
Abstract
Nipped-B-like protein plays a pivotal role as a cohesin loading factor in the segregation of chromosomes when cells divide. Accumulating evidence indicates that alterations of this protein are involved in human carcinogenesis, especially in the regulation of chemotherapeutic drug response. However, the role of Nipped-B-like protein in esophageal squamous cell carcinoma remains unknown. In this study, we investigated the relevance of Nipped-B-like protein in the regulation of cisplatin sensitivity in esophageal squamous cell carcinoma. Ectopic expression of Nipped-B-like protein inhibited the growth of COLO-680N cells with low endogenous expression levels of Nipped-B-like protein, and increased sensitivity to cisplatin, a commonly used chemotherapy drug for patients with esophageal squamous cell carcinoma. In contrast, loss of Nipped-B-like protein stimulated the growth of EC9706 and Eca-109 cells with high levels of the protein, and resulted in resistance to cisplatin. P53-upregulated modulator of apoptosis, which is essential in the modulation of cisplatin sensitivity in a variety of cancers, acts as a downstream effector of Nipped-B-like protein. Restoration of this pro-apoptotic protein in Nipped-B-like protein-overexpressing esophageal squamous cell carcinoma cells effectively increased cisplatin sensitivity. Conversely, the silencing of P53-upregulated modulator of apoptosis in Nipped-B-like protein-depleted esophageal squamous cell carcinoma rendered cells resistant to cisplatin. Moreover, Nipped-B-like protein could bind directly to the promoter region of P53-upregulated modulator of apoptosis. In summary, our study addresses the involvement of Nipped-B-like protein in the development of esophageal squamous cell carcinoma, and the modulation of cisplatin sensitivity via regulation of P53-upregulated modulator of apoptosis.
Introduction
Esophageal cancer is not only one of the most common cancers of the digestive system, but also the sixth leading cause of cancer-related mortality worldwide. Esophageal squamous cell carcinoma (ESCC) is one of the main sub-types of esophageal cancer, with half of the total ESCC cases occurring in China. 1 ESCC usually develops as a consequence of smoking and excessive alcohol consumption, which induces multiple genetic and epigenetic changes, leading to aberrant activation of oncogenes, and inactivation of tumor suppressor genes. 2 -4
In most cases, cisplatin is widely used, along with surgery, for the treatment of patients with ESCC. 5 The mechanism of action of cisplatin has been linked to its ability to crosslink with purine bases on the DNA, interfere with repair, and cause DNA damage, subsequently inducing apoptosis. 6 However, cancer cells can develop drug resistance, leading to recurrence and metastasis. 7 As a result, the identification of a new therapeutic target to sensitize ESCC cells to cisplatin will contribute to the improvement of chemotherapy in such patients.
Cohesin, a conserved ring-shaped protein complex, encircles sister chromatids, and ensures correct chromosome segregation during mitosis and meiosis. Cohesin has recently been implicated in the transcriptional regulation of gene expression, as well as DNA condensation and repair. 8 -10 The cohesin complex consists of 4 core subunits: 2 structural maintenance of chromosomes (SMC) proteins (SMC1 and SMC3), the kleisin subunit RAD21 (Double-strand-break repair protein rad21 homolog), and stromal antigen (SA). SMC1 and SMC3 bind to each other to form a ring structure. Subsequently, RAD21 and SA bind the ATPase domains of SMC1 and SMC3 to stabilize the ring structure. Nipped-B-like protein (NIPBL) forms a cohesin loading complex with MAU2 (MAU2 chromatid cohesion factor homolog) to facilitate the loading of cohesin onto chromatin at specific chromosomal sites. 8
NIPBL has been implicated in transcriptional regulation, and shows mutations in the majority of individuals afflicted with Cornelia de Lange syndrome (CdLS), a developmental disorder characterized by dysmorphic facial features, growth delay, limb reduction defects, and mental retardation. Heterozygous mutations of NIPBL account for 65% of the total cases of CdLS. 11,12 Notably, NIPBL was identified as a critical transcription factor in recruiting the cohesin complex, and mediator of RNA polymerase II transcription (Mediator), which is a large complex with modular organization, to enforce long-range chromosomal interactions (via looping) that are essential for enhancer-driven pol II transcription. 13 -16 Increasing evidence has shown that alterations of NIPBL expression are involved in human carcinogenesis, especially in the regulation of chemotherapy sensitivity. Genome-wide functional profiling has shown that the silencing of NIPBL renders breast cancer cells resistant to tamoxifen. 17 In contrast, high levels of NIPBL were reported to be associated with poor prognosis and chemotherapy resistance in patients with non-small cell lung cancer. 18 The function of NIPBL in the modulation of chemosensitivity is context dependent, and depends on the tissue types and pathological statuses. In this study, we observed that NIPBL sensitizes ESCC cells to cisplatin through regulation of p53-upregulated modulator of apoptosis (PUMA).
