aggravated prognosis.

The protein expression of m6A regulators in LUAD tissues and paired non-tumor samples(400X).

Survival analysis of patients with lung adenocarcinoma. (A) Kaplan-Meier survival curves of OS for high- and low-METTL3 expression groups; (B) Kaplan-Meier survival curves of OS for high- and low-IGF2BP2 expression groups; (C) Kaplan-Meier survival curves of OS for high-and low-HNRNPC expression groups; (D) Kaplan-Meier survival curves of OS for high-and low-HNRNPA2B1 expression groups; (E) Kaplan-Meier survival curves of PFS for high- and low-METTL3 expression groups; (F) Kaplan-Meier survival curves of PFS for high- and low-IGF2BP2 expression groups; (G) Kaplan-Meier survival curves of PFS for high-and low-HNRNPC expression groups; (H) Kaplan-Meier survival curves of PFS for high-and low-HNRNPA2B1 expression groups.
Clinical data of LUAD patients.
Exploration of Target Genes of m6A Regulators and Prospective Drug Prediction
Validation indicated the four genes used to construct the risk model (writer: METTL3 and three readers: IGF2BP2, HNRNPC, and HNRNPA2B1) may be important in lung adenocarcinoma. Further, we utilized the m6A2Target website, from which the downstream genes that these four M6a key molecules may act on, were explored, and we represented them separately from three major aspects (validated targets, binding, and perturbation) (Table S2). The results not only included the downstream target genes with different expression levels, methylation levels, and translation levels, but also predicted the target genes of the interactions between protein and DNA, protein and RNA, and protein and protein, and obtained many experimentally confirmed downstream binding genes. Figure S2 showed the downstream genes and possible binding sites. Furthermore, using the CMap database, prospective small molecule drugs that may inhibit the development of lung adenocarcinoma were predicted, we obtained 31 prospective drugs (Table S3), and selected 10 of them for display (Table 4). We explored their probable pathways, and found that these drugs may not only be related to olfactory transduction pathway, neuroreceptor interaction, and systemic lupus erythematosus, but also to some tumor-related pathways such as MAPK, JAK-STAT, and B cell signaling pathways (Figure 8E).

The drug-pathway network to indicate pathways significantly affected by potential therapy drugs of LUAD. The red nodes represent drugs, and the orange nodes represent the pathways affected by drugs.
10 prospective small molecule drugs.
Discussion
The underlying reason for the poor prognosis of non-small cell lung cancer is the difficulty in early diagnosis. Over 70% patients are often diagnosed at an advanced and usually fatal stage, when therapeutic measures are usually not very effective. 27 Lung adenocarcinoma accounts for a large proportion of non-small cell lung cancer; hence, it is essential to understand the development mechanism for its proper diagnosis and treatment, in particular, finding the key molecules that can help to predict prognosis. 28 Classical molecules such as TP53 and EGFR have been discovered, and various targeted drugs have been developed with excellent technical and clinical success. 29 Emerging evidence, however, has indicated that lung adenocarcinoma development is affected by both epigenetic and genetic variations. 30 Gene expression is regulated by epigenetics from multiple levels, including DNA methylation, RNA regulation, histone modification, and chromosome remodeling. 31 M6A is a very important part of epigenetics and extensively occurs in different types of more than 7600 genes and over 300 non-coding RNAs, the abundance of which varies from 0.1% to 0.4% of total adenosine residues.32–35 It is formed in the nucleus with the activity of writers and can also be removed by erasers. The readers will subsequently bind to the m6A modification sites and produce corresponding biological effects. 36 M6A RNA methylation becomes a new frontier research hotspot,37–40 which is closely associated with cancer pathogenesis and has extensive therapeutic implications in cancer. However, in lung cancer, including lung adenocarcinoma, the molecular mechanism of action of m6A and its diagnostic value need to be further elucidated.
