
Abstract
Colorectal cancer is one of the most prevalent malignancies worldwide. ENKUR is a transient receptor potential canonical–binding protein that acts as a potential regulator or effector of transient receptor potential canonical channels. It also directly interacts with the p85 regulatory subunit of phosphoinositide 3-kinase. However, the role of ENKUR in colorectal cancer remains unclear. In the present study, the expression profiles of ENKUR in the The Cancer Genome Atlas and ONCOMINE databases were analyzed. Significant downregulation of ENKUR was observed in clinical tumor samples of various cancer types, including colorectal cancer. Decreased ENKUR messenger RNA expression and ENKUR protein level were detected in 6 human colorectal cancer cell lines. Silencing of ENKUR in colorectal cancer cells led to enhanced cell proliferation, migration, and invasion, while the opposite effects were achieved in ENKUR-overexpressing cells. Furthermore, ENKUR-underexpressing cells exhibited increased activity of phosphoinositide 3-kinase /Akt signaling pathway. Downregulation of the epithelial marker, E-cadherin, and upregulation of the mesenchymal markers, vimentin and N-cadherin, were detected in ENKUR-underexpressing cells, suggesting the induction of epithelial–mesenchymal transition. In conclusion, the present study demonstrates that ENKUR may be responsible for alterations in the proliferative, migratory, and invasive potential of colorectal cancer cells through possible involvement in the phosphoinositide 3-kinase /Akt signaling pathway.
Introduction
At present, colorectal cancer (CRC) has become one of the most common cancer responsible for more than 500 000 estimated annual mortality worldwide. 1 Despite significant advances in CRC treatment in the past decade, the curative rate for CRC remains unsatisfactory due to frequent local recurrence and distant metastases, which account for approximately 50% of death in patients with CRC. 2,3 Therefore, prevention and early diagnosis are essential for reducing the morbidity and mortality of CRC.
The pathogenesis of CRC is a complex process involving progressive disruption of homeostatic mechanisms mainly caused by genetic mutation or epigenetic alteration. 4,5 Up to now, a number of oncogenes and tumor suppressor genes have been linked with CRC initiation and progression, including p53, Ras, c-erbB-2, SFRP1, and PTCH gene family. 6 -10 Many of these genes encode components of pathways controlling intestinal epithelial cell proliferation, differentiation, and programmed cell death. A better understanding of molecular mechanisms underlying colon tumourigenesis and CRC metastases may have pathogenic and therapeutic implications for clinical treatment of CRC.
Located at chromosomal band 10p12.1, the ENKUR (Enkurin) gene encodes a CaM-binding protein that was originally identified using yeast 2-hybrid screening in a study of transient receptor potential canonical (TRPC). 11 Direct interactions were found between ENKUR protein and several TRPC proteins. Three potential protein–protein interaction sites in ENKUR were identified from the deduced amino acid sequences and confirmed by GST pull-down assay. These 3 interaction domains include a C-terminal region involved in channel interaction, an IQ motif related to the Ca2+ sensor, and an N-terminal region. Among these, the proline-rich N-terminal region is the key site that allows ENKUR to interact with the p85 regulatory subunit of phosphoinositide 3-kinase (PI3K). It was thus hypothesized that ENKUR may act as a bridge connecting PI3K signaling and TRPC channel. 11 However, so far few studies have focused on the function of ENKUR in human cancer.
In this study, we examined the expression profiles of ENKUR in clinical CRC samples from the ONCOMINE and The Cancer Genome Atlas (TCGA) databases. ENKUR messenger RNA (mRNA) expression and ENKUR protein level in CRC cells were validated using quantitative real-time polymerase chain reaction (qRT-PCR) and western blot analyses. We also investigated the effects of ENKUR overexpression and silencing on the cellular biological behavior of CRC cells and presented the first evidence of ENKUR involvement in the PI3K/Akt signaling pathway.
Materials and Methods
Ethical Statement
This study did not require an ethical board approval because it did not contain human or animal trials. The protocol of cellular experiments was approved by the Ethic Committee of Southeast University (Nanjing, Jiangsu, China) and Zhejiang Shuren University (Hangzhou, Zhejiang, China).
ONCOMINE Data Set
The expression level of ENKUR in CRC was analyzed using ONCOMINE (https://www.oncomine.org/resource/login.html), a web-based data-mining platform aimed at facilitating discovery from genome-wide expression analyses. Clinical cases in the ONCOMINE data set with statistical significance (P < .05) were enrolled in this study. Expression data sets were screened to examine the differences of ENKUR expression in CRC tumor tissues and normal colon tissues.