Materials and Methods
Cell Lines and Drug Treatment
Human ESCC cell lines, including COLO-680N, KYSE-140, KYSE-150, KYSE-180, KYSE-450, TE-10, and TE-13, were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Eca-109 and EC9706 cells were obtained from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (CBTCCCAS, Shanghai, China). These cells were cultured in 1640 medium (Gibco, Waltham, MA, USA) supplemented with 10% (v/v) fetal bovine serum (Gibco) at 37°C, 5% CO2, and 95% humidity. FuGENE HD (Promega, Madison, WI, USA), was used for transfection according to the manufacturer’s protocol. Total RNA was extracted using Trizol RT reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. The concentrations of RNA were quantified by NanoDrop 2000 spectrophotometry (Nanodrop, Wilmington, DE, USA). ESCC cells were cultured overnight and treated with cisplatin (Sigma, St. Louis, MO, USA) for 72 h, and subsequently collected for flow cytometry and analysis of cell viability.
Plasmids and Antibodies
NIPBL ORF (1-8,094 bp) was cloned into the pEGFP-N1-FLAG vector (Addgene, Watertown, Massachusetts, USA). PUMA ORF (1-786 bp) inserted into a neomycin-resistant mammalian expression vector (EX-H3633-M14) was obtained from GeneCopoeia (GeneCopoeia, Rockville, MD, USA). NIPBL antibody was purchased from Sigma-Aldrich Chemicals (Sigma). PUMA and GAPDH-HRP conjugated antibodies were obtained from Abcam Biotechnology (Abcam, Cambridge, UK).
siRNA Transfection
NIPBL and PUMA knock down was performed by using tranfected siRNAs (Genepharma, Shanghai, China). The sequences of the siRNAs used are listed in Table 1. Cells were seeded in 6-well plates (3.0 × 10 5 /well) for 24 h, and subsequently transfected with siRNA duplexes (10 nM) using LipofectamineTM 3000 transfection reagent (Invitrogen) following the manufacturer’s instructions.
Sequence of siRNAs.

Quantitative Real-Time PCR
Reverse transcription was performed with PrimeScriptTM Reverse Transcriptase Kit (Takara, Kusatsu, Shiga Prefecture, Japan) using 500 ng of total RNA. Gene expression was assessed by quantitative real-time PCR by using the SYBR Green Master Mix Kit (Promega), and the ABI 7500 Real-time PCR System (Applied Biosystems, Foster City, CA, USA). Human Actin was used as an internal control. Primers used for NIPBL, PUMA, and HDACs are listed in Table 2.
Sequence of Primers.

Cell Growth Assay (MTS)
Cell growth was assessed by using the CellTiter 96® Aqueous NonRadioactive Cell Proliferation Assay Kit (Promega). After transfection, cells were treated with G418 (400 μg/mL) and re-plated in a 96-well plate for 2 additional days. The cell growth was determined following the manufacturer’s instructions. Samples were prepared in triplicates, and cell viability was analyzed as the mean± standard deviation.