In our study, based on bioinformatics approaches, we analyzed the expression of 18m6A regulators in lung adenocarcinoma versus normal tissues and discovered that the majority of the studied genes showed considerable alterations in gene expression levels of LUAD tumors when compared with normal tissues, however two readers: YTHDC1 (p = .419) and YTHDC2 (p = .339) and one eraser: ALKBH5 (p = .235), did not reach statistical significance in terms of their expression in lung adenocarcinoma data. Previous studies have shown that the YTH domain family and its RNA-binding domain are the first readers to be recognized. YTHDC1 regulates mRNA splicing, whereas YTHDC2 binds to certain noncoding RNAs to perform its function.41,42 The binding sites of each protein are known, all the YTH domain-containing proteins bind to m6A, except YTHDC2. Lobo et al. stated that YTHDC1 expression was relatively higher in seminomas than embryonal carcinomas. 43 Some researchers have found that in gynecologic tumor cell lines, reduction of YTHDC1 protein levels, due to hypoxia, alters RNA splicing. 44 ALKBH5 was the second m6A demethylase identified, both in vitro and in vivo, and it may exhibit higher RNA-binding affinity than FTO. 45 The expression of ALKBH5 is activated in a hypoxia-inducible factor (HIF)-dependent manner and results in a reduction of the total RNA m6A level. Hypoxia can induce breast cancer in an HIF-dependent manner, suggesting that ALKBH5 may play an important role in oncogenesis. 46
These studies suggest that YTHDC1, YTHDC2, and ALKBH5 have an important role in tumors; however, they are not well studied in lung cancer and need to be elucidated in future studies. In our study, consensus clustering based on expression profiles of the 15m6A-related genes could stratify the LUAD cohort into two non-overlapping subgroups regarding OS. There were significant differences in sex (p < .05), M (p < .05), and survival status (p < .05) between the two subgroups. Moreover, according to the combined algorithm of lasso regression and multivariate Cox regression, four key genes were selected (writer: METTL3 and three readers: IGF2BP2, HNRNPC and HNRNPA2B1) to construct a survival risk prediction model in the training cohort. Overall, as with some other cancers, the expression of m6ARNA methylation regulators is closely related to LUAD malignant clinicopathological features. Moreover, these findings also contribute to the development of novel therapeutic approaches by characterizing the expression of each individual m6A methylation regulator in LUAD, since chemicals targeting m6A methylation are potential cancer therapies.
METTL3 is the most important catalytic enzyme in the m6A methyltransferase system and can act as a proto-oncogene or tumor suppressor gene involved in biological processes such as tumorigenesis, proliferation, invasion, migration, cell cycle, and differentiation.47,48
METTL3 functions as a writer to control eukaryotic mRNA translation during post-transcriptional methylation. It increases the translation of certain mRNA, including epidermal growth factor receptor (EGFR) and the Hippo pathway effector TAZ in human cancer cells. 49 Studies have shown that knockdown of METTL3 can inhibit the survival and proliferation of lung cancer A549 and H1299 cells, induce apoptosis, and change the protein expression and phosphorylation of PI3K/AKT signaling pathway members, thus exerting the tumor suppressor effect. 50 In our research, we found that METTL3 expression was increased in lung adenocarcinoma samples compared with normal samples, which may be consistent with conclusions of many previous studies and requires the support of subsequent experiments. Consistent with our findings, Sun et al. found that NSCLC patients with high METTL3 expression were associated with better OS. 51 However, Gregory et al. indicated that the upregulation of METTL3 promotes the growth, survival, and invasion of human lung cancer cells. 52 JIN et al. found that METTL3 enhanced translation efficiency and promoted metastasis of NSCLC by interacting with eIF3h to cyclize mRNA and improve the efficiency of ribosome recycling and reuse. 