UALCAN Analysis Based on TCGA Data Set
The web-portal UALCAN (http://ualcan.path.uab.edu/index.html) 12 was accessed to obtain in-depth analyses of ENKUR mRNA expression profiles from the TCGA database (cancergenome.nih.gov). 13 Expression data sets with statistical significance (P < .05) were obtained. The expression of ENKUR was examined in various types of clinical tumor samples, including colon adenocarcinoma, liver hepatocellular carcinoma, lung adenocarcinoma, lung squamous cell carcinoma, prostate adenocarcinoma, and thyroid carcinoma. The expression profile of CRC includes 286 samples of CRC tumor tissues and 41 nontumor colorectal tissues.
Cell Line Preparation
Six human CRC cell lines (HT29, HCT116, SW620, LS174T, RKO, and LOVO) were obtained from the American Type Culture Collection (Manassas, Virginia). One normal human colon mucosal epithelial cell line NCM460 was obtained from INCELL (San Antonio, Texas). All cell lines used in this study were authenticated using short tandem repeat profiling provided by the cell bank. Cells were cultivated in Roswell Park Memorial Institute 1640 medium containing 10% heat-inactivated fetal bovine serum (FBS) at 37°C in a humidified atmosphere with 5% CO2. Cells were used for all experiments within 6 months upon receipt or resuscitation.
RNA Extraction and qRT-PCR
Total RNA was extracted from CRC cells using Trizol reagent (Invitrogen, Carlsbad, California) according to the manufacturer’s instructions. Complementary DNAs (cDNAs) were synthesized by reverse transcription using the RevertAid first-strand cDNA synthesis kit (Fermentas, Ontario, Canada). Quantitative real-time PCR was conducted using the Platinum SYBR green master mix (Invitrogen) on an ABI StepOne Plus RT-PCR system (Applied Biosystems, Foster City, California). Amplification was performed in 20 μL reactions containing 10 μL of Supermix, 0.8 μM of each primer, and 0.1 to 0.5 μg of template cDNA. The primer sequences are provided in Table 1. The amplification procedure includes an initial denaturation step for 2 minutes at 95°C, followed by 40 cycles of denaturation for 30 seconds at 95°C, annealing for 45 seconds at 55°C, extension for 30 seconds at 72°C, and a final extension stage for 10 minutes at 72°C. Expression of β-ACTIN was quantified as internal control. Relative quantification of mRNA expression was calculated using the ΔΔCt method. 14 Each experiment was conducted in triplicates.
Primer Sequences Used for qRT-PCR Analysis.

Abbreviation: qRT-PCR, quantitative real-time polymerase chain reaction.
Immunoblotting Analysis
For protein extraction, approximately 1 × 107 cells were collected and rinsed with ice-cold phosphate-buffered saline (PBS). The cells were resuspended and lysed on ice in radio immunoprecipitation assay buffer. Protein concentrations were determined using the Bradford reagent (BioRad, Hercules, California). Protein lysates were then resolved on 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred to polyvinylidene fluoride membranes (ImmobilonP; Millipore, Bedford, Massachusetts). After blocking with 5% nonfat dry milk in Tris-buffered saline, the membranes were immunoblotted with the following antibodies: PI3K p85 (1:1000, #4292, Cell Signaling, Danvers, Massachusetts); phospho-PI3K p85 (Tyr458; 1:1000, #4228, Cell Signaling); Akt (1:1000, #9272, Cell Signaling); pAkt (Ser473; 1:2000, #4060, Cell Signaling); E-cadherin (1:500, #SAB4503751; Sigma Aldrich, St. Louis, Missouri); N-cadherin (1:5000, #SAB2702400; Sigma Aldrich); vimentin (1:200, #V6630; Sigma Aldrich); β-ACTIN (1:5000, #A5441; Sigma Aldrich); and ENKUR (1:500, #SAB1103399; Sigma Aldrich). Then, the appropriate secondary antibodies (Horseradish Peroxidase [HRP]-conjugated anti-rabbit immunoglobulin G [IgG], 1:2000, #7074; HRP-conjugated anti-mouse IgG, 1:2000, #7076; Cell Signaling) were applied. Finally, signals were detected by enhanced chemiluminescence (Pierce, Rockford, Illinois) following the manufacturer’s instructions.
Subcellular Fractionation and Co-Immunoprecipitation
Subcellular fractionation was conducted using the NE-PER kit according to the manufacturer’s instructions (Thermo Fisher Scientific, Rockford, Illinois). For co-immunoprecipitation, lysates of the total cell and cell cytoplasm were cleared by centrifugation at 15 100g for 10 minutes at 4°C. Then the lysates were incubated with preblocked protein A Sepharose beads (Zymed, San Francisco, California) and the following individual antibodies (PI3K p85, 1:1000, #4292, Cell Signaling; ENKUR, 1:500, #SAB1103399, Sigma Aldrich; normal rabbit IgG, #2729, Cell Signaling) were added. After overnight incubation at 4°C, complexes with protein A Sepharose (Zymed, San Francisco, California) were harvested and brief centrifuged. Bound proteins were separated with SDS-PAGE and visualized using Western blotting.