IC50 Detection
KYSE-150 and TE-10 cells (5 × 10 3 cells/well) were seeded in 96-well plates with 6 replicate wells and cultured overnight, and subsequently incubated with fresh medium containing cisplatin at different concentrations (0, 0.23, 0.47, 0.94, 1.88, 3.75, 7.5, 15, 30, 60 μg/mL) for 48 h. Cell viability was tested by using the CellTiter 96® Aqueous NonRadioactive Cell Proliferation Assay Kit (Promega) following the manufacturer’s instructions.
Flow Cytometry Analysis
FITC Annexin V Apoptosis Detection Kit I (BD Bioscience, Bedford, MA, USA) was used to determine cell apoptosis by flow cytometry-based on the following protocol as stated by the manufacturer. Briefly, after washing twice with cold 1× PBS, the transfected cells were suspended in 1× binding buffer at a concentration of 1 × 10 6 cells/mL. Following this, 100 µl of the cell suspension was mixed with 5 µl of FITC Annexin V and 10 µl 0.05% propidium iodide, and incubated for 15 min at room temperature in the dark. The samples were submitted for analysis as described above.
Western Blotting Analysis
Quantitation of total protein was performed by using Bio-Rad protein assay kit II (Bio-Rad Laboratories, Hercules, CA, USA). The same amount of protein from each sample was resolved by SDS-PAGE and transferred to PVDF membranes. Membranes were incubated with the primary antibody, washed with PBS-T (PBS with 0.1% of Tween-20), incubated with HRP-conjugated secondary antibody, and autoradiographed with chemiluminescence (Thermo Fisher Scientific, Waltham, MA, USA).
Chromatin Immunoprecipitation (ChIP)
Chromatin immunoprecipitation was performed by using a ChIP assay kit (MilliporeSigma, Burlington, MA, USA) according to the manufacturer’s instructions. Cells were cross-linked with 1% formaldehyde (Sigma) in culture medium at room temperature for 10 min, washed twice with cold PBS, suspended in SDS lysis buffer (50 mM Tris pH 8.0, 1% SDS, 10 mM EDTA, and 1× protease inhibitor). The samples were then sonicated on ice. Subsequently, the chromatin solution was precleared, incubated with the NIPBL antibody (Sigma), and the immune complexes were eluted. PCR was performed to assess the occupancy of NIPBL around the promoter region of PUMA. Primers used for ChIP are listed in Table 1.
Statistical Analysis
SPSS version 25.0 software (SPSS, USA) was used for data analysis. Student’s t-tests were used to assess the differences between the 2 groups. All tests were 2-sided and p < 0.05 was considered statistically significant.
Results
The Relevance of NIPBL on the Growth of ESCC Cells
We searched The Cancer Genome Atlas (TCGA, https://cancergenome.nih.gov) to assess the possible involvement of NIPBL expression in esophageal carcinogenesis, and found that esophageal cancer patients with high levels of NIPBL exhibited better survival than those with low levels of NIPBL. These results indicate that NIPBL expression might be positively correlated to the prognosis of patients with esophageal cancer (Supplementary Figure 1). However, the sample size of this study was small; therefore, additional studies using large cohorts to assess the relevance of NIPBL expression on patient prognosis is required.
Furthermore, we determined NIPBL levels in ESCC cell lines by western blotting to address its function in the development of ESCC. Interestingly, NIPBL was downregulated in the majority of ESCC cell lines (Figure 1A) compared to normal esophageal squamous epithelial tissue samples. To investigate the relevance of NIPBL downregulation in ESCC, we tested the effect of NIPBL on the growth of COLO-680N cells with low levels of the protein. Ectopic NIPBL expression dramatically inhibited the growth of COLO-680N cells (Figure 1B, C). In contrast, the growth of EC9706 cells with high levels of NIPBL was promoted significantly after NIPBL depletion (Figure 1D, E, F). Likewise, the loss of NIPBL also promoted cell growth in Eca-109, one of the ESCC cell lines with medium levels of the protein (Figure 1A, F). These results indicate that NIPBL plays an important role in the development of ESCC.