53 The study also found that inhibition of METTL3 may not only be a target for the treatment of NSCLC, but may also enhance chemosensitivity. Thus, it is believed that METTL3 may be a potential diagnostic and therapeutic target for NSCLC. The oncogenic property of IGF2BP2 has been well documented in various types of solid cancers, for instance, augmented levels of IGF2BP2 have been detected in acute myelocytic leukemia primary cells. 54 Moreover, existing literature has indicated that IGF2BP2 is often amplified in pancreatic cancer tissues versus normal pancreatic tissues and is associated with a poor survival rate. 55 Analysis of the Cancer Genome Atlas (TCGA) revealed that the expression level of IGF2BP1 in NSCLC tissues was significantly higher than that in normal tissues, and downregulation of IGF2BP1 inhibited NSCLC cell proliferation, 56 which was consistent with our conclusion. HNRNPC and HNRNPA2B1 are hnRNPs superfamily proteins associated with m6A modification, we performed univariate Cox regression analysis of each selected m6A RNA methylation genes, therefore, we performed univariate Cox regression analysis of each selected m6A RNA methylation genes, and discovered that HNRNPC (HR: 1.8, 95% CI:1.22-2.656; p = .003) and HNRNPA2B1 (HR: 1.8, 95% CI:1.22-2.656; p = .003) were also a hazard of OS. For breast cancer cells, the repression of HNRNPC could inhibit cell proliferation and tumor development. 57 In gastric cancer, HNRNPC has also been identified as a prognostic and therapeutic marker. 58 However, it has been rarely reported that HNRNPC and HNRNPA2B1 were involved in the prognosis of LUAD. Our findings suggest an important role for hnRNPs superfamily proteins in lung adenocarcinoma, and in previous studies, we found a very interesting phenomenon that m6A methylation modification of RNA can cause changes in the secondary structure of RNA. Some m6A recognition proteins do not directly recognize and bind m6A methylation sites, but recognize RNA structures with changed conformations, and then regulate gene expression, this phenomenon is called “m6A switch”. Crystal structure analysis and bioinformatics confirmed that HNRNPA2B1 does not contain a hydrophobic pocket that binds to the m6A methylation site, possibly by using the m6A switch mechanism to recognize the m6A modification site. 59 Similarly, the recognition binding of m6A modification by ribonucleoprotein HNRNPC/G is also indirect; HNRNPC/G is involved in the processing and maturation of target mRNAs through the m6A switch mechanism. 60
TP53, EGFR, and other prognostic markers have been identified in previous studies, and because of the heterogeneity of NSCLC, the genes that are involved, however, might vary greatly among different individuals. 61 Therefore, it prompted researchers to build gene expression profiles that were composed of multiple prognostic genes for use in patient risk stratification, and a four-gene risk score model was built. It stratified the OS of patients with LUAD into high-and low-risk groups, which exhibited entire differences in survival, and the risk score was used as an independent predictor of lung adenocarcinoma.
At present, TNM staging is commonly performed to assess patient prognosis. Because of the limited risk factors included in the TNM staging system, it is not possible to conduct an accurate prediction for NSCLC. In the present study, based on characteristics of diagnostic age, gender, TNM stage, and risk score, a nomogram was built to facilitate clinicians to predict outcomes. To some extent, it was expected to address the issue of prognostic heterogeneity caused by a single factor or insufficient risk factor analysis. Further, our study utilized tissue samples from a realistic population to validate the protein levels of four key genes; similar to previous studies, our analysis revealed that the expression of the four m6A regulatory genes was mostly related to the prognosis of LUAD patients. Patients with high expression of METTL3 had greater OS and PFS, and as for the remaining three readers, IGF2BP2, HNRNPC, and HNRNPA2B1, their high expression was associated with an aggravated prognosis; however, owing to the small number of our external validation cohort, conclusions of causality are limited, and further large cohorts in the future are needed for validation.