Lentiviral Transduction and Transfection
Overexpression of exogenous ENKUR and short hairpin RNA (shRNA)-induced silencing of endogenous ENKUR were achieved using the pEZ-Lv105 lentiviral ORF expression plasmid and psi-LVRH1GP lentiviral shRNA expression plasmid (GeneCopoeia, Rockville, Maryland). The silencing efficiencies of 2 different shRNA sequences: CCGGCCAACCTCGATACTCTTATTTCTCGAGAAATAAGAGTATCGAGGTTGGTTTTTTG and CCGGCCAACCTCGATACTCTTATTTCTCGAGAAATAAGAGTATCGAGGTTGGTTTTTTG were tested. Lentiviral particles were generated at Aoqian Bio-Tech Cooperation (Hangzhou, Zhejiang, China) using the Lenti-Pac HIV expression packaging kit (GeneCopoeia, Rockville, Maryland). Human embryonic kidney cells HEK293T were used as the packaging cell line. Specifically, about 1.5 × 106 HEK293T cells were seeded in a 10-cm dish containing 10 mL of Eagle’s minimal essential medium (DMEM) supplemented with 10% heat-inactivated FBS 2 days before transfection. Cells were transfected with 2.5 μg of the corresponding lentiviral ORF/shRNA expression plasmids and 2.5 μg of Lenti-Pac HIV mix containing packaging plasmids. After incubation for 10 hours at 37°C with 5% CO2, the transfection medium was removed and replaced with fresh DMEM supplemented with 5% FBS. After 48 hours of transfection, the pseudovirus-containing medium was collected and centrifuged at 500g for 10 minutes. Then, cell debris was removed and the supernatant containing the lentiviral particles was filtered and harvested. For transfection of CRC cells, LS174T and SW620 cells were plated in 24-well plates containing DMEM with 5% heat-inactivated FBS. After 24 hours, the old culture medium was removed and replaced with 0.5 mL of virus suspension diluted in complete medium with 6 μg/mL polybrene. Cells were transfected overnight, reseeded onto 6-well plates, and incubated in medium containing puromycin. Stably transduced cells were selected after 48 hours incubation and samples of mRNA and protein were taken for qRT-PCR and immunoblotting analyses.
Cell Proliferation Assay
Cells were seeded at a density of 1 × 103 on 96-well plates containing 100 μL of culture medium and shaken on a microplate shaker for 1 to 7 days at 37°C in a humidified atmosphere supplied with 5% CO2. To assess the number of cells at each time point, 20 μL of 5 g/L 3-(4,5-dimethylthiazol-z-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma Aldrich) was added into each well. The plate was incubated for a further 4 hours before removal of MTT and extraction of the formazan into 150 μL of dimethyl sulphoxide (Sigma Aldrich). The absorbance of the wells was measured at 450 nm using a microplate autoreader (BioRad). The experiment was performed in triplicates.
Colony Formation Assay
Cells were seeded at a density of 2 × 102 on 6-well culture plates and incubated at 37°C for 2 weeks. Then the cells were washed twice with PBS and fixed and stained with hematoxylin. The number of colonies containing ≥50 cells was counted using a microscope. The experiment was performed in triplicates for each cell line.
Cell Wound-Healing Assay
Cells were seeded on 6-well tissue culture plates at a density of 1.25 × 106 per well and incubated for 24 hours until grown to 80% to 90% confluence. Then, the cells were washed twice with PBS and a uniform scratch was created in straight line across each well using a 10 μL standard pipette tip. The plates were washed twice with PBS to remove the detached cells. Cells were allowed to grown for 48 hours, during which time wound margins were photographed and migration was monitored using an inverted microscope. Images were captured using an image-analyzing frame-grabber card (LG-3 Scientific Frame Grabber; Scion, Frederick, Maryland) and analyzed with the image analysis software NIH Image 1.55. The unfilled scratched zones were quantified by measuring the distance between the advancing margins of cells in 3 randomly selected microscopic fields (×200) at each time point. The experiment was performed in triplicates.
Transwell Migration Assay
About 5 × 104 cells suspended in serum-free medium were seeded on the upper compartment of 8-μm-pore transwells (Costar, Corning, Cambridge, Massachusetts). The lower compartment was supplied with 300 μL of 10% FBS in free medium (Gibco; Invitrogen). Cells were allowed to migrate for 24 hours. Then cells in the lower chamber were fixed with 1% paraformaldehyde and stained with hematoxylin after removal of cells inside the chamber with cotton swabs. The number of cells migrated through the chamber was counted in 5 randomly selected fields (×200) under a microscope. The experiment was performed in triplicates.
Statistical Analysis
Statistical analyses were performed using SPSS version 20 (SPSS Inc, Chicago, Illinois). Cell proliferative and migratory rate, cell invasive ability, and ENKUR mRNA expression and protein level were compared using Student t test between 2 groups and 1-way analysis of variance followed by Tukey post hoc test among 3 groups. A P value of <.05 was considered to indicate statistically significant difference.