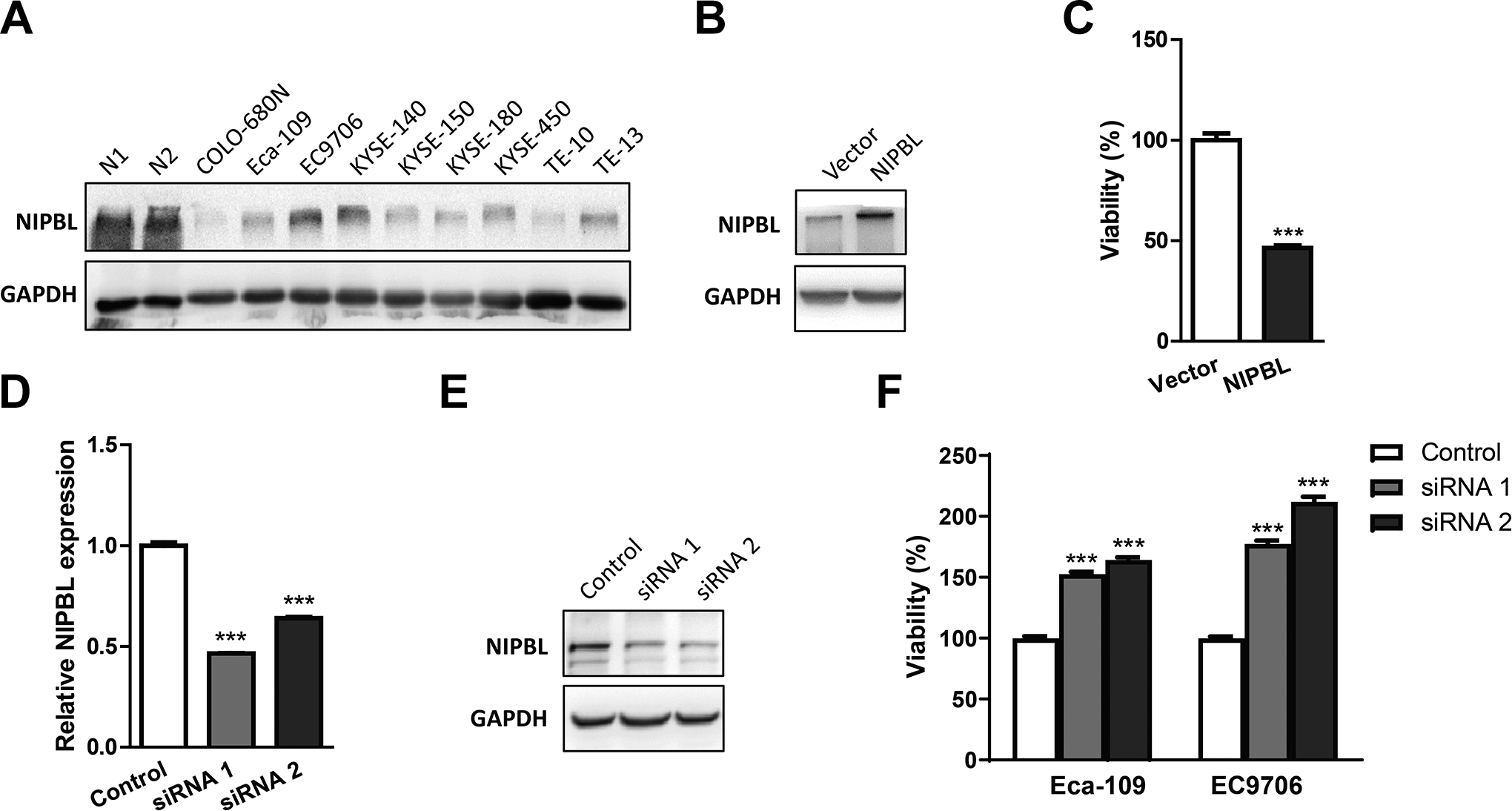
NIPBL is relevant to the growth of ESCC cells. A, Western blotting analysis of NIPBL expression in ESCC cell lines. GAPDH is shown as loading control. Normal esophageal squamous epithelial tissue from 2 patients, N1 and N2, were used as the control. B, Western blotting analysis of NIPBL expression in COLO-680N cells transfected with the NIPBL overexpressing vector. GAPDH is shown as loading control. C, Relative cell proliferation of COLO-680N with NIPBL overexpression was determined by the MTS assay. Cells were transfected with pEGFP-N1-FLAG vector or NIPBL recombinant vector respectively, and the relative cell proliferation was determined by MTS assay after transfection for 72 h. All experiments were repeated thrice and the representative results are shown. The statistical significance is p < 0.001 (Student’s t-test, *** represents p < 0.001). NIPBL expression in EC9706 cells transfected with NIPBL siRNA was determined by quantitative real-time PCR (D) and western blotting analysis (E). siRNA 1 and siRNA 2 are 2 different NIPBL siRNAs, whereas control is a non-targeting scrambled control siRNA. F, Relative cell proliferation in Eca-109 and EC9706 cells with NIPBL depletion was determined by MTS assay after transfection with NIPBL siRNA for 72 h. All experiments were repeated thrice and the representative results are shown. The statistical significance is p < 0.001 (Student’s t-test, *** represents p < 0.001).