Epigenetics plays an important role in the occurrence, development, diagnosis, and treatment of malignant tumors. Multiple new drugs targeting DNA methylases or histone-modifying enzymes have been used for the treatment of malignant tumors. For example, the DNA methyltransferase inhibitors azacitidine and decitabine have been approved by the US Food and Drug Administration (FDA) for the clinical treatment of myelodysplastic syndrome.62,63 Chidamide, a histone deacetylase inhibitor, has been approved by the China Food and Drug Administration (CFDA) for the treatment of peripheral T. lymphocytoma, and the clinical trial for non-small cell lung cancer has entered phase III. 64 In recent years, targeted therapy with m6A modification as the core content has become a research hotspot. Bu et al. found that when the lung adenocarcinoma cell line A549 was treated with the combination of FTO inhibitor rhein and pemetrexed, the IC50 was reduced from 2.05 µmol/L to 0.62 µmol/L, and the antitumor activity was significantly improved. 65 Duffy et al. found that the cell survival rate was 65% when lung adenocarcinoma cell line A549 was treated with adriamycin, while the cell survival rate was 16% when FTO specific inhibitor MA was combined with adriamycin. 66 The phenomenon indicated that MA significantly increased the sensitivity of lung adenocarcinoma cells to adriamycin. Han et al. found that subcutaneous injection of melanoma cells in YTHDF1 knockout mice, combined with programmed cell death ligand 1 (PD-L1) immune checkpoint blocking therapy, achieved better therapeutic effects. 67 The above studies suggest that we can find new cancer treatments by exploring the m6A-related drugs. In our study we used bioinformatics methods to identify 31 prospective drugs by compound structure comparison and compound functional analysis, and selected the most representative 10. Furthermore, the possible pathways were explored, and it was found that these drugs may not only be related to olfactory transduction, neuroactive ligand–receptor interaction, and systemic lupus erythematosus, but also related to tumor-related pathways such as the MAPK signaling pathway, JAK-STAT signaling pathway, and B cell signaling pathway.
A previous study by Wang et al. 68 has explored five m6A regulatory factors closely related to the overall survival (OS) of patients with lung adenocarcinoma and these exhibited potential prognostic value. Zhang et al. 51 found that m6A-related genes were differentially expressed in LUAD compared to normal patients; however, these studies have only preliminarily explored the possible relationship between m6a-related factors and lung adenocarcinoma, hence the possibility of use of m6a-related genes as new prognostic markers, remains to be elucidated. However, based on previous studies, we further conducted preliminary observational validation with realistic clinical samples and explored the possible downstream binding sites and predicted possible small-molecule drugs. Our findings may provide new insights for us to further explore the possible mechanism of action of these m6A key molecules; however, our analysis is based on the prediction of bioinformatics data and our sample was too small to be representative, and we conducted an observational rather than a controlled study. Further relevant experimental and clinical studies are therefore required.
Conclusion
In conclusion, we explored the expression differences, clinicopathological characteristics, and prognostic value of widely reported m6A RNA regulators in LUAD, established a prognostic prediction model, used patient tissues for immunohistochemical expression verification, explored the potential mechanism and related drugs of these m6A key molecules for the first time, and found that the expression of m6A RNA methylation regulators may play a significant role in LUAD, making them effective diagnostic and prognostic factors. However, more prospective studies are needed to validate their prognostic function.
Supplemental Material
sj-docx-1-tct-10.1177_15330338221085373 - Supplemental material for Four m6A RNA Methylation Gene Signatures and Their Prognostic Values in Lung Adenocarcinoma
Supplemental material, sj-docx-1-tct-10.1177_15330338221085373 for Four m6A RNA Methylation Gene Signatures and Their Prognostic Values in Lung Adenocarcinoma by Yuzhu Wang, Xu Zhao, Jing Li, Xuan Wang, WeiBin Hu and Xiaozhi Zhang in Technology in Cancer Research & Treatment
Footnotes
Abbreviations
Acknowledgments
The authors acknowledge TCGA database for providing their platforms and contributors for uploading their meaningful datasets.
Author Contributions
Xiaozhi Zhang and Yuzhu Wang conceived and designed the research. Yuzhu Wang,Xu Zhao, downloaded and collated the data. Yuzhu Wang, Jing Li, Xuan Wang and WeiBin Hu performed bioinformatics analysis and statistical analysis on the data. Yuzhu Wang wrote the first draft of the article. Yuzhu Wang, Xiaozhi Zhang reviewed and edited the manuscript. All authors have read and agreed to the final version of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval Statement
Our study was approved by the Ethics Committee of the First Affiliated Hospital of Xi’an Jiaotong University (Approval number: XJTU1AF2020LSK-169).Verbal informed consent was obtained from the patient(s) for their anonymized clinical information to be published in this article.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the National Natural Science Foundation of China (grant number 81773239).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.