Results
Expression Profile of ENKUR in Patients With CRC and Other Cancer Types
In the ONCOMINE database, ENKUR expression was significantly downregulated in 60 analyses and upregulated in 43 analyses in different cancer types (P < .05; Figure 1A). An expression profile of CRC including 101 clinical cases of colon adenocarcinoma, 22 cases of cecum adenocarcinoma, 22 cases of colon mucinous adenocarcinoma, and 60 cases of rectal adenocarcinoma showed significantly lower ENKUR expression as compared with 22 samples of normal colon tissue (P < .001; Figure 1B). The expression level of ENKUR in normal tissues was about 2-fold higher than that in CRC tissues (Figure 1B). However, although ENKUR expression was lower in tumor tissues with higher pathological stage, no statistically significant difference was detected.
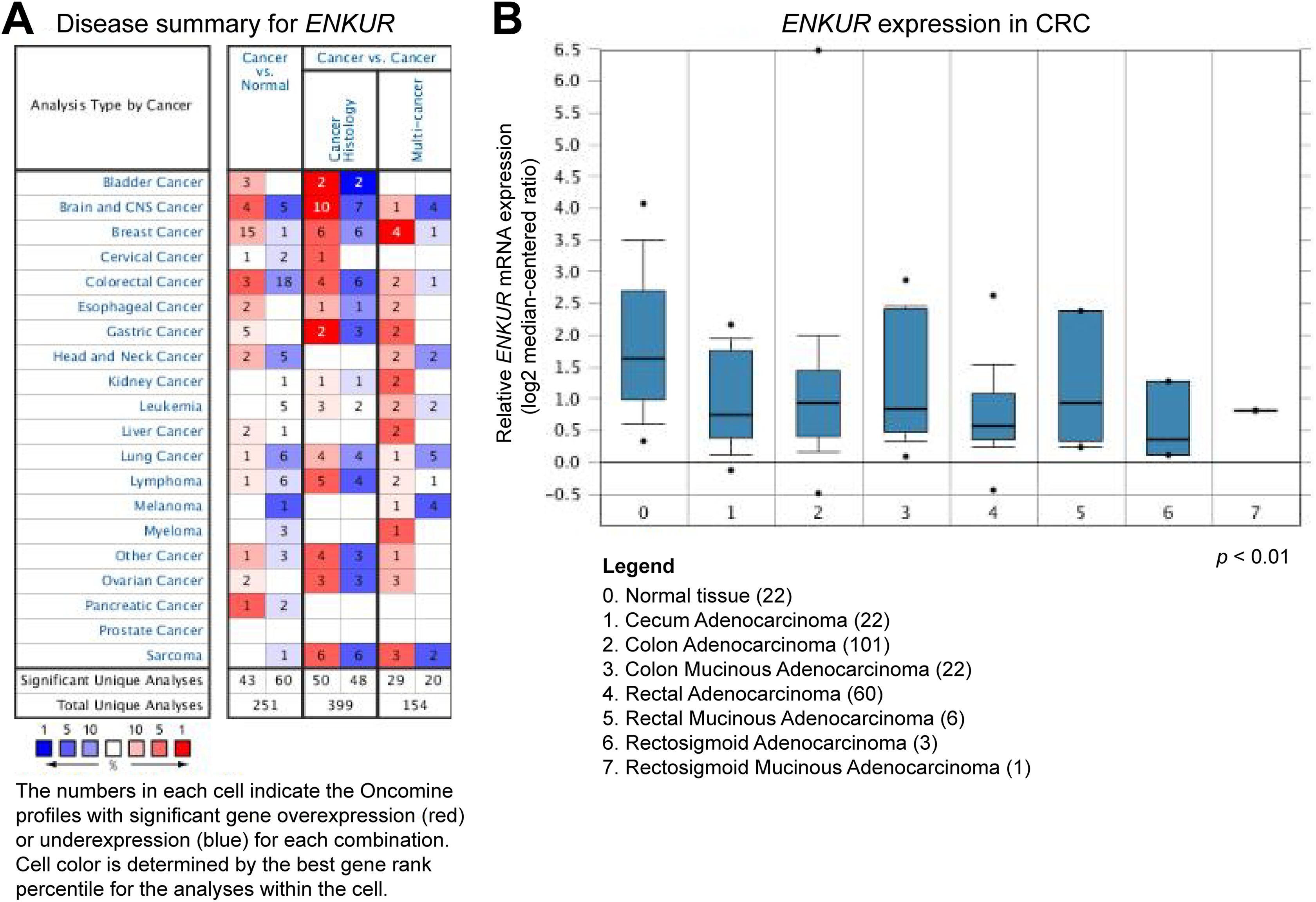
Expression profiling of ENKUR in the ONCOMINE database. A, Summary of ENKUR expression in different cancer types. B, ENKUR expression in 215 clinical CRC tumor tissues and 15 normal tissues. All the clinical cases demonstrated statistically significant differences in the expression level of ENKUR between tumor tissues and normal tissues with a P value less than .001. CRC indicates colorectal cancer.