NIPBL Positively Correlates With Cisplatin Sensitivity in ESCC Cells
Cisplatin is one of the most commonly used chemotherapeutic drugs in the treatment for ESCC patients, and we wanted to determine the role of NIPBL in sensitivity to cisplatin in ESCC cells. Takashima et al. previously measured the IC50 of cisplatin in different kinds of ESCC cell lines, including KYSE-140, KYSE-150, TE-1, TE-4, TE-8, TE-10, TE-11, TE-12, and TE-15. 19 These ESCC cell lines were divided into 2 groups, low sensitivity, and high sensitivity, based on the IC50 of cisplatin. We analyzed the relative mRNA expression levels of NIPBL in these ESCC cell lines based on the Catalogue of Somatic Mutations in Cancer (COSMIC) (https://cancer.sanger.ac.uk/cell_lines/download) (Figure 2A), and found that ESCC cell lines with higher NIPBL levels are more sensitive to cisplatin than cell lines with lower expression level of NIPBL (Figure 2B). The observations suggest that NIPBL expression might be positively correlated with cisplatin sensitivity in ESCC. To validate this conclusion, we tested NIPBL levels and IC50 of cisplatin in KYSE-150 and TE-10 cells. We found that NIPBL levels in the TE-10 cell line were 10-fold higher than that in KYSE-150 (Figure 2C), which is consistent with the data from the COSMIC. The IC50 of cisplatin in TE-10 cells (1.90 ± 0.11 μg/mL) was much lower than that in KYSE-150 (4.16 ± 0.12 μg/mL), which indicates that TE-10 is more sensitive to cisplatin than KYSE-150 (Figure 2D).

NIPBL is positively correlated with cisplatin sensitivity in ESCC cells. A, Relative levels of NIPBL mRNA in ESCC cell lines downloaded from the COSMIC database. B, Relative expression of NIPBL in low and high cisplatin sensitive ESCC cell lines. C, Relative expression of NIPBL in KYSE-150 and TE-10 was determined by quantitative real-time PCR. D, The IC50 of cisplatin in KYSE-150 and TE-10 was determined by MTS assay. All experiments were repeated thrice and the representative results are shown. Statistical significance was determined using the Student’s t-test (*** represents p < 0.001).
To further confirm the role of NIPBL in the regulation of cisplatin sensitivity in ESCC, we overexpressed NIPBL in COLO-680N cells and found that NIPBL-overexpressing COLO-680N were prone to cell death following cisplatin treatment (Figure 3A, B), indicating that NIPBL overexpression increases the sensitivity of ESCC cells to cisplatin. On the contrary, NIPBL knockdown decreased cisplatin sensitivity of EC9706 and Eca-109 cells, as measured using flow cytometry (Figure 3C, D) and a cell viability assay (Figure 3E, F), respectively. These results suggest that NIPBL is important in the regulation of cisplatin sensitivity in ESCC.