The expression levels of ENKUR in various types of clinical tumor tissues were also obtained from the UALCAN portal based on the TCGA database. Compared with the corresponding normal nontumor samples, ENKUR showed significant downregulation in colon adenocarcinoma, liver hepatocellular carcinoma, lung adenocarcinoma, lung squamous cell carcinoma, prostate adenocarcinoma, and thyroid carcinoma (P < .005; Figure 2). Specifically, the expression profile of clinical CRC showed that ENKUR was downregulated by nearly 50% in 286 clinical samples of CRC tumor tissues compared with 41 samples of normal colorectal tissues (P < .005; Figure 2A).

Expression profiling of ENKUR in different cancer types from UALCAN based on the TCGA database. A, ENKUR expression was significantly downregulated in clinical tumor tissues (n = 286) compared with normal tissues (n = 41) in colon adenocarcinoma. B, ENKUR expression was significantly downregulated in clinical tumor tissues (n = 471) compared with normal tissues (n = 50) in hepatocellular carcinoma. C, ENKUR expression was significantly downregulated in clinical tumor tissues (n = 515) compared with normal tissues (n = 59) in lung adenocarcinoma. D, ENKUR expression was significantly downregulated in clinical tumor tissues (n = 503) compared with normal tissues (n = 52) in lung squamous cell carcinoma. E, ENKUR expression was significantly downregulated in clinical tumor tissues (n = 497) compared with normal tissues (n = 52) in prostate adenocarcinoma. F, ENKUR expression was significantly downregulated in clinical tumor tissues (n = 505) compared with normal tissues (n = 59) in thyroid carcinoma. *P < .005. TCGA indicates The Cancer Genome Atlas.
ENKUR is Downregulated in CRC Cells
To validate the expression profiles of ENKUR, immunoblotting and qRT-PCR analyses were performed to examine ENKUR mRNA expression and ENKUR protein level in all the 6 CRC cell lines, HT29, HCT116, SW620, LS174T, RKO, and LOVO, as well as the normal human colon mucosal epithelial cell line NCM460. Varying levels of ENKUR expression were detected in different CRC cell lines. However, all the 6 CRC cell lines showed decreased expression of ENKUR compared with the human colon mucosal epithelial cell line (Figure 3). Among the 6 cell lines, ENKUR expression was the highest in LS174T cells and the lowest in SW620 cells (Figure 3).

ENKUR mRNA expression and ENKUR protein level in 6 CRC cell lines and the human colon mucosal epithelial cell line. A, ENKUR expression relative to β-ACTIN as revealed by qRT-PCR analysis. B, Western blot and related densitometric analysis of ENKUR protein level relative to β-ACTIN. Each bar represents the mean (SD) of 3 replicates. CRC indicates colorectal cancer; mRNA, messenger RNA; qRT-PCR, quantitative real-time polymerase chain reaction; SD, standard deviation.
Silencing of ENKUR Promotes the Proliferation, Migration, and Invasion of Human CRC Cells
To investigate whether ENKUR plays a role in the biological behavior of CRC cells, we silenced endogenous ENKUR expression in CRC cells LS174T and SW620 using a lentiviral vector carrying a specific shRNA. Colorectal cancer cells LS174T and SW620 transduced with lentiviral vectors carrying a scrambled shRNA were used as negative controls. The silencing efficiencies of 2 shRNA sequences were tested and the one with higher efficiency was selected for further transduction of CRC cells (Figure 4A). A considerable reduction of ENKUR expression was achieved in LS174T and SW620 cells after transduction (P < .005; Figure 4B). Silencing of ENKUR in CRC cell lines provoked accelerated cell growth and stronger migratory ability. In the colony formation assay and MTT assay, the LS174T-shRNA-ENKUR and SW620-shRNA-ENKUR cells demonstrated a significant growth advantage over the control cells (P < .001; Figures 5 and 6). Meanwhile, remarkably higher migration rates and more rapid wound closures were observed in LS174T-shRNA-ENKUR and SW620-shRNA-ENKUR cells, as revealed by transwell migration assay and wound-healing assay (P < .001; Figures 5 and 6). At 48 hours, the extent of wound closure was 70.0% (7.50%) and 96% (1.26%) for LS174T-shRNA-ENKUR and SW620-shRNA-ENKUR cells, respectively, but was only 28.3% (3.90%) and 36.8% (6.37%) for cells from the corresponding control groups.

Silencing and overexpression efficiencies of ENKUR in CRC cells. A, Silencing efficiencies of 2 different shRNAs (cloned in psi-LVRH1GP lentiviral plasmid) against ENKUR mRNA were measured in HEK293T cells. B, Expression level of ENKUR mRNA relative to β-ACTIN in ENKUR-underexpressing LS174T cells (shRNA) and control cells (NC) assessed by qRT-PCR. C, Expression level of ENKUR mRNA relative to β-ACTIN in ENKUR-overexpressing LS174T cells (ENKUR) and control cells (NC). D, Expression level of ENKUR mRNA relative to β-ACTIN in ENKUR-underexpressing SW620 cells (shRNA) and control cells (NC). E, Expression level of ENKUR mRNA relative to β-ACTIN in ENKUR-overexpressing SW620 cells (ENKUR) and control cells (NC). Each bar represents the mean (SD) of 3 replicates. *P < .005; **P < .001. CRC indicates colorectal cancer; mRNA, messenger RNA; MTT, 3-(4,5-dimethylthiazol-z-yl)-2,5-diphenyltetrazolium bromide; qRT-PCR, quantitative real-time polymerase chain reaction; SD, standard deviation; shRNA, short hairpin RNA.

Aberrant ENKUR expression leads to changes in cellular behavior of CRC cells LS174T. A, Cell proliferation capacity was significantly enhanced in ENKUR-underexpressing cells LS174T-shRNA-ENKUR compared with the control groups (NC) as determined by the MTT assay. B, Cell proliferation capacity was significantly reduced in ENKUR-overexpressing cells LS174T-ENKUR compared with the control groups. C, CRC cells LS174T-shRNA-ENKUR demonstrated increased migration and invasion through ECM as indicated by the transwell migration assay. Representative photographs (left) and quantification (right) are shown. The number of cells migrated through the ECM after 24 hours was counted in 5 randomly selected (×200) microscopic fields. D, Overexpression of ENKUR in LS174T cells results in reduced migration rate and invasion through ECM as revealed by the transwell migration assay. E, Silencing of ENKUR provoked stronger invasive ability in CRC cells as revealed by the cell wound-healing assay. Images were taken at 0 and 48 hours. F, Overexpression of ENKUR results in decreased invasive ability in CRC cells as revealed by the cell wound-healing assay. Images were taken at 0 and 48 hours. G, Silencing of ENKUR promoted cell growth as determined by the colony formation assay. Both the number of colonies and colony formation efficiency were significantly higher in LS174T cells transfected with ENKUR shRNA than those in the corresponding control groups. Formation of plate colonies in CRC cell lines after 2-week incubation was shown on the left. H, Overexpression of ENKUR inhibited LS174T cell growth as determined by the colony formation assay. Each error bar represents the mean (SD) of 3 replicate samples. *P < .005; **P < .001. CRC indicates colorectal cancer; ECM, extracellular matrix; MTT, 3-(4,5-dimethylthiazol-z-yl)-2,5-diphenyltetrazolium bromide; SD, standard deviation; shRNA, short hairpin RNA.

Aberrant ENKUR expression leads to changes in cellular behavior of CRC cells SW620. A, Cell proliferation capacity was significantly enhanced in ENKUR-underexpressing cells SW620-shRNA-ENKUR compared with the control groups (NC) as determined by the MTT assay. B, Cell proliferation capacity was significantly reduced in ENKUR-overexpressing cells SW620-ENKUR compared with the control groups. C, CRC cells SW620-shRNA-ENKUR demonstrated increased migration and invasion through extracellular matrix as indicated by the transwell migration assay. Representative photographs (left) and quantification (right) are shown. The number of cells migrated through the extracellular matrix (ECM) after 24 hours was counted in 5 randomly selected (×200) microscopic fields. D, Overexpression of ENKUR in SW620 cells resulted in reduced migration rate and invasion through extracellular matrix as revealed by the transwell migration assay. E, Silencing of ENKUR provoked stronger invasive ability in CRC cells as revealed by the cell wound-healing assay. Images were taken at 0 and 48 hours. F, Overexpression of ENKUR results in decreased invasive ability in CRC cells as revealed by the cell wound-healing assay. Images were taken at 0 and 48 hours. G, Silencing of ENKUR promoted cell growth as determined by the colony formation assay. Both the number of colonies and colony formation efficiency were significantly higher in SW620 cells transfected with ENKUR shRNA than those in the corresponding control groups. Formation of plate colonies in CRC cell lines after 2-week incubation was shown on the left. H, Overexpression of ENKUR inhibited SW620 cell growth as determined by the colony formation assay. Each error bar represents the mean (SD) of 3 replicate samples. *P < .005; **P < .001. CRC indicates colorectal cancer; MTT, 3-(4,5-dimethylthiazol-z-yl)-2,5-diphenyltetrazolium bromide; SD, standard deviation; shRNA, short hairpin RNA.