NIPBL levels affect the cisplatin sensitivity of ESCC cells. A, Apoptosis of COLO-680N cells transfected with control and NIPBL overexpressing plasmids after cisplatin treatment was evaluated by flow cytometry. B, The apoptosis index is presented as the mean± standard deviation of triplicate experiments. C, The apoptosis of EC9706 and Eca-109 cells transfected with control siRNA and NIPBL siRNA after cisplatin treatment was evaluated by flow cytometry. D, The apoptosis index is presented as the mean± standard deviation of triplicate experiments. E, Relative cell proliferation of EC9706 and Eca-109 cells transfected with control siRNA and NIPBL siRNA was determined after cisplatin treatment for 72 h by MTS assay. F, Relative cell proliferation of Eca-109 cells transfected with control siRNA and NIPBL siRNA was determined after treatment with different concentrations of cisplatin by MTS assay. All experiments were repeated thrice and the representative results are shown. Statistical significance was determined using Student’s t-test (** represents p < 0.01, *** represents p < 0.001).
NIPBL Modulates Cisplatin Sensitivity Through Upregulation of PUMA
PUMA, also known as Bcl-2 binding component 3 (BBC3), belongs to the Bcl-2 family and induces apoptosis via a caspase cascade by interacting with Bcl-2/Bcl-xl and Bax/Bak. 20,21 Downregulation of PUMA has been observed in various human cancers, and associated with cisplatin resistance. 22 -24 To address the role of PUMA in the regulation of NIPBL-induced cisplatin sensitivity, PUMA levels were determined by quantitative real-time PCR and western blotting. Interestingly, PUMA expression in COLO-680N cells was remarkably upregulated after ectopic expression of NIPBL (Figure 4A, B). In contrast, PUMA levels were downregulated dramatically by NIPBL depletion (Figure 4C, D), suggesting that PUMA probably is a downstream effector of NIPBL in ESCC. Furthermore, we found that the silencing of PUMA rescues the effect of NIPBL overexpression on cell viability after cisplatin treatment. On the other hand, ectopic expression of PUMA reverses the effect of NIPBL depletion on cell growth after cisplatin treatment (Figure 4E, F). These results indicate that NIPBL modulates cisplatin sensitivity in ESCC cells via regulation of PUMA.

NIPBL modulates the response to cisplatin via the upregulation of PUMA. Expression of PUMA in COLO-680N cells transfected with the NIPBL overexpressing vector was determined by quantitative real-time PCR (A) or western blotting analysis (B), respectively. PUMA expression in EC9706 cells transfected with NIPBL siRNA was determined by quantitative real-time PCR (C) or western blotting analysis (D), respectively. E, The growth of COLO-680N cells co-transfected with NIPBL overexpressing plasmids and/or PUMA siRNA after cisplatin treatment for 72 h was determined by the MTS assay. F, The growth of ESCC cells, including EC9706, Eca-109, and KYSE-140, co-transfected with NIPBL siRNA and/or PUMA overexpressing plasmids after cisplatin treatment for 72 h was determined by the MTS assay. All experiments were repeated thrice and representative results are shown. Statistical significance was determined using Student’s t-test (* represents p < 0.05, ** represents p < 0.01, *** represents p < 0.001).
Transcription of PUMA Is Regulated by NIPBL
To gain insight into the relevance of NIPBL to PUMA transcription, we performed a ChIP assay, and found that the promoter region of PUMA co-precipitated with NIPBL, implying direct transcriptional regulation of PUMA by NIPBL (Figure 5A). NIPBL has been reported to recruit histone deacetylases (HDACs) to modulate local chromatin modifications, and the transcription of PUMA was reported to be regulated by HDACs. 25,26 To further elucidate the mechanism, we tested the expression levels of several HDACs by quantitative real-time PCR before and after NIPBL knockdown. Interestingly, several HDACs, including HDAC1, HDAC3, HDAC6, SIRT2, SIRT3, SIRT5, SIRT6, and SIRT7, were significantly downregulated following silencing of NIPBL (Figure 5B). Searching the GEPIA server (Gene Expression Profiling Interactive Analysis, http://gepia.cancer-pku.cn/), we found that HDAC levels positively correlated with NIPBL and PUMA in patients with esophageal cancer (Figure 5C, D), implying that the regulation of PUMA by NIPBL might occur in a histone acetylation-dependent manner. However, further studies are required to fully delineate the definitive mechanism of regulation of PUMA by NIPBL.