Overexpression of ENKUR Suppresses the Proliferation, Migration, and Invasion of Human CRC Cells
We next established stable ENKUR-overexpressing CRC cells LS174T-ENKUR and SW620-ENKUR. The CRC cells LS174T and SW620 transduced with empty lentiviral vectors were used as negative controls. Quantitative real-time PCR analysis proved a significant increase in ENKUR expression in LS174T-ENKUR and SW620-ENKUR cells (P < .001; Figure 4C). Overexpression of ENKUR significantly suppressed cell growth and inhibited cell proliferation ability (P < .001; Figures 5 and 6). Meanwhile, transwell migration assay and wound-healing assay revealed remarkably reduced migration rates and slower wound closure in LS174T-ENKUR and SW620-ENKUR cells (P < .001; Figures 5 and 6). At 48 hours, the extent of wound closure was only 6.2% (3.6%) and 5.7% (2.66%) for LS174T-shRNA-ENKUR and SW620-shRNA-ENKUR cells, respectively, but was 30.8% (4.60%) and 35.8% (7.93%) for cells from the corresponding control groups.
ENKUR Is Involved in the PI3K/Akt Signaling Pathway
We also investigated the level of protein markers associated with epithelial–mesenchymal transition (EMT) in ENKUR-overexpressing and ENKUR-underexpressing CRC cells. Western blot analyses showed that silencing of ENKUR led to a remarkably decreased level of E-cadherin and increased level of N-cadherin and the intermediate filament vimentin. On the contrary, overexpression of ENKUR resulted in increased E-cadherin level and decreased N-cadherin and vimentin levels (Figure 7A). In addition, silencing of ENKUR induced noninvasive epithelial cells to acquire a mesenchymal, spindle cell phenotype, suggestive of EMT (Figure 7B).

ENKUR associates with EMT in CRC cells. A, Silencing of ENKUR induces hallmarks of the EMT, including loss of E-cadherin and accumulation of N-cadherin and vimentin in CRC cells LS174T and SW620. B, The spindle cell phenotype of LS174T-shRNA-ENKUR and SW620-shRNA-ENKUR cells and the epithelial phenotype of their corresponding control groups. CRC indicates colorectal cancer; EMT, epithelial–mesenchymal transition; shRNA, short hairpin RNA.
Next, we investigated the possible pathway that may involve the function of ENKUR. Western blot analysis revealed remarkably decreased ENKUR protein levels in LS174T-shRNA-ENKUR and SW620-shRNA-ENKUR cells and increased ENKUR protein levels in LS174T-ENKUR and SW620-ENKUR cells (Figure 8A). Downregulation of ENKUR in CRC cells led to sustained phosphorylation of Akt. In contrast, overexpression of ENKUR caused significantly decreased phosphorylation levels of PI3K and Akt as compared with the control group (Figure 8A). Co-immunoprecipitation analysis revealed protein–protein interaction between ENKUR and PI3K in CRC cells LS174T, indicating that ENKUR was physically associated with PI3K (Figure 8B). The cytoplasmic fraction derived from LS174T cells was further analyzed for the location of interaction between ENKUR and PI3K. Our results showed direct interaction of the 2 proteins in the cytoplasm (Figure 8B).

Correlation between ENKUR and the PI3K/Akt signaling pathway. A, ENKUR silencing is associated with activation of the PI3K/Akt signaling pathway. Activation of Akt and PI3K can be observed in ENKUR-underexpressing CRC cell lines. Overexpression of ENKUR, on the contrary, led to reduced downstream activities of PI3K/Akt. B, Co-immunoprecipitation of total cell and the cytoplasmic fraction for ENKUR in CRC cell line LS174T. Lysates were immunoprecipitated with ENKUR antibody or control IgG and detected with PI3K p85 antibody on a Western blot, then immunoprecipitated with PI3K p85 antibody or control IgG and detected with ENKUR antibody on a Western blot. CRC indicates colorectal cancer; IgG, immunoglobulin G; PI3K indicates phosphoinositide 3-kinase.
Discussion
In patients with CRC, early-developed dissemination of metastatic cells often leads to tumor relapse after potentially curative resection, which signifies a poor prognosis. 15 Studies have shown that cancer metastasis arises as a consequence of multiple pathogenic steps, including invasion, detachment, adhesion, extravasation, proliferation, and growth in distant organs. 16 These steps are closely related to functions of many genes participating in intracellular signaling pathways. 8,17
The PI3K/Akt pathway is commonly activated in most cancers. 18 As a regulatory subunit of PI3K, p85 can increase PI3K signaling and initiate a signal transduction cascade that promotes cancer cell growth, survival, and metabolism which further keenly protect the cells from undergoing apoptotic mode of cell death. 19 -21 Similarly, an oncogenic function of protein members from the TRPC family has also been reported in lung cancer, breast cancer, ovarian cancer, and colorectal carcinoma. 22 -25 Given the importance of P13K/Akt signaling pathway and TRPC channels in the development and chemoresistance of various cancer types, it is possible that ENKUR, as an interaction partner of these signaling molecules, is also involved in the regulation of pathogenic processes that can finally lead to cancer initiation and progression.
In this study, we examined the expression profiles of ENKUR in CRC as well as some other cancer types in the TGCA and ONCOMINE databases. ENKUR expression at the mRNA level was starkly downregulated in clinical samples of CRC, liver hepatocellular carcinoma, lung adenocarcinoma, lung squamous cell carcinoma, prostate adenocarcinoma, and thyroid carcinoma as compared with normal nontumor samples (P < .05; Figures 1 and 2). Here, we showed that ENKUR mRNA expression and ENKUR protein level were downregulated in 6 CRC cell lines, HT29, HCT116, SW620, LS174T, RKO, and LOVO, as compared with the normal human colon mucosal epithelial cell line NCM460 (Figure 3). In addition, silencing of ENKUR promoted CRC cell proliferation, migration, and invasion, while overexpression of ENKUR led to the opposite effects (Figures 5 and 6). Together, our results implicated that ENKUR may have an antitumor function through regulating the proliferative, migratory, and invasive properties of CRC cells.
The pivotal role of PI3K/Akt signaling pathway in CRC has long been well established and inhibitors of PI3K/Akt signaling have been suggested as potential therapeutic agents of CRC. 19,26 Under normal conditions, PI3K/Akt activation is tightly controlled and dependent on extracellular and intracellular growth signals, which are the fundamental premises for proper cell function, while genetic aberrations can cause PI3K/Akt hyperactivation that ultimately leads to reduced apoptosis and enhanced cell growth. 26 Studies have shown that inactivating PI3K phosphorylation (Tyr458) reduced the expression of pAkt (Ser473), while activation of both PI3K and Akt phosphorylation contributes to cell growth and cancer metastasis. 27 In this study, we also investigated changes in the activation of PI3K/Akt signaling pathway after ENKUR overexpression or silencing. Increased activity of PI3K/Akt signaling pathway was detected in ENKUR-underexpressing CRC cells, while decreased PI3K/Akt activity was detected in ENKUR-overexpressing cells (Figure 8). Meanwhile, co-immunoprecipitation analysis revealed protein–protein interaction between ENKUR and PI3K p85. This confirms the previous finding that ENKUR binds to the p85 subunit of PI3K. 11 Our results also demonstrated that silencing of ENKUR expression in CRC cells leads to decreased level of E-cadherin and increased level of N-cadherin and vimentin accompanied with changes in cell phenotype, suggesting possible induction of EMT (Figure 7). Since the PI3K/Akt signaling pathway can affect EMT in a variety of ways to influence tumor aggressiveness, 28 silencing of ENKUR may cause an invasion-promoting effect on CRC cells through abnormal activation of PI3K/Akt signaling. On the other hand, studies have shown that the activation of several TRPC channels, including TRPC1, TRPC5, and TRPC6, is mediated through the PI3K/Akt signaling pathway. 29,30 It was also speculated that the physical interaction between ENKUR and TRPC channels is determined by channel opening and the subsequent influx of Ca2+. 11 Hence, ENKUR may also affect TRPC-mediated Ca2+ influx and its interaction with TRPC channels by interfering with PI3K/Akt signaling, preventing CRC cell migration and tumor metastasis. 31,32 However, so far the interaction between ENKUR and TRPC channels has only been reported in sperm. Whether ENKUR functions in affecting TRPC protein levels in tumor tissues remains unclear. Based on the current results, we postulate that ENKUR may be involved in the regulation of PI3K/Akt signaling pathway, which is among the major drivers for cellular proliferation, migration, and invasion of CRC.
In conclusion, our study provides evidence for a potential role of ENKUR in the regulation of cellular behavior of CRC. We propose that ENKUR is downregulated in CRC and silencing of ENKUR promotes CRC cell proliferation, invasion, and migration through possible involvement in the PI3K/Akt signaling pathway. However, the correlation between ENKUR and PI3K/Akt signaling, including how ENKUR can affect the activation status of PI3K/Akt signaling and whether activation of PI3K/Akt is dependent on alterations in ENKUR level, needs further investigation. Future molecular studies of the genetic basis underlying aberrant ENKUR expression and identification of direct targets of ENKUR will help unravel the intrinsic mechanism of ENKUR-mediated carcinostatic effect and develop novel gene therapies for patients with CRC.
Footnotes
Authors’ Note
Ye Gu is now affiliated with Department of Gastroenterology, Affiliated Hangzhou First People's Hospital, Zhejiang University School of Medicine. Hangzhou, Zhejiang, People's Republic of China.
Acknowledgments
The authors thank Josie Losh from Zhejiang University for English editing and proof reading of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Natural Science Foundation of China (31600257, 31800187), Zhejiang Provincial Natural Science Foundation of China (LQ19C020005), Zhejiang Provincial Public Welfare Technology and Application Research Project (2016C32022), Southeast University Fundamental Research Funding Project (3224008704), and the National Undergraduate Innovation and Entrepreneurship Training Program (201811842025).