The transcriptional regulation of PUMA by NIPBL. A, The interaction of NIPBL with the promoter region of PUMA in ESCC cells was determined by ChIP. B, The expression of HDACs in KYSE-140 cells transfected with NIPBL siRNAs was determined by quantitative real-time PCR. All experiments were repeated thrice, and representative results are shown. Statistical significance was determined using Student’s t-test (** represents p < 0.01, *** represents p < 0.001). C, NIPBL expression correlated with HDACs in patients with esophageal cancer. D, The expression of PUMA correlated with HDACs in patients with esophageal cancer. All the data is summarized from the GEPIA database. E, A proposed model of the mechanism of NIPBL-mediated cisplatin sensitivity in ESCC.
Discussion
PUMA, which is also known as BBC3, is a pro-apoptotic protein. As a pro-apoptotic member of the Bcl-2 family, PUMA was first identified as a critical modulator of apoptosis induced by the tumor suppressor gene p53 and DNA damage agents. PUMA plays an important role in p53-dependent and -independent apoptosis induced by a variety of signals, and is regulated by transcription factors, and not by post-translational modifications. 27 -30 PUMA expression has been reported to be associated with cisplatin sensitivity in different types of human cancers. 22 -24 In this study, we found that NIPBL-induced dysregulation of PUMA affects cisplatin sensitivity in ESCC.
Increasing evidence has shown that NIPBL is involved in the transcriptional regulation of downstream genes by recruiting the cohesin complex and CCCTC-binding factor (CTCF) to maintain the 3-dimensional structure and stability of genomes, and long-range interactions of chromatin. 31 -33 Furthermore, NIPBL has been shown to recruit the cohesin and Mediator complexes to enforce long-range chromosomal interactions to initiate enhancer-driven pol II transcription. 14,15 In this study, we demonstrated that NIPBL binds to the promoter region of PUMA and affects its expression at the transcriptional level. However, more studies are needed to delineate the definitive mechanism of how NIPBL modulates the transcription of PUMA.
In addition to expression alterations, somatic mutations of NIPBL are also implicated in various types of human cancers. The first NIPBL genetic mutation in cancer was reported in 2008 when Barber et al. identified heterozygous somatic missense mutations in the SMC1A, SMC4, STAG3, and NIPBL genes in 9 out of 132 patients with colorectal adenocarcinomas. The study stated that the chromosomal instability present in the majority of patients with colorectal cancer could be attributed to chromatid cohesion defects. 34 Subsequently, NIPBL mutations were identified with high microsatellite instability in gastric and colorectal cancers. 35 Recent cancer genomic analyses discovered a high frequency of NIPBL mutations in a select subset of cancers, including gliomas, endometrial carcinoma, and acute megakaryoblastic leukemia, suggesting that these mutations may underlie the development of human cancers. 36 -38 We searched the COSMIC database and found recurrent NIPBL mutations in patients with esophageal cancer (26/1513, 1.72%), with 14 of these mutations occurring in patients with ESCC (Supplementary Figure 2). The majority of NIPBL mutations in ESCC are missense. It is unclear whether NIPBL mutations are passenger mutations, and whether these mutations affect signaling pathways. The clinical relevance of NIPBL mutations in the pathogenesis of ESCC remains to be determined. In order to exclude the effect of NIPBL mutations in this study, we analyzed the NIPBL mutational status of ESCC cell lines in the COSMIC database. The only ESCC cell line used in this study that had a NIPBL mutation was KYSE-450. The mutation in KYSE450 is silent, and does not affect the amino acid sequence of NIPBL.
Although cisplatin has demonstrated clinical success in ESCC patients, drug resistance remains a major challenge. Multiple mechanisms of resistance have been established in tumor cells, including decreased drug accumulation, increased detoxification activity, facilitation of DNA repair, and inactivated cell death signaling. Recently, increasing evidence has shown that the tumor microenvironment (TME) also plays an important role in the development of resistance to cisplatin. In general, components of TME have been reported to affect cisplatin response through decreased drug delivery, increased acidity, cell adhesion, and immunosuppressive activity. Combination treatments with cisplatin and novel agents targeting components in TME have resulted in major clinical successes. 39 -41 There are many contributors to cisplatin resistance in tumor cells, and additional studies are needed to develop novel approaches to overcome this issue.
NIPBL is crucial for the loading of the cohesin complex onto chromatin at specific sites to modify gene-specific transcription. The cohesin and Mediator complexes co-occupy different promoter regions to enforce cell-type-specific DNA loops to control gene expression. Mediator plays an important role in the modulation of chemosensitivity in different types of cancers. 16 Consequently, Mediator is likely important in the regulation of cisplatin sensitivity induced by NIPBL in ESCC. Although we demonstrate the relevance of NIPBL to cisplatin sensitivity in ESCC cell models, validation of the findings in clinical tissue samples is necessary, especially correlation analysis between NIPBL levels and multiple clinicopathological parameters and prognosis. We speculated that NIPBL modulates the transcription of PUMA by recruitment of multiple HDACs; however, the mechanism needs to be further elucidated in future studies.
Collectively, we found that NIPBL sensitizes ESCC cells to cisplatin through the upregulation of PUMA (Figure 5E). Although we could not exclude other mechanisms by which NIPBL modulates cisplatin sensitivity, the positive transcriptional correlation of NIPBL and PUMA clearly implicates PUMA as a major downstream effector that mediates NIPBL-specific drug response. NIPBL might serve as an independent prognostic biomarker to predict cisplatin sensitivity, and a therapeutic target for patients with ESCC.
Supplementary Material
Supplemental Material, Supplementary_Figure_1 - Nipped-B-like Protein Sensitizes Esophageal Squamous Cell Carcinoma Cells to Cisplatin via Upregulation of PUMA
Supplemental Material, Supplementary_Figure_1 for Nipped-B-like Protein Sensitizes Esophageal Squamous Cell Carcinoma Cells to Cisplatin via Upregulation of PUMA by Shengjie Zhang, Yun Zhou, Qinchuan Wang, Kristine Donahue, Jianguo Feng, Yinli Yao, Aiping Chen, Xia Li and Lianlian Hong in Technology in Cancer Research & Treatment
Supplementary Material
Supplemental Material, Supplementary_Figure_2 - Nipped-B-like Protein Sensitizes Esophageal Squamous Cell Carcinoma Cells to Cisplatin via Upregulation of PUMA
Supplemental Material, Supplementary_Figure_2 for Nipped-B-like Protein Sensitizes Esophageal Squamous Cell Carcinoma Cells to Cisplatin via Upregulation of PUMA by Shengjie Zhang, Yun Zhou, Qinchuan Wang, Kristine Donahue, Jianguo Feng, Yinli Yao, Aiping Chen, Xia Li and Lianlian Hong in Technology in Cancer Research & Treatment
Footnotes
Abbreviations
Authors’ Note
Shengjie Zhang and Yun Zhou contributed to the work equally. Our study did not require ethical board approval because it did not contain human or animal trials.
Acknowledgments
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project is supported by National Natural Science Foundation of China (81502603), Natural Science Foundation of Zhejiang Province (LGF18H160016) and Medicine and Health Science Fund of Zhejiang Province (2018RC021) to Zhang, S., Natural Science Foundation of Zhejiang Province (LQ19H160005) and Medicine and Health Science Fund of Zhejiang Province (2019ZD025) to Hong, L., National Natural Science Foundation of China (81802472) Natural Science Foundation of Zhejiang Province (LQ18H160020) to Chen, A.
Supplementary